
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization of urea hydrolysis in fresh human urine and inhibition by chemical addition†
Hannah
Ray
 *ab,
Daniella
Saetta
ab and
Treavor H.
Boyer
a
*ab,
Daniella
Saetta
ab and
Treavor H.
Boyer
a
aSchool of Sustainable Engineering and the Built Environment (SSEBE), Arizona State University, P.O. Box 873005, Tempe, Arizona 85287-3005, USA. E-mail: hgray3@asu.edu; Tel: +1 418 6899
bDepartment of Environmental Engineering Sciences, Engineering School of Sustainable Infrastructure & Environment (ESSIE), University of Florida, P.O. Box 116450, Gainesville, Florida 32611-6450, USA
First published on 3rd November 2017
Abstract
Urea hydrolysis is a chemical reaction that occurs in soils, the human body, and in wastewater urine diversion systems. The reaction, which transforms the urea in urine into ammonia and bicarbonate, results in ammonia volatilization and mineral scaling in bathroom fixtures, piping, and storage tanks. Urea hydrolysis is inhibited through different chemical additions that affect the function of the urease enzyme. Bench-scale batch experiments were performed where urea hydrolysis was simulated by adding Jack bean urease to both synthetic and real, fresh human urine. Urea hydrolysis was characterized by measurements of urea concentration, ammonia concentration, conductivity, and pH over time. Conductivity was positively correlated with ammonia concentration and negatively correlated with urea concentration making conductivity a simple, surrogate measurement for tracking the extent of urea hydrolysis. Acetic acid, citric acid, and vinegar were effective at inhibiting urea hydrolysis at concentrations varying from 3.2 × 101 to 1.6 × 102 meq L−1 in both synthetic and real, fresh urine as indicated by the conductivity and pH remaining constant throughout the experiments. Fluoride did not inhibit urea hydrolysis in real, fresh urine at concentrations of 3.2 × 10−2, 3.2 × 10−1, and 3.2 meq L−1. Ionic zinc and ionic silver were ineffective inhibitors of urea hydrolysis due to interactions with phosphate and chloride in urine, respectively, which caused precipitative loss of the metals from solution.
Water impactAs urine diversion gains support as a novel process to save potable water and recycle the nutrients in urine, the necessity for working fixtures becomes paramount. Precipitation and odor occur in the fixtures due to urea hydrolysis. Characterizing urea hydrolysis and investigating the inhibition of the urease enzyme assists in the operation and maintenance of the essential urine diversion fixtures. |
1. Introduction
The urease enzyme is found in the environment and in humans and occurs in many forms such as bacterial, plant, fungal, and soil.1,2 Urea hydrolysis via the urease enzyme is also the cause of operating problems with urine-diverting toilets and nonwater urinals.3–7 The reaction involves urea, an abundant compound found in the environment and human urine,8 hydrolyzing to form ammonia and bicarbonate resulting in an elevated pH of the surrounding soil or solution.9 Hydrolysis requires the urease enzyme, which is a metalloenyzme with two nickel ions per catalytic unit in the active site.10,11 Due to its bi-nickel active sites, one is responsible for binding and activating the substrate, urea, and the other is responsible for the binding and activating of the water molecule.12 Urease's bi-nickel active site selectively binds with the urea to stabilize a tetrahedral transition state in an orientation-specific mode.13 Through the binding, urea collapses into ammonia and is released from the bond along with the carbamate due to unfavorable interactions.12 Carbamate spontaneously decomposes to produce one molecule each of ammonia and carbonic acid. The latter is in equilibrium with its deprotonated form of bicarbonate.14Urea is a stable compound with a decomposition half-life in aqueous media of 3.6 years.12,15 However, hydrolysis of urea is 104 times faster when the urease enzyme is present.12 The time of urea hydrolysis depends on the amount of urease in the surrounding environment and, in the case of urine diversion systems, in the urine. The time of urea hydrolysis depends on the amount of urease in the urine and surrounding environment. Liu et al. (2008) reported that the pH and ammonium concentrations of fresh urine became stable at 72 and 84 h, respectively.9 However, the time of natural urea hydrolysis is dependent on the specific conditions of the environment and will vary in different environments.
Urease plays a vital role in the nitrogen cycle through nitrogen assimilation.2 However, extensive urea hydrolysis in soils can have a detrimental effect to the surrounding environment.1 Over fertilization of urea coupled with an abundance of urease in the soil can cause loss of nitrogen through ammonia volatilization. Ammonia toxicity and an elevated pH in the soil are toxic factors for seedling germination and seed growth.1,2 Eutrophication, addition of greenhouse gases, and acidification are other impacts that result from the volatilization of ammonia into the atmosphere.16 Urea hydrolysis can also occur within the human body causing serious health issues such as kidney stone formation, urolithiasis, ammonia encephalopathy, and urinary catheter encrustation.1,12Helicobacter pylori, an etiologic agent for gastric and peptic ulceration, contains the protein urease which it uses as a defense for colonization.14
Urine diversion processes are susceptible to the urea hydrolysis reaction due to urine's high urea content, and the ubiquitous presence of bacteria and bacterial urease on bathroom fixtures and on the human body.3,5,17 Urea hydrolysis in urine diversion systems is especially detrimental due to the formation of calcium and magnesium phosphates that form after hydrolysis has occurred in the urinals, pipes, and storage tanks of undiluted urine.4,5,9 The precipitates that form are hard minerals that are not easily removed.18,19 This results in frequent and expensive cleaning and maintenance. The ammonia produced also causes strong odors to persist in restrooms and storage tanks.19 An effective cleaning agent is one that not only cleans the urinal but prevents urea hydrolysis from occurring. However, common urinal maintenance has not been effective due to the inability of cleaning agents to inhibit the urease enzyme. If a cleaning agent inhibits the enzyme, then the reaction will not occur and the precipitation and smell should desist.
Metals, fluoride, and acids are the three types of inhibitors tested in this study. The mechanisms of inhibition can vary for each type of inhibitor: metals inhibit hydrolysis through binding with the enzyme's functional groups, which are necessary for the catalytic function of the enzyme;2,20,21 fluoride binds directly with the active nickel site;2,22,23 acids inhibit hydrolysis through alteration of the pH which affects the protonation states on the active site.24–26 Bases, thiols, boron compounds, sulfur compounds, and natural urease inhibitors such as garlic, onion, or cabbage extract, are just a few of the many different inhibitors for urease that previous research has identified.2,27,28 When comparing different inhibitors for urea hydrolysis, choosing the most effective inhibitor depends on the ultimate goals for urine diversion or urease inhibition in general. For example, when comparing acid and base addition, acid addition has the advantage of inhibition of ammonia volatilization in the fixtures and pipes while base addition is favorable if precipitation is the goal due to the elevated pH. Therefore, it is necessary to know the ultimate goal of a process to determine an effective inhibitor.
Previous research on urea hydrolysis has predominantly focused on soil science and gastric/urinary human health. There is very little research on urea hydrolysis and urea hydrolysis inhibition with regard to urine diversion systems. There is also no consensus on the most accurate and efficient way to track the progression of urea hydrolysis. For instance, measuring substrate, products, or changes in solution chemistry, and which measurements are the most effective for engineered systems and not just laboratory research. The soil science and human health fields have done previous research on the urease enzyme and the effectiveness of inhibitors in their respective environments. However, how urine and the urease inhibitors that previous research has recommended will interact together when mixed, and how this will affect urea hydrolysis is largely unknown.
The goal of this research was to provide an improved understanding of urea hydrolysis in human urine and its inhibition in the context of urine diversion systems. The specific objectives for this research were to (1) characterize urea hydrolysis in synthetic, fresh human urine through the measurement of urea, ammonia, conductivity, and pH; (2) inhibit urea hydrolysis in synthetic, fresh human urine via chemical addition; and (3) confirm the results for urea hydrolysis characterization and inhibition via chemical addition in real, fresh human urine. Batch experiments were performed where urea hydrolysis was simulated by addition of Jack bean urease to either synthetic or real, fresh human urine and the extent of hydrolysis was monitored though different measurements. Once hydrolysis was measured and understood, different chemicals were added during the batch experiments to inhibit urea hydrolysis and the inhibition was quantified.
2. Materials and methods
2.1. Fresh urine
Both synthetic, fresh human urine and real, fresh human urine were used in this research. The synthetic, fresh human urine used for experiments was prepared based on previous literature.29,30 The composition of this urine is reported in Table S1† with the concentrations of individual species in the urine reported in Table S2 (ESI†). The pH of the synthetic, fresh urine was adjusted to 6 using a sodium hydroxide solution.30,31 The real, fresh human urine was collected by volunteers in plastic collection trays, without dilution, stored in bottles in the refrigerator, and then combined before the start of the experiment. The real, fresh urine was collected and utilized for the experiment within 24 h and the pH was tested to ensure it was in the range for fresh urine reported in the literature (pH 6–6.5).30,31 The age range of the donors varied from 18–45 and included females and males. Human urine collection was approved as exempt by the University of Florida Institutional Review Board.2.2. Urease
Jack bean urease (CAS 9002-13-5, Fisher Scientific) was used as the source of urease in all experiments at a dose of 0.533 g L−1. This was based on preliminary experiments where four concentrations of urease were used to simulate hydrolysis, 0.267, 0.533, 0.800, and 1.07 g L−1 of urine (refer to Fig. S1 in the ESI†).2.3. Inhibiting chemicals
Seven different chemicals, silver nitrate (CAS 7761-88-8, Fisher Scientific), zinc nitrate (CAS 7779-88-6, Fisher Scientific), sodium fluoride (CAS 7681-49-4, Fisher Scientific), glacial acetic acid (CAS 64-19-7, Fisher Scientific), vinegar, citric acid (CAS 77-92-9, Fisher Scientific), and sulfuric acid (CAS 7664-93-9, Fisher Scientific), were used to inhibit urea hydrolysis. Three different types of vinegar were used: white distilled vinegar (White House White Distilled Vinegar) which has a 5% acetic acid content, cleaning vinegar (White House All Nature Cleaning Vinegar Lemon Scent) which has a 6% acetic acid content, and champagne wine vinegar (Colavita Champagne Wine Vinegar) which has a 7% acetic acid content.2.4. Experimental methods
| Inhibitora | Synthetic urine, urease added before inhibitor | Synthetic urine, urease added after inhibitor | Real urine, urease added after inhibitor |
|---|---|---|---|
| a All concentrations represent the concentration of the inhibitor in the beaker. | |||
| Acetic acid (meq L−1) | 1.6 | 1.6 | 3.2 × 101 |
| 1.6 × 101 | 1.6 × 101 | ||
| — | 3.2 × 101 | — | |
| — | 8.1 × 101 | 8.1 × 101 | |
| 1.6 × 102 | 1.6 × 102 | 1.6 × 102 | |
| Sulfuric acid (meq L−1) | 1.6 | 1.6 | — |
| 1.6 × 101 | 1.6 × 101 | — | |
| — | 3.2 × 101 | — | |
| — | 8.1 × 101 | — | |
| 1.6 × 102 | 1.6 × 102 | — | |
| Citric acid (meq L−1) | — | 3.2 × 101 | 3.2 × 101 |
| — | 8.1 × 101 | 8.1 × 101 | |
| — | 9.7 × 101 | 9.7 × 101 | |
| Vinegar (meq L−1) | — | 2.8 × 101 | — |
| — | 3.4 × 101 | 3.4 × 101 | |
| — | 3.9 × 01 | — | |
| Zinc nitrate (meq L−1 as Zn2+) | — | 2.2 × 10−1 | — |
| — | 2.2 | — | |
| — | 2.0 × 101 | — | |
| Silver nitrate (meq L−1 as Ag+) | — | 3.2 × 10−4 | — |
| — | 3.2 × 10−3 | — | |
| — | 3.2 × 10−2 | — | |
| Sodium fluoride (meq L−1 as F−) | — | 3.2 × 10−2 | 3.2 × 10−2 |
| — | 3.2 × 10−1 | 3.2 × 10−1 | |
| — | 3.2 | 3.2 | |
2.5. Analytical methods
The pH and conductivity experiments were performed in batch triplicate experiments using an Orion Dual Star Multiparameter Meter, an Orion 9156BNWP Combination pH probe, and Orion Star A212 conductivity probe. The pH and conductivity were both calibrated following the instructions detailed in the pH probe and conductivity manuals. The ammonia concentration experiments were performed using an Orion Dual Star Multiparameter Meter, the Orion Combination pH probe, and Standard Ammonia Ion Selective Electrode and concentrations were measured as NH3 and then converted to nitrogen concentrations for reporting. The urea concentration was measured using UV absorbance on a U-2900 UV-visible spectrophotometer (Hitachi High Technologies) and 1 cm quartz cuvette. The Watt and Chisp (1954) method that utilized a modified Ehlich reagent detailed in With et al. (1961) was followed.32 All experiments were conducted in triplicate to ensure precision with analytical measurements made singly. The mean and standard deviation of the triplicate data was calculated and represented on the graphs.2.6. Data analysis
The theoretical extent of urea hydrolysis was calculated for the ammonia concentration experiments and urea concentration experiments by dividing the final ammonia and urea concentration readings at 240 min by the total nitrogen (TN) in the synthetic urine.33 Urea hydrolysis completes when all of the urea is converted to ammonium/ammonia which would be reported as 100% extent of urea hydrolysis. However, in a natural system, 100% extent of hydrolysis may not occur due to the lack of enough urease available to convert all of the urea to ammonia/ammonium. Tang et al. (2013) reported 89.5% extent of hydrolysis as measured by ammonium and total nitrogen over a 160–240 h period with seawater addition to real, fresh urine as a urease source.33 Two samples (one from the control and one from the sample simulating hydrolysis) at the end of two different urea concentration experiments were analyzed for TN by a TOC-TN (Shimadzu TOC-TN). The average of the samples was 6210 mg L−1 as N and the average of the controls was 6740 g L−1 as N.Visual MINTEQ 3.1, a chemical equilibrium software, was used to determine saturation indices for the chemical addition experiments. The components of urine were entered at their appropriate concentrations as well as the chemical inhibitors. Saturation indices provided by the software were used to determine oversaturation of minerals and thermodynamically favorable precipitations that would occur within the solutions.
3. Results and discussion
3.1. Characterization of urea hydrolysis
The urea hydrolysis equation progresses as the substrate, urea, is hydrolyzed to form ammonia and bicarbonate, which results in an elevated pH of the solution and an increase in ionic strength/charged species due to the transformation of neutral urea to charged compounds.| NH2(CO)NH2 + 3H2O → 2NH4+ + HCO3− + OH− | (1) |
Fig. 1 shows the results of the measurements recorded as urea hydrolysis was simulated in both synthetic, fresh urine and real, fresh urine. Urea, which can be seen in eqn (1), is the substrate in urea hydrolysis so its concentration should decrease as it is transformed. Fig. 1(a) shows a decreasing trend for urea concentration over the 240 min experiment. Fig. 1(b) shows the ammonia concentration increasing throughout the entire experiment in both synthetic and real, fresh urine following the opposite trend as urea concentration because it is the product of urea hydrolysis. The extent of hydrolysis for the ammonia monitoring during urea hydrolysis experiments was 20% while the extent of hydrolysis for the urea monitoring during urea hydrolysis experiments was 25%. Thus, the ammonia concentration increased a relatively similar amount that the urea concentration decreased in agreement with eqn (1).
 | ||
| Fig. 1 Urea hydrolysis simulation utilizing 0.533 g L−1 of Jack bean urease. Open squares represent experiments using synthetic, fresh urine and solid squares represent experiments using real, fresh urine. 4 h duration, mixing at 350 rpm. pH and conductivity experiments were performed using 75 mL of either synthetic or real, fresh urine. Ammonia and urea experiments were performed using 500 mL of urine. (a) pH (b) conductivity (c) ammonia concentration (d) urea concentration data are mean ± one standard deviation for triplicate samples. | ||
The conductivity, a measurement of the sum of the current-carrying capacity of each ion in solution, was expected to follow an increasing trend for hydrolysis since the reaction produces the ammonium and bicarbonate ions from neutral urea which is depicted in eqn (1). In Fig. 1(c), the conductivity increased throughout the entire experiment in both synthetic and real, fresh urine following a similar trend to the ammonia concentration. The pH was expected to increase as well due to the production of hydroxide through the hydrolysis reaction (see eqn (1)). Fig. 1(d) shows the pH increased from 6 to 9, but the pH plateaued relatively early in the experiment, around 90 min in both the synthetic and real, fresh urine. The pH plateaus due to the bicarbonate equilibrium and the ammonia/ammonium in the system, which buffers the system and prevents the pH from increasing above pH 9 although the hydrolysis reaction has not reached completion.34
From these experiments, it can be concluded that urea concentration and ammonia concentration are accurate measurements of urea hydrolysis because they are the substrate and product, respectively, of the reaction. However, these measurements require more time and effort than pH and conductivity, and are not as easily automated as pH and conductivity. Hence, ammonia concentration was plotted against pH and conductivity to see if a correlation could be made that would support either pH and/or conductivity as surrogate measurements for tracking urea hydrolysis. Fig. 2(a) shows the correlation between ammonia concentration and pH in both real and synthetic, fresh urine. In the beginning of hydrolysis, which corresponds to acidic pH and lower ammonia concentration, there was a trend as pH increased so did ammonia concentration. However, once the pH reached 9, there was not a correlation between ammonia concentration and pH due to buffering of the system. This trend was observed for both the synthetic, fresh urine and the real, fresh urine. Therefore, pH is a useful measurement for testing if urea hydrolysis is occurring and whether or not the urine is fresh or partially hydrolyzed but it is not an effective measurement for tracking the progression of the reaction due to its ability to be buffered, which can cause the reaction to appear completed.
 | ||
| Fig. 2 (a) Correlation between pH and ammonia concentration. (b) Correlation between conductivity and ammonia concentration. | ||
Fig. 2(b) shows the correlation between ammonia concentration and conductivity. For both the synthetic, fresh urine and real, fresh urine, as the ammonia concentration increased there was an increase in conductivity. Conductivity is a simple and quick measurement to make, and is an effective surrogate measurement of hydrolysis due to its correlation to ammonia concentration and the understanding that if ammonia concentration increases, it is due to urea hydrolysis. Grau et al. (2012) used conductivity as an effective measurement to control dosing in struvite precipitation reactors which shows ability of conductivity to be applied in urine diversion systems as a control measurement.35 Moving forward into the inhibition phase of the project, pH was measured to understand if hydrolysis was occurring and conductivity was used to track the extent of hydrolysis and if inhibition was effective or not. In this work, inhibition is quantified indirectly by whether or not urea hydrolysis occurred with its consequent changes in solution chemistry.
3.2. Inhibition of urea hydrolysis through chemical additions
Fig. 3 shows the conductivity of two different types of synthetic, fresh urine mixed with ionic zinc and two different types of synthetic, fresh urine mixed with ionic silver as urea hydrolysis was simulated (see Fig. S2 in the ESI† for pH data). Two different types of synthetic, fresh urine were created for each inhibitor due to chemical equilibrium calculations (using Visual MINTEQ 3.1) that showed the thermodynamically favorable precipitation in urine of ionic zinc with phosphate and ionic silver with chloride (refer to Table S3† for saturation indices). This is problematic due to the presence of phosphate and chloride in urine, which would lead to the ionic metals precipitation out of solution. Once precipitated out of solution, the ionic metals are no longer available to inhibit the urease enzyme and would thus be ineffective urease inhibitors. Testing the ionic zinc and ionic silver in synthetic, fresh urine with and without either phosphate or chloride would help determine the role of phosphate and chloride on the ionic metal's effectiveness in urine (i.e. if there is precipitation or not) and therefore if ionic metals are practical options for urine diversion processes due to the presence of phosphate and chloride in fresh urine. Thus, ionic zinc, in the form of soluble zinc nitrate, was added to the standard phosphate-containing synthetic, fresh urine and urease, and the inhibition measured by conductivity and pH was investigated. The experiment was then repeated except the synthetic, fresh urine was altered where the sodium phosphate was replaced by sodium chloride and the inhibition was investigated. The ionic silver was tested in the same manner as the ionic zinc, where the ionic silver, in the form of soluble silver nitrate, was tested in the standard chloride-containing synthetic, fresh urine as well as in the altered synthetic, fresh urine where all chloride-containing compounds were replaced by corresponding nitrate compounds.
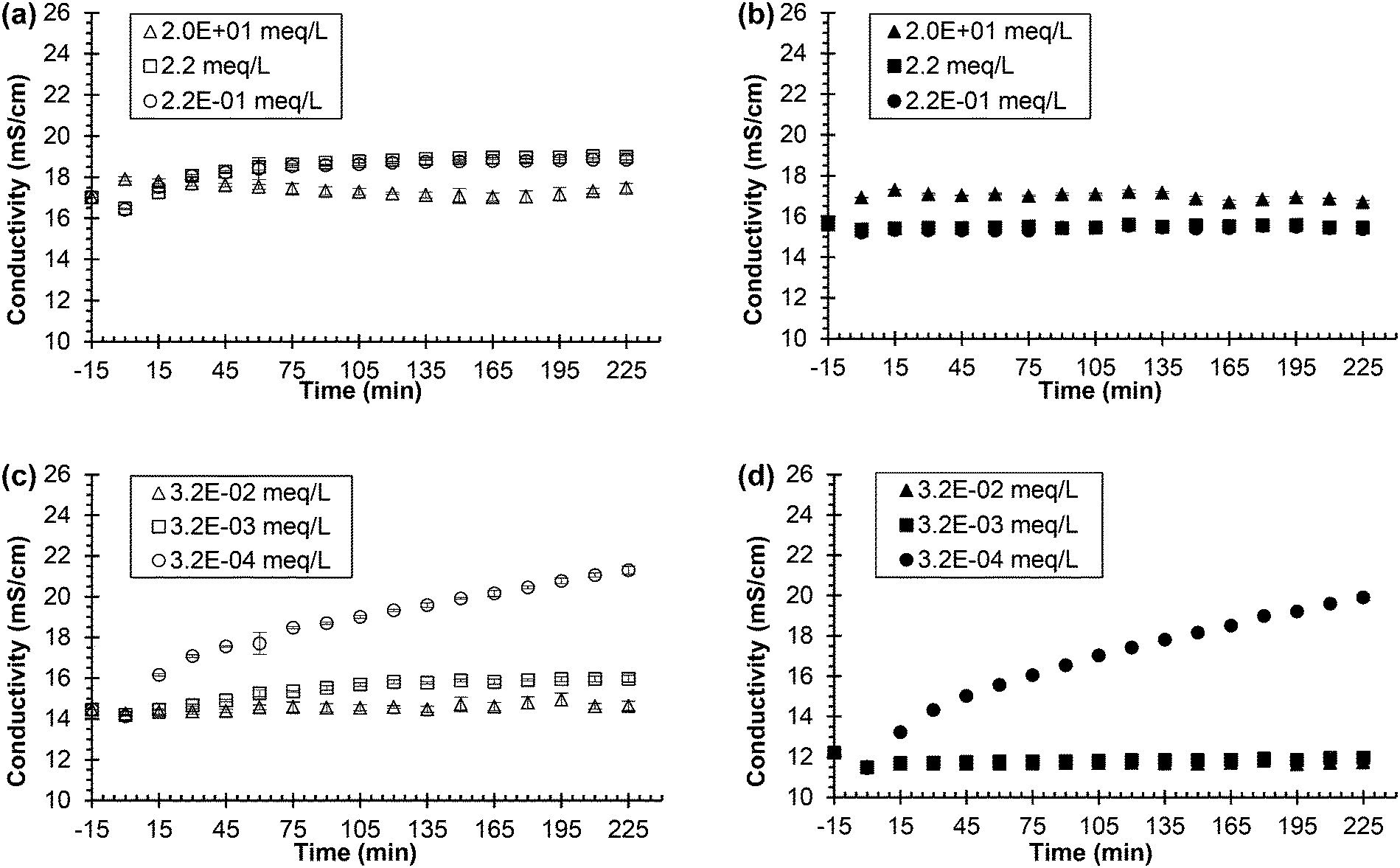 | ||
| Fig. 3 Urea hydrolysis inhibition utilizing 0.533 g L−1 of Jack bean urease and zinc nitrate or silver nitrate as the inhibitor. Legend indicates the concentration of added chemical inhibitor in the different synthetic, fresh urine types. All experiments performed using 75 mL of synthetic, fresh urine. Experiments start with urine and chemical inhibitor mixing at time equal to −15 min. Time equal to 0 min represents the time when urease is added. (a) Conductivity vs. time for the zinc nitrate in synthetic, fresh urine containing phosphate and (b) conductivity vs. time for zinc nitrate in synthetic, fresh urine with phosphate replaced by sodium chloride. (c) Conductivity vs. time for the silver nitrate in synthetic, fresh urine containing chloride and (d) conductivity vs. time for silver nitrate in synthetic, fresh urine with chloride replaced by nitrate. The additional sodium chloride concentration used in the zinc nitrate experiment was determined by adding the corresponding chloride molar concentration of phosphate that was removed. For the silver nitrate experiment, all chloride compounds were replaced with corresponding nitrate compounds (i.e., sodium nitrate for sodium chloride). Data are mean ± one standard deviation for triplicate samples. Corresponding pH plots in ESI,† Fig. S1. | ||
Fig. 3(a), the synthetic urine with phosphate, shows the conductivity of urine containing 2.2 and 2.2 × 10−1 meq L−1 Zn2+ increasing throughout the entire experiment inferring hydrolysis. Thus, the ionic zinc at the middle and lower concentrations was unable to inhibit hydrolysis, which is supported by the pH increase from 6 to 9 for both concentrations of Zn2+ (see Fig. S2(a)†). However, the conductivity of urine for the highest concentration of 2.0 × 101 Zn2+ stayed constant over the 240 min. The pH initially decreased at the beginning of the experiment from 6 to ∼4 due to the precipitation of zinc phosphate, which is supported by a high saturation index for Zn3(PO4)2·4H2O (see Table S3†) derived by the equilibrium calculations, causing the solution to become more acidic. The pH remained constant after the initial decrease for about 160 min and slowly increased to 6.9, inferring inhibition of urea hydrolysis was effective for this concentration of ionic zinc.
The results for the synthetic urine without phosphate, Fig. 3(b) and Fig. S2(b),† show the conductivity remaining constant for all concentrations of ionic zinc and the pH increasing very little, ∼0.5, for all concentrations of ionic zinc throughout the experiment, besides the initial increase in pH at the beginning of the experiment due to the unstable pH of the urine. These results confirm that the ionic zinc was effective at inhibiting urea hydrolysis as measured by conductivity and pH when the phosphate was removed. Thus, ionic zinc was more effective at inhibiting the urease enzyme in the absence of phosphate than presence of phosphate presumably due to loss of ionic zinc via precipitation.
The pH for the urine without phosphate in Fig. S2(b)† increased very little for all three concentrations of the ionic zinc, the 2.0 × 101 meq L−1 Zn2+ remained fairly constant at pH ∼ 8.7, the 2.2 meq L−1 Zn2+ remained fairly constant at pH ∼ 7.4, and the 2.2 × 10−1 meq L−1 Zn2+ remained fairly constant at pH ∼ 6.6. The elevated pH would normally infer hydrolysis. However, this can be explained by the removal of phosphate from the synthetic urine. Phosphate provides pH buffering in the synthetic urine and with that removed there is nothing to buffer changes in the pH. At this condition the urine had an unstable pH and changes in pH occurred when the pH was adjusted to 6 using sodium hydroxide. Thus, when the experiment began and zinc nitrate and urease were added, the pH initially increased but hydrolysis was not occurring as confirmed by the stable conductivity and pH over time and the absence of ammonia odor that is normally observed.
Fig. 3(c), the synthetic urine with chloride, shows the conductivity of the lowest concentration of ionic silver, 3.2 × 10−4 meq L−1 Ag+, increased throughout the experiment suggesting hydrolysis. The pH, shown in Fig. S2(c),† increased from 6 to 9 confirming hydrolysis for the lowest concentration of ionic silver, 3.2 × 10−4 meq L−1 Ag+. The conductivity of urine for the 3.2 × 10−3 meq L−1 Ag+ concentration increased very slowly over the experiment by less than 2 mS cm−1. The pH also slowly increased over the entirety of the experiment for the 3.2 × 10−3 meq L−1 Ag+ concentration confirming that while hydrolysis is occurring to some extent, there is also inhibition occurring as the conductivity and pH did not increase at the rate of the lowest dose of ionic silver, 3.2 × 10−4 meq L−1 Ag+. The conductivity and pH of the 3.2 × 10−2 meq L−1 Ag+ concentration remained constant throughout the entire experiment, which implies urea hydrolysis inhibition was effective at this concentration.
Thus, the highest concentration of ionic silver, 3.2 × 10−2 meq L−1 Ag+, was effective for inhibition, the middle concentration, 3.2 × 10−3 meq L−1 Ag+, greatly slowed the rate of hydrolysis but was not effective for complete inhibition, and the lowest concentration, 3.2 × 10−4 meq L−1 Ag+, was ineffective for inhibition.
The conductivity and pH for the experiments where chloride was removed are shown in Fig. 3(d) and Fig. S2(d).† For the 3.2 × 10−4 meq L−1 Ag+ concentration, the conductivity and pH increased quickly over time implying inhibition was ineffective. The conductivity and pH remained constant for both the 3.2 × 10−3 and 3.2 × 10−2 meq L−1 Ag+ concentrations inferring inhibition was effective for both concentrations.
The lowest dose of ionic silver was not sufficient to inhibit as it was ineffective for inhibition when chloride was present and when chloride was removed. The highest dose of ionic silver was sufficient to inhibit regardless if chloride was present or not. However, the middle concentration, 3.2 × 10−3 meq L−1 Ag+, shows the impact of chloride on silver as inhibition was less effective when chloride was present compared to when chloride was removed due to presumed precipitation.
Previous literature reports that ionic silver is the most effective metal inhibitor of urease and that various other metals such as ionic zinc are also effective albeit to a lesser extent.2,21,28,37 In the context of synthetic urine, ionic silver was also more effective than ionic zinc as indicted by a lower concentration of silver than zinc being able to maintain constant conductivity and constant pH. However, the results in Fig. 3 and Fig. S2† also show that previous research on urea hydrolysis and urease from other fields such as soil chemistry, while helpful, cannot be directly applied to human urine without first considering the composition of urine and unintended chemical reactions. For instance, given that ionic silver was less effective in synthetic urine containing chloride than synthetic urine in which chloride was replaced with nitrate, it was decided not to test ionic silver addition to real, fresh urine because of the high concentration of chloride.
 | ||
| Fig. 4 Urea hydrolysis inhibition utilizing 0.533 g L−1 of Jack bean urease and sodium fluoride as the inhibitor. Legend indicates the concentration of added chemical inhibitor in either synthetic or real, fresh urine. All experiments were performed using 75 mL of either synthetic or real, fresh urine. Experiments start with urine and the chemical inhibitor mixing at time equal to −15 min. Time equal to 0 min represents the time when urease is added. (a) Conductivity vs. time in synthetic, fresh urine and (b) conductivity vs. time in real, fresh urine. Data are mean ± one standard deviation for triplicate samples. Corresponding pH plots in ESI,† Fig. S2. | ||
Further analysis suggested that the metabolites in urine and thermodynamically favorable precipitation of calcium fluoride played a role in the effectiveness of fluoride as an inhibitor. Metabolites, which exist in urine in concentrations similar to the concentration of fluoride inhibitor used (refer to Table S4†),38 could interfere with the fluoride's ability to inhibit the enzyme. Precipitation of calcium fluoride (refer to Table S3† for saturation indices) would also remove fluoride from solution and thus cause the inhibitor to be ineffective. Additional experiments were performed in synthetic, fresh urine where calcium was removed (refer to Fig. S4†) as well as an experiment where metabolites were added to the synthetic urine (refer to Fig. S5†). The experimental data did not support the original ideas and suggest that a combination of factors affect the effectiveness of the fluoride in urine due to the complex nature of urine.
Fig. 5(a)–(f) show the trend of conductivity remaining constant at both synthetic and real, fresh urine conditions for most of the experiment for all concentrations of acids. The only exception is the lowest concentration of acetic acid for the synthetic, fresh urine experiment, which slowly increased in conductivity as the experiment progressed. The real, fresh urine results for all three acids at all three concentrations were effective for inhibition by keeping the conductivity and pH constant (see Fig. S8† for pH data). At the start of each experiment, the acid addition results in an initial decrease in the pH of the solution. The acidified pH remained constant for each acid for the low, medium, and high concentrations further implying inhibition. The synthetic, fresh urine and real, fresh urine experiments followed very similar trends. The constant conductivity infers that hydrolysis was being inhibited due to the lack of production of charge that would normally occur during hydrolysis. The pH also remaining constant further supports the inhibition by no addition of hydroxide into solution that comes from hydrolysis.
 | ||
| Fig. 5 Urea hydrolysis inhibition utilizing 0.533 g L−1 of Jack bean urease and acid as the inhibitor. Legend indicates the concentration of added chemical inhibitor in either synthetic or real, fresh urine. All experiments were performed using 75 mL of either synthetic or real, fresh urine. Experiments start with urine and the chemical inhibitor mixing at time equal to −15 min. Time equal to 0 min represents the time when urease is added. Open symbols represent experiments using synthetic, fresh urine and solid symbols represent experiments using real, fresh urine. Each row represents a different chemical acid inhibitor. (a) and (b) acetic acid, (c) and (d) 6% cleaning vinegar, (e) and (f) citric acid. Data are mean ± one standard deviation for triplicate samples. Corresponding pH plots in ESI,† Fig. S3. | ||
Fig. S9† shows the conductivity and pH results for the three different types of vinegar used in synthetic, fresh urine. The lowest concentration vinegar, the distilled vinegar, started hydrolyzing around 105 min, as determined by the increasing pH and conductivity. The middle and higher concentration vinegars, the cleaning vinegar and white wine vinegar respectively, were effective inhibitors for urea hydrolysis as determined by the constant conductivity and pH.
Fig. S6(a–d)† compares the effectiveness of acetic acid and sulfuric acid over a concentration range of three orders of magnitude where the two acids displayed similar results for the highest and lowest concentrations, 1.6 and 1.6 × 102 meq L−1. The concentration range of the inhibitors was decreased to a factor of 10, 3.2 ×101–1.6 × 102 meq L−1, which can be seen for both the conductivity and pH for sulfuric acid in Fig. S6(e–f).† This same concentration range for acetic acid can be seen in Fig. 5(a) for the conductivity results and in Fig. S8(a)† for the pH results. These results show acetic acid as a more effective inhibitor than sulfuric acid at lower concentrations due to acetic acid being a stronger acid compared to sulfuric acid.
Acetic acid, vinegar, and citric acid were the most desirable options considering safety, low-cost, and easily accessible. For instance, vinegar, citric acid, and acetic acid are commercially available products. Vinegar is a cleaning agent that can be bought at any supermarket for a low price while citric acid is used in many cleaning products as well as a food preservative and can also be bought at a supermarket. USP grade citric acid can be bought on a larger scale for $1.03–3.20 per kg depending on the desired form, and glacial acetic acid can be bought commercially at $1.50 per kg.40
The mechanism of acid inhibition of urease can be explained by the protonation state of functional groups on the enzyme. Jack bean urease has an optimal pH range of 7–7.5.2 According to Krajewska and Ciurli (2005), the urease enzyme has two ionizable groups on the active site responsible for catalysis with pKa1 = 8.67 and pKa2 = 5.34.25 pKa1 is attributed to the Ni–Ni bridging hydroxide and pKa2 is attributed to an imidazole of a histidine residue.24 For optimum catalysis, the groups must be in opposite protonation states with pKa1 in a protonated state and pKa2 in a deprotonated state.25 Each acid lowered the pH to the range of 3–4.5, which would put both ionizable groups in a protonated state and thus hinder the enzyme's ability to function optimally. Following similar logic, Randall et al. (2016) demonstrated that raising the pH above 11 was an effective mode to inhibit urea hydrolysis.26 At pH 11, both ionizable groups would be in a deprotonated state also hindering urease's ability to function, similar to lowering the pH. The effect of pH on the active site of the urease enzyme is further supported by acid addition experiments in which the enzyme needed to enter a low pH environment for inhibition to be effective (see Fig. S5 in ESI†). When acid was added 15 min after the urine and urease had been mixing, inhibition was ineffective. In this case, the ionizable groups are not accessible as they are already taking part in hydrolysis and thus cannot be altered by the pH to inhibit hydrolysis.
Urease inhibition by acid addition is a reversible form of inhibition. After 120 min of stable conductivity and pH, the conductivity and pH began to increase for 3.2 × 101 meq L−1 of acetic acid (refer to Fig. 5) and 2.8 × 101 meq L−1 of distilled vinegar (see Fig. S9 in ESI†) inferring hydrolysis was inhibited but then over time hydrolysis began to occur. Thus, if the pH were to be raised back to the optimal range for the urease function (i.e., pH 7–7.5), hydrolysis is presumed to occur.
4. Conclusions
- Conductivity, due to its correlation with ammonia concentration, was an effective, real time measurement for tracking urea hydrolysis.- Effective urease inhibitors for urine diversion processes were identified as chemicals that maintained constant conductivity and pH in fresh urine in the presence of the urease enzyme. The results from this work showed the following order of decreasing effectiveness: citric acid > acetic acid > vinegar > sulfuric acid > ionic silver > ionic zinc > sodium fluoride.
- Of the chemical inhibitors tested in this study, acids proved to be the most effective inhibitors that can be easily implemented into the daily maintenance of urine diversion systems. Previous soil science and medical research on urea hydrolysis cannot be applied directly to urine diversion systems due to the composition of urine that leads to precipitation of metals with chloride and phosphate. Other metals known to inhibit urease would be expected to precipitate with chloride, phosphate, sulfate, and/or carbonate, and therefore not be effective inhibitors. Similarly, ligands such as fluoride can interact with constituents in urine (e.g., calcium) rendering the ligand ineffective as an inhibitor.
- The results displayed the validity and reliability of using synthetic, fresh human urine to simulate urea hydrolysis in real, fresh human urine. Synthetic, fresh urine and real, fresh urine followed similar trends consistently throughout the experiments.
- Inhibition of hydrolysis was most effective when the urease entered into a low pH environment. This was determined through experiments in which the urease was added both 15 min before the acid inhibitor and 15 min after the acid inhibitor. Therefore, having nonwater urinals release the acid starting early morning and then periodically, based on usage throughout the day could be an effective strategy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was made possible by USEPA grant 83556901. Its contents are solely the responsibility of the grantee and do not necessarily represent the official views of the USEPA. Further, USEPA does not endorse the purchase of any commercial products or services mentioned in the publication.References
- H. L. T. Mobley and R. P. Hausinger, Microbiol. Rev., 1989, 53, 85–108 CAS.
- B. Krajewska, J. Mol. Catal. B: Enzym., 2009, 59, 9–21 CrossRef CAS.
- K. M. Udert, T. A. Larsen and W. Gujer, Water Sci. Technol.: Water Supply, 2003, 3, 71–78 CAS.
- K. M. Udert, T. A. Larsen, M. Biebow and W. Gujer, Water Res., 2003, 37, 2571–2582 CrossRef CAS PubMed.
- T. Ohki, N. Nishikawa, T. Hasegawa, T. Okano and Y. Tanizawa, J. Surfactants Deterg., 2010, 13, 19–26 CrossRef CAS.
- H. Jonsson and B. Vinneras, Water Sci. Technol., 2007, 56, 71–76 CrossRef CAS PubMed.
- J. F. Zhang, A. Giannis, V. W. C. Chang, B. J. H. Ng and J. Y. Wang, J. Air Waste Manage. Assoc., 2013, 63, 472–481 CAS.
- P. M. Glibert, J. Harrison, C. Heil and S. Seitzinger, Biogeochemistry, 2006, 77, 441–463 CrossRef CAS.
- Z. G. Liu, Q. L. Zhao, K. Wang, D. J. Lee, W. Qiu and J. F. Wang, J. Environ. Sci., 2008, 20, 1018–1024 CrossRef CAS.
- N. E. Dixon, C. Gazzola, R. L. Blakeley and B. Zerner, J. Am. Chem. Soc., 1975, 97, 4131–4133 CrossRef CAS PubMed.
- M. J. Todd and R. P. Hausinger, J. Biol. Chem., 1989, 264, 15835–15842 CAS.
- Z. Amtul, R. Atta ur, R. A. Siddiqui and M. I. Choudhary, Curr. Med. Chem., 2002, 9, 1323–1348 CrossRef CAS PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Miletti, S. Ciurli and S. Mangani, Structure with Folding & Design, 1999, 7, 205–216 CAS.
- H. L. T. Mobley, M. D. Island and R. P. Hausinger, Microbiol. Rev., 1995, 59, 451–480 CAS.
- B. Zerner, Bioorg. Chem., 1991, 19, 116–131 CrossRef CAS.
- K. C. Cameron, H. J. Di and J. L. Moir, Ann. Appl. Biol., 2013, 162, 145–173 CrossRef CAS.
- K. Oki, K. Washio, D. Matsui, S. Kato, Y. Hirata and M. Morikawa, Biosci., Biotechnol., Biochem., 2010, 74, 583–589 CrossRef CAS PubMed.
- M. Maurer, W. Pronk and T. A. Larsen, Water Res., 2006, 40, 3151–3166 CrossRef CAS PubMed.
- S. Blume and M. Winker, Water Sci. Technol., 2011, 64, 579–586 CrossRef CAS PubMed.
- B. Krajewska, J. Enzyme Inhib. Med. Chem., 2008, 23, 535–542 CrossRef CAS PubMed.
- G. Behbehani, A. A. Saboury, A. Taherkhani, L. Barzegar and A. Mollaagazade, J. Therm. Anal. Calorim., 2011, 105, 1081–1086 CrossRef.
- N. E. Dixon, R. L. Blakeley and B. Zerner, Can. J. Biochem., 1980, 58, 481–488 CrossRef CAS PubMed.
- A. A. Saboury and A. A. Moosavi-Movahedi, J. Enzyme Inhib., 1997, 12, 273–279 CrossRef CAS PubMed.
- B. Krajewska, J. Mol. Catal. B: Enzym., 2016, 124, 70–76 CrossRef CAS.
- B. Krajewska and S. Ciurli, Plant Physiol. Biochem., 2005, 43, 651–658 CrossRef CAS PubMed.
- D. G. Randall, M. Krahenbuhl, I. Kopping, T. A. Larsen and K. M. Udert, Water Res., 2016, 95, 361–369 CrossRef CAS PubMed.
- L. V. Modolo, A. X. de Souza, L. P. Horta, D. P. Araujo and A. de Fatima, J. Adv. Res., 2015, 6, 35–44 CrossRef CAS PubMed.
- L. S. B. Upadhyay, Indian J. Biotechnol., 2012, 11, 381–388 CAS.
- K. A. Landry, P. Sun, C. H. Huang and T. H. Boyer, Water Res., 2015, 68, 510–521 CrossRef CAS PubMed.
- J. A. Wilsenach, C. A. H. Schuurbiers and M. C. M. van Loosdrecht, Water Res., 2007, 41, 458–466 CrossRef CAS PubMed.
- D. P. Griffith, D. M. Musher and C. Itin, Invest. Urol., 1976, 13, 346–350 CAS.
- T. K. With, B. Petersen and T. D. Petersen, J. Clin. Pathol., 1961, 14, 202–204 CrossRef CAS PubMed.
- W. T. Tang, J. Dai, R. L. Liu and G. H. Chen, Water Res., 2015, 87, 10–19 CrossRef CAS PubMed.
- S. Dupraz, M. Parmentier, B. Menez and F. Guyot, Chem. Geol., 2009, 265, 44–53 CrossRef CAS.
- B. E. Maximilian Grau, K. M. Udert, C. J. Brouckaert and C. A. Buckley, Development and operation of struvite reactors to recover phosphorus from source separated urine in Ethekwini, Cape Town, South Africa, 2012 Search PubMed.
- J. F. Ambrose, G. B. Kistiakowsky and A. G. Kridl, J. Am. Chem. Soc., 1951, 73, 1232–1236 CrossRef CAS.
- W. H. R. Shaw, J. Am. Chem. Soc., 1954, 76, 2160–2163 CrossRef CAS.
- S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem, P. Liu, Z. T. Dame, J. Poelzer, J. Huynh, F. S. Yallou, N. Psychogios, E. Dong, R. Bogumil, C. Roehring and D. S. Wishart, PLoS One, 2013, 8, 28 Search PubMed.
- X. Z. Li and Q. L. Zhao, Ecol. Eng., 2003, 20, 171–181 CrossRef.
- ICIS, Indicative Chemical Prices A-Z, https://www.icis.com/chemicals/channel-info-chemicals-a-z/, (accessed 02/23/17, 2017).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ew00271h |
| This journal is © The Royal Society of Chemistry 2018 |