
Assessing the interactions of metal nanoparticles in soil and sediment matrices – a quantitative analytical multi-technique approach†
Hind
El Hadri
 *,
Stacey M.
Louie
and
Vincent A.
Hackley
*
*,
Stacey M.
Louie
and
Vincent A.
Hackley
*
National Institute of Standards and Technology, Materials Measurement Science Division, 100 Bureau Drive, Gaithersburg, MD 20899-8520, USA. E-mail: h.el.hadri@hotmail.fr; vince.hackley@nist.gov
First published on 19th December 2017
Abstract
The impact and behavior of engineered nanomaterials (ENMs) entering the environment is an important issue due to their growing use in consumer and agricultural products. Their mobility and fate in the environment are heavily impacted by their interactions with natural particle components of saturated sediments and soils. In this study, functionalized gold nanoparticles (AuNPs – used as model ENMs) were spiked into complex solid-containing media (standard soils and estuarine sediment in moderately hard water). AuNPs were characterized in the colloidal extract (<1 μm) following centrifugal separation of the non-colloidal phase, using different analytical techniques including asymmetric-flow field-flow fractionation and single particle inductively coupled plasma mass spectrometry. Attachment of functionalized AuNPs to the soil particles did not significantly depend on their concentration or surface coating (citrate, bPEI, PVP, PEG). Similarly, UV degradation of coatings did not substantially alter their recovery. Conversely, the presence of natural organic matter (NOM) is a key factor in their adhesion to matrix particles, by decreasing the predicted influence of native surface chemistry and functional coatings. A kinetic experiment performed over 48 h showed that attachment to soil colloids is rapid and that hetero-aggregation is dominant. These results suggest that transport of ENMs away from the point of discharge (or entry) could be limited in soils and sediments, but additional experiments under more realistic and dynamic field conditions would be necessary to confirm this more generally. Transport properties may also differ substantially in matrices where NOM is largely absent or otherwise sequestered or when dissolution of ENMs is an important factor.
Environmental significanceThe transport and interaction of engineered nanoparticles in complex solids containing media, such as saturated soils, will largely determine their fate and impact once they enter the environment. The presence of natural organic matter (NOM), natural colloids and more massive soil particles, as well as the functional coatings on the nanoparticles themselves and the potential impact of sunlight-derived UV radiation, combine to create substantial complexity, which in turn creates measurement challenges for the characterization, quantification and monitoring of these systems. This study utilizes model functionalized gold nanoparticles to develop quantitative and qualitative methodology to interrogate the association of engineered nanoparticles with colloidal and non-colloidal components of two standard agricultural soils and a standard estuarine sediment. The combination of multi-detector asymmetrical flow field-flow fractionation with single particle ICP-MS allow us to assess the relative dominance of homo- and hetero-aggregation, to quantify recoveries and attachment kinetics, and to evaluate the effect of surface coatings and NOM on these processes. |
1 Introduction
Because of the wide range of applications for engineered nanomaterials (ENMs) in industry, medicine, consumer products and agriculture, the nanotechnology field has witnessed rapid development. Consequently, the number of studies focused on ENMs is increasing rapidly. However, knowledge of the physico-chemical state and reactivity in environmentally relevant media and validated tools to measure those properties remain a significant challenge.1,2Soils (and sediments) are rich environmental matrices with respect to naturally occurring colloids (nano- to micro-scale). Soil colloids (operationally defined by a size between 1 nm and 1 μm (ref. 3)) typically carry a net electrostatic charge (negative in most cases) and/or have hydrophobic characteristics that may promote interactions with natural or anthropogenic species (e.g., trace elements, ENMs, etc.). These colloids are composed of mineral (clay, iron oxides, etc.) and/or organic (natural organic matter – NOM) components, and play a central role in determining the behavior, mobility and fate of ENMs that enter the environment. Correspondingly, colloidal transport through soils has been widely studied for metallic trace element fate.4–8 The most important sources of entry for ENMs into soils arise from application of wastewater sludge as a soil amendment, intentional releases for environmental applications (e.g., zero valence iron nanoparticles9,10) and accidental spillages;11 ENMs may also enter the environment via aerosols that subsequently deposit into soil or aquatic systems and from end-of-life disposal in landfills.
A number of studies have examined the behavior of ENMs in environmental matrices and more particularly in soils.12–18 The aggregation rate including homo-aggregation (ENM–ENM) and hetero-aggregation (ENM–natural colloid) is a key parameter to assess the reactivity, transport and bioavailability of ENMs in the soil compartment and their potential impact on the biota and groundwater.19 Here we use the term ‘aggregation’ in the generic form (i.e., equivalent to agglomeration), due to its historical use in this context.
The principal methods used to detect and characterize ENMs in soils and sediments include transmission electronic microscopy (TEM),15 hydrodynamic chromatography (HDC),14 asymmetrical flow field-flow fractionation (AF4)12 and dynamic light scattering (DLS).13 Both HDC and AF4 are typically coupled in tandem to specific detectors, such as DLS, inductively coupled plasma mass spectrometry (ICP-MS), or fluorescence. The benefits of stable isotope detection with ICP-MS for quantifying and differentiating ENMs from naturally occurring species has also been investigated.20
Given the low environmental concentrations of ENMs, pre-concentration methods are often needed for analytical techniques with higher limits of quantification (LOQ). Thus, our previous study focused on cloud point extraction applied to a complex soil matrix (extract) spiked with gold nanoparticles (AuNPs).21 In the present study, however, spiked analyte concentrations are sufficiently high (above expected environmental levels) to negate the need for pre-concentration, as the purpose here is to validate the general analytical approach and to demonstrate its potential to interrogate ENM–matrix interactions. This work builds on our previous studies by assessing the behavior and fate of model AuNPs in soil and sediment matrices under different conditions using a standard leaching test procedure. AuNPs were chosen because they are relatively stable and easy to detect. Moreover, surface coatings tend to play a determinant factor with respect to the interaction of metal NPs with complex media (unless the cores are significantly unstable).22–24 To this end, a method of extraction for complex solid matrices (soils and sediments), derived from the US Environmental Protection Agency25 and the International Organization for Standardization 18772:2008,26 was applied. EPA 1316 and ISO 18772:2008 are guidance methods on leaching procedures. They provide information on the liquid/solid partitioning of contaminants (inorganic, non-volatile organic and natural radionuclides). For comparison purposes the leaching procedure is generally standardized by setting conventional operating parameters, allowing a higher degree of robustness, repeatability, reproducibility and a broad applicability to different types of soils.26 The leaching method was used as a means to obtain an extract (eluate) from a solid matrix, which may be used to estimate the release of the most available fraction of soil colloids from spiked samples under laboratory conditions. Laboratory conditions do not aim to fully simulate actual field conditions, but rather this approach offers improved insight into the behavior of ENMs in complex solid matrices and provides proof of principle for the combined analytical methodology demonstrated here. It is important to note that batch methods represent a given time frame (which may vary) in which equilibrium is not necessarily reached.27–29 The relative merits of batch versus dynamic column tests in predicting transport and fate is beyond the scope of the present work.
Analytically, we employ hyphenation of AF4 with tandem detection methods, including ultra-violet visible (UV-vis) absorption, multi-angle light scattering (MALS) and ICP-MS, which have proven particularly valuable and flexible in our previous work.20,21 This coupling provides information about the size, but also about the nature and composition of NPs under relevant conditions, and with minimal perturbation of the analyte. Additionally, other complementary techniques were used, such as single particle (sp) ICP-MS, in order to determine the AuNP size distribution and to assess the homo-aggregation rate. In this study, AuNPs with different surface coatings and concentrations were spiked into the matrix before extraction to assess the Au distribution between the more massive soil particles and the colloidal fraction (nominally <1 μm for the present work). A kinetic study was also performed on soil samples spiked with citrate-capped AuNPs, up to 48 h. Finally, the potential role of surface coating degradation on the AuNP–soil interaction was evaluated using UV exposure as a model approach to induce coating degradation of polyethylene glycol (PEG) capped AuNPs.
2 Materials and methods
2.1 Chemicals
Deionized (DI) water (>18 MΩ cm) was supplied by a type II biological grade water purification system (Aqua Solutions, Jasper, GA, USA)‡ and was utilized for all sample preparation and dilution. AuNPs coated with PVP (10 kDa), PEG (5 kDa) and bPEI (25 kDa) with a nominal size of 30 nm were purchased from Nanocomposix (San Diego, CA, USA). 30 nm and 60 nm citrate (Cit) AuNPs were obtained from Ted Pella (Redding, CA, USA). To prepare AuNPs (from Cit-AuNPs) coated with NOM, commercial humic acid (Suwannee River Humic Acid Standard II, HA) from the International Humic Substances Society (IHSS, St. Paul, MN, USA) and HA extracted directly from the soil sample (see protocol in ESI†) were used. Ammonium nitrate (NH4NO3, 99%) was used for the AF4 mobile phase after dilution with DI water (to reach a concentration of 0.5 mmol L−1) and filtration through a 0.2 μm regenerated cellulose membrane. Concentrated nitric, HNO3, (≥69%) and hydrochloric, HCl, (≥37%) acids (Fluka TraceSelect, Sigma-Aldrich, St. Louis, MO, USA) were used to digest AuNPs prior to ICP-MS experiments for trace analysis. Elemental calibration standards were prepared from NIST SRM 3121 Au and SRM 3140 Pt standard solutions.2.2 Sample preparation
- Nebraska SONE-1 (internal reference agricultural soil) obtained from the US Geological Survey (USGS); this sample was used for most experiments reported in the present study,
- NIST SRM 2709a (San Joaquin Soil), an agricultural soil collected by the USGS from a fallow field in central California's San Joaquin Valley,30
- NIST SRM 1646a (Estuarine Sediment) dredged from the Chesapeake Bay.31
Information provided by the producer on the corresponding certificates indicated that the matrix materials were sieved at 2 mm for the two soil samples and at 1 mm for the sediment, to remove the coarsest fraction; materials were then ground to a particle size below 100 μm (<90 μm for SONE-1 and 75 μm for the two SRMs). In general, particulates with a higher specific surface area are likely to present a more reactive material and to represent the greater portion of available surface area (on a mass basis) within the matrix.
Fig. 1 schematically shows the extraction procedure based on EPA 1316 and ISO 18772:2008 leaching methods and previous work performed in our laboratory.20,21,25,26 Moderately hard water (MHW) was used as the leachant (i.e., the liquid used in a leaching test) and was prepared according to the EPA recommendation i.e., 96 mg L−1 NaHCO3, 60 mg L−1 CaSO4·H2O, 60 mg L−1 MgSO4 and 4 mg L−1 KCl.32 It is generally preferable to use leachant that provides an ionic strength closer to typical environmental media rather than DI water alone, and MHW has been used extensively for environmental studies. The calculated ionic strength is 3.3 mmol L−1. A liquid/solid (L/S) ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used, containing 1 g of solid matrix and 10 g of leachant. This ratio is somewhat arbitrary in nature, but has been commonly used in previous studies and proved both workable and reproducible. AuNPs were spiked into soil samples by addition of a small volume (100 μL) into the solid-leachant system, to obtain concentrations in the range from (10 to 5000) μg kg−1. The resulting preparations were agitated for 24 h using an end-over-end rotator.
:1 was used, containing 1 g of solid matrix and 10 g of leachant. This ratio is somewhat arbitrary in nature, but has been commonly used in previous studies and proved both workable and reproducible. AuNPs were spiked into soil samples by addition of a small volume (100 μL) into the solid-leachant system, to obtain concentrations in the range from (10 to 5000) μg kg−1. The resulting preparations were agitated for 24 h using an end-over-end rotator.
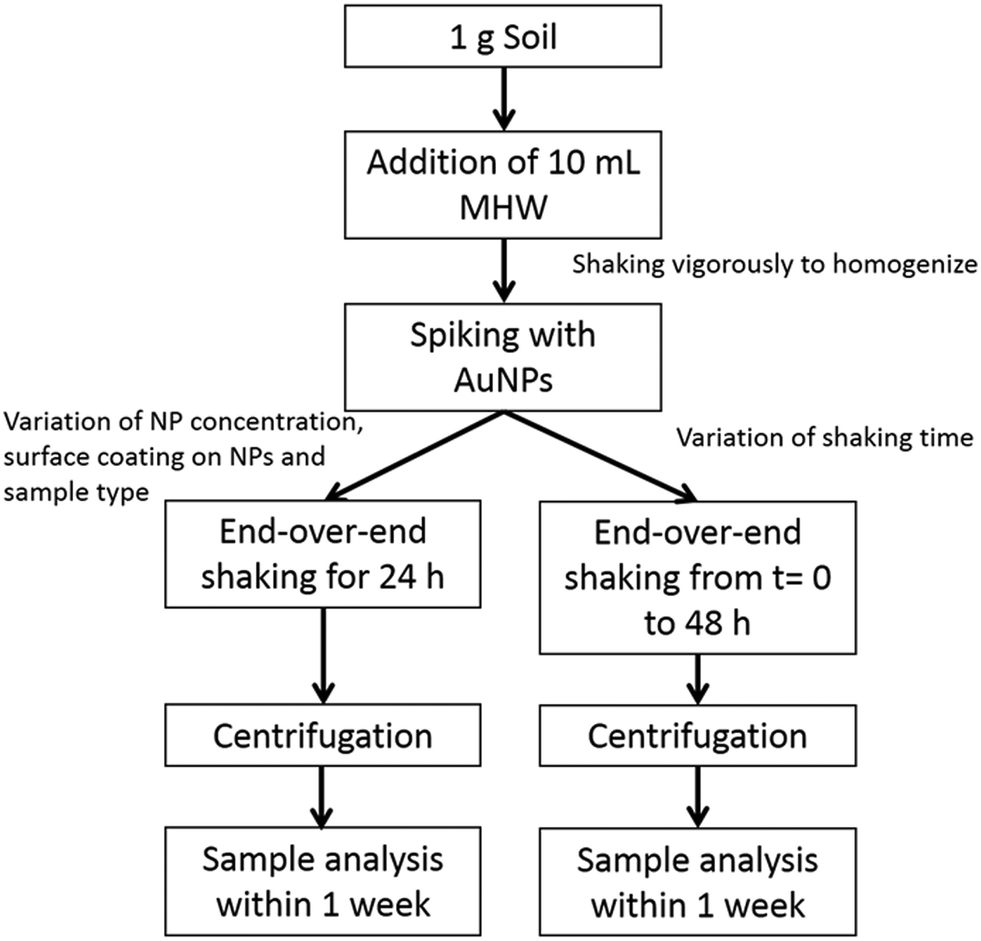 | ||
| Fig. 1 Flowchart showing the soil preparation and extraction procedures used in this study. | ||
A centrifugation technique was used to isolate soil colloids <1 μm (total range of the colloidal fraction) and <0.45 μm (commonly used threshold). The settling time to obtain the two different size fractions was determined using the equation: 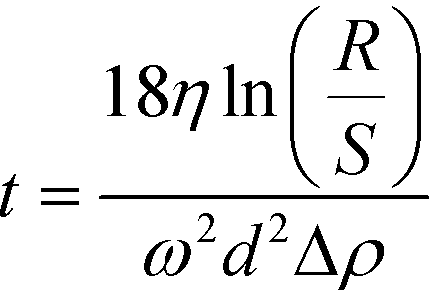 where Δρ is the density difference between the particles and the medium, R (115 mm) and S (minimum 64 mm) represent the distances from the axis of rotation at the bottom and at the top of the solution, respectively, w is the angular velocity of the centrifuge, d the particle (equivalent spherical) diameter and η is the viscosity of the suspension medium (i.e., water). The densities considered are 2.65 g cm−3 for soil (an estimated mean value) and 19.3 g cm−3 for Au.33,34 This method assumes that the particles in suspension are spheres and behave according to Stokes law; therefore, an unavoidable uncertainty on the size fraction exists and the obtained fractions should be viewed as nominal approximations. A Beckman J2-HC centrifuge (Beckmann Coulter Life Sciences, Indianapolis IN, USA) using a J20.1 fixed angle rotor (15 mL tube volume) was used. The centrifugation speed was set at 2000 rpm (515 g at R). In a first step, a centrifugation time of 2 min was applied to remove material >1 μm. The supernatant was collected and a small part of it was put aside for analysis. In a second step, the fraction ≤1 μm was centrifuged for 10 min to remove material >0.45 μm. Under these conditions Au particles smaller than 300 nm and 140 nm remain in suspension for the soil colloidal thresholds of 1 μm and 0.45 μm, respectively.
where Δρ is the density difference between the particles and the medium, R (115 mm) and S (minimum 64 mm) represent the distances from the axis of rotation at the bottom and at the top of the solution, respectively, w is the angular velocity of the centrifuge, d the particle (equivalent spherical) diameter and η is the viscosity of the suspension medium (i.e., water). The densities considered are 2.65 g cm−3 for soil (an estimated mean value) and 19.3 g cm−3 for Au.33,34 This method assumes that the particles in suspension are spheres and behave according to Stokes law; therefore, an unavoidable uncertainty on the size fraction exists and the obtained fractions should be viewed as nominal approximations. A Beckman J2-HC centrifuge (Beckmann Coulter Life Sciences, Indianapolis IN, USA) using a J20.1 fixed angle rotor (15 mL tube volume) was used. The centrifugation speed was set at 2000 rpm (515 g at R). In a first step, a centrifugation time of 2 min was applied to remove material >1 μm. The supernatant was collected and a small part of it was put aside for analysis. In a second step, the fraction ≤1 μm was centrifuged for 10 min to remove material >0.45 μm. Under these conditions Au particles smaller than 300 nm and 140 nm remain in suspension for the soil colloidal thresholds of 1 μm and 0.45 μm, respectively.
To digest AuNPs in soil extracts, either in solution or attached to soil colloids, undiluted aqua regia (1:3; HNO3:HCl) was added to the aliquots of the supernatants. These solutions were then left at room temperature overnight in a fume hood. This was followed by dilution with DI water (containing 0.1% thiourea to avoid memory effects in the instrument and to stabilize Au) to reach 2% aqua regia. Total gold concentration was then determined by ICP-MS. The digestion procedure was verified by spiking a known mass quantity of AuNPs in a soil extract (<0.45 μm) followed by ICP-MS analysis; this yielded total Au content within experimental uncertainty. The potential for AuCl3 precipitation was mitigated due to the large excess of Cl (from aqua regia) over Au in all samples.
To verify that the AuNPs did not adhere substantially to the centrifuge tube walls (polypropylene), AuNPs were agitated in the absence of the soil matrix under the same conditions. Results (not shown) indicate the percentage of Au remaining in solution after agitation is >95% for Cit-, PVP- and PEG- coatings. bPEI-AuNPs (positively charged) presented a significant loss (70%). The affinity of bPEI-AuNPs for the soil particles is relatively high (compared to other coatings) due to the soil's net negative charge, whereas the container surface is neutral and has a total area that is relatively small compared with the colloidal fraction. Based on these results, we conclude that analyte loss to the container surface represents a relatively small effect, of order 5% or less; this effect may be measurably higher for bPEI coated particles, but the soil colloids should outcompete the container surface for adsorption in an agitated system.
2.3 Instrumentation
A Zetasizer Nano ZS (Malvern, Worcestershire, UK) was used offline to determine the hydrodynamic diameter (z-average) and the zeta-potential (Smoluchowski limit) of the NP suspensions and soil extracts. The ionic strength and pH for these measurements were established by the combination of MHW and the soil samples; no additional adjustments were made. Conductivity and polydispersity index were also determined using this instrument.Total organic carbon (TOC) was determined by UV/persulfate oxidation using a Phoenix 8000 (Teledyne Tekmar, Mason, OH). The default instrument method for TOC concentrations from (0.1 to 20) mg L−1 as C was used: 4.0 mL of sample was mixed with 0.5 mL of 21% phosphoric acid and sparged with N2 for 120 s to remove inorganic carbon; then, the acidified sample and 1.0 mL of 10% persulfate/5% phosphoric acid reagent were injected into the UV reactor and sparged with N2. CO2 from oxidation of the organic carbon was monitored by a nondispersive infrared detector. Concentrations as mass fraction C were determined against potassium hydrogen phthalate calibration solutions.
Total Au concentrations were determined with an ICP-MS model 7900 from Agilent Technologies (Santa Clara, CA, USA). The instrument was equipped with a concentric nebulizer and a refrigerated Scott chamber (2 °C). To obtain maximum sensitivity, the tune solution provided by Agilent containing multi-element standards (1 μg L−1 each of 7Li, 89Y, 140Ce and 205Tl in 2% v/v HNO3) was measured before analysis. The instrument was optimized for minimum oxide (156CeO/140Ce) and doubly charged (70Ce++/140Ce+) level (<2%). Platinum (Pt) was added as an internal standard to both the Au solution calibration standards and to the samples. Data were collected at m/z 197 for Au and m/z 195 for Pt. Dissolved Au calibration standards were prepared over a mass fraction range from (0.05 to 5) μg kg−1 in aqua regia (2.0% v/v). Single particle (sp) ICP-MS measurements were also performed on the Agilent 7900 using the time resolved analysis (TRA) mode, with a dwell time of 10 ms and measurement time of 300 s. The transport efficiency was determined each day using dissolved Au calibration standards and NIST Reference Materials 8012 and 8013, with nominal AuNP size of 30 nm and 60 nm, respectively.37,38 Samples were diluted to appropriate levels (less than 15000 particles per mL). Serial dilutions were performed to exclude coincident artefacts. Additionally, standard suspensions containing only monomers were evaluated at different concentrations to test for onset of significant coincidence.
The AF4 model used in this study was an Eclipse 3+ system from Wyatt Technology (Santa Barbara, CA). It was equipped with an 1100 series isocratic pump (Agilent Technologies) to generate mobile phase flow and a degasser (Gastorr TG-14, Flom Co., Ltd., Tokyo, Japan). All injections were performed with an Agilent Technologies 1260 ALS series autosampler. The detection system was formed by a 1200 series UV-vis absorbance diode array detector (Agilent Technologies) and a multi-angle laser light scattering (MALS) detector (DAWN HELEOS, Wyatt Technology). Due to the large size range present in these samples, a 250 μm spacer (with dimensions of 26.5 cm length and narrowing width from 2.1 to 0.6 cm) was used to set the AF4 channel height. The main flow rate and the cross flow rate were set at 0.5 mL min−1 and 0.3 mL min−1, respectively.21 The retention time (tR) starts after the focus step (flow of 2 mL min−1). Polyethersulfone (PES) 10 kDa membranes were purchased from Wyatt Technology and used for the accumulation wall. Data from the AF4 detectors were processed using Astra ver. 6.1.4.25 (Wyatt Technology) and OpenLab CDS Rev. C.01.06 (Agilent Technologies) software. The Berry formalism (first order) was used to determine the radius of gyration (Rg) from MALS data.39 The AF4 selectivity was determined using polystyrene latex beads (Thermo Fisher Scientific, Waltham, MA, USA) of known size and the relationship 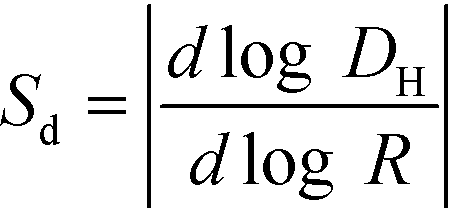 . For the AF4 conditions used in this study, a selectivity of 0.9 ± 0.1 was calculated, which indicates high quality separation (i.e., value close to unity).40 During hyphenation with the ICP-MS (Agilent 7900), in addition to 197Au, characteristic isotopes were monitored to track the natural colloids: 27Al, 55Mn and 57Fe. The collision cell was used in these measurements, with He at 3.5 mL min−1, to mitigate interferences from polyatomic species (see ESI,† Table S1).
. For the AF4 conditions used in this study, a selectivity of 0.9 ± 0.1 was calculated, which indicates high quality separation (i.e., value close to unity).40 During hyphenation with the ICP-MS (Agilent 7900), in addition to 197Au, characteristic isotopes were monitored to track the natural colloids: 27Al, 55Mn and 57Fe. The collision cell was used in these measurements, with He at 3.5 mL min−1, to mitigate interferences from polyatomic species (see ESI,† Table S1).
2.4 Uncertainty analysis
Uncertainty intervals and error bars reported in this study, unless otherwise noted, represent one standard deviation about the mean determined under repeatability conditions with measurements on at least two replicate samples.3 Results and discussion
3.1 Sample characterization
Table 1 summarizes the sample characteristics of the solid (soils and sediment) extracts (below 0.45 μm) and the AuNP samples. The hydrodynamic diameter and zeta-potential (ZP) of the natural solid colloid samples were measured for the <0.45 μm fraction after extraction with MHW without modification. Prior to measurements, AuNP solutions were diluted in MHW (leachant), with a pH of 7.8. pH was also measured in the solid extracts and was 6.8, 8.0 and 8.1 for SONE-1, San-Joaquin and Estuarine sediment, respectively. Given that the same leachant was used (MHW), the difference in pH, is attributed to the soil characteristics (e.g., SONE-1 is the most acidic solid). ZP is pH dependent, therefore ZP of SONE-1 is not directly comparable to the other natural samples. However, it appears that all colloids are negatively charged, especially in the estuarine sediment with −24.2 ± 0.8 mV. For the AuNPs, the ZP is negative for all samples except for bPEI, which is positive as expected. Cit-AuNPs and HA-AuNPs are the most negatively charged with a ZP near −20 mV. PEG-AuNPs exhibit a small residual negative ZP of −2.2 mV, which has been commonly observed for this coating. The dH (z-average) determined by DLS shows that the PEG coating increases the size of the nominally 30 nm AuNPs to 51 nm, compared to 42 nm and 33 nm, for PVP and HA coatings, respectively. The soils/sediment extracts contain polydisperse colloids, therefore the measured sizes are at best rough estimates of the mean size. The average dH is below 450 nm for all extracted matrices, though the estuarine sediment, at 400 nm, is significantly larger than the soils. We note that after centrifugation the sediment derived particles were more easily resuspended relative to the soil particles.| Zeta-potential (mV) | Average dH (nm) | |
|---|---|---|
| a Uncertainties represent the standard deviation of five replicate measurements. σ is conductivity and PDI is polydispersity index. | ||
| SONE-1 | −14.2 ± 0.6 (σ = 0.451 ± 0.002 mS cm−1) | 299 ± 5 (PDI = 0.14 ± 0.03) |
| San-Joaquin | −16.0 ± 0.6 (σ = 0.33 ± 0.02 mS cm−1) | 291 ± 2 (PDI = 0.14 ± 0.02) |
| Estuarine sediment | −24.2 ± 0.8 (σ = 2.61 ± 0.07 mS cm−1) | 406 ± 7 (PDI = 0.35 ± 0.05) |
| Cit-AuNPs | −21.1 ± 0.7 | 29.2 ± 0.7 |
| bPEI-AuNPs | 12.8 ± 2.2 | 36.0 ± 0.2 |
| PVP-AuNPs | −10.8 ± 1.4 | 41.7 ± 0.2 |
| PEG-AuNPs | −2.2 ± 0.3 | 50.9 ± 0.4 |
| SRHA-AuNPs | −19.2 ± 0.8 | 33.3 ± 0.3 |
| SONEHA-AuNPs | −20.6 ± 0.6 | 33.0 ± 0.5 |
TOC was measured in the natural samples after extraction in the <0.45 μm fraction, it was higher in SONE-1 and the estuarine sample with (44 ± 3 and 40 ± 1) mg C kg−1, respectively. The San-Joaquin soil contained (29.3 ± 0.4) mg C kg−1. The estimated carbon content in the initial solid soil and sediment samples was 1.9%, 1.2% and 1.6% mass fraction for SONE-1, San Joaquin and estuarine sediment, respectively (Table S2†).41,42 For AuNPs coated by HA, the TOC in the HA solution was set at 20 mg C kg−1, prepared by dilution from the stock solution.
3.2 Influence of AuNP concentration, coating and sample type
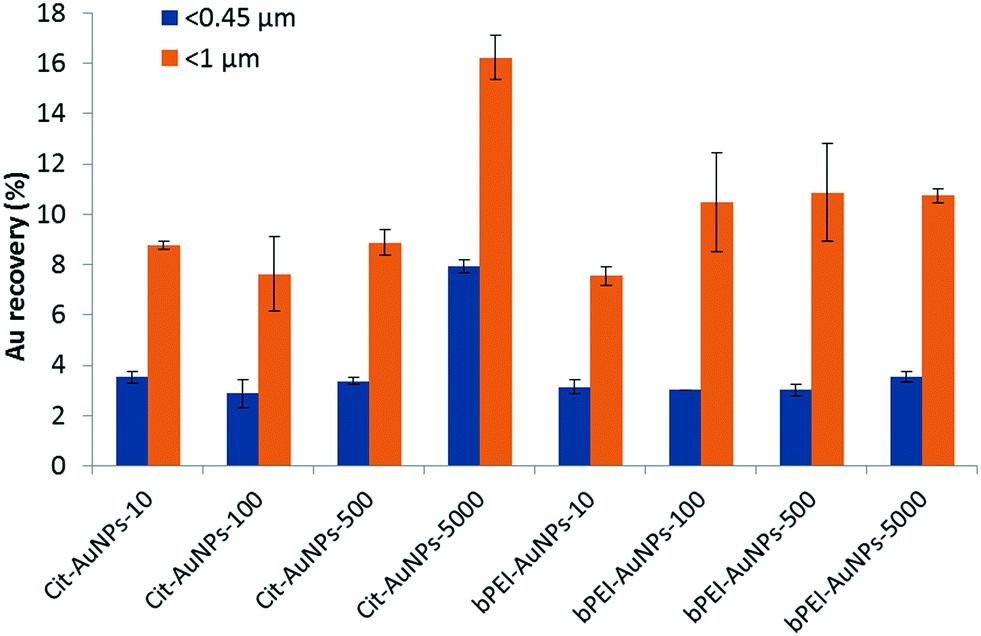 | ||
| Fig. 2 Extracted Au recovery after acid digestion and ICP-MS analysis for Cit-AuNPs and bPEI-AuNPs at different concentrations. The agitation time was 24 h. | ||
For the fraction below 0.45 μm, the extracted Au recovery is around 3% except for Cit-AuNPs at 5000 μg kg−1, which is (7.9 ± 0.3) %. The AuNPs are either free or adsorbed onto the sub-0.45 μm soil colloids in the extracted suspension. Extracted Au recovery for <1 μm fraction varies from (7.5 ± 0.4) % to (16.2 ± 0.9) %. For Cit-AuNPs, it is comparable at low concentrations (≤500 μg kg−1). However, as for the <0.45 μm fraction, the recovery for 5000 μg kg−1 is higher with (16.2 ± 0.9) %. For bPEI-AuNPs, the recovery is similar from 100 μg kg−1 at around (10.5 ± 2.2) % and is lower (7.5 ± 0.4) % for 10 μg kg−1. These observed differences in recovery are not substantial, and the attachment may be similar, but appears to vary somewhat between the larger and colloidal (<1 μm) soil particles.
In both fractions <0.45 μm and <1 μm, the recoveries of Au as a function of the concentration are similar for the Cit-AuNPs (up to 500 μg kg−1), and for the bPEI-AuNPs (at all concentrations tested), which is consistent with basic hetero-aggregation theory as described by a modified Smoluchowski model (eqn (1)),29,43 assuming that the processes of homo-aggregation and breakup of hetero-agglomerates are insignificant:
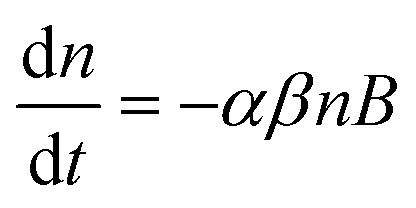 | (1) |
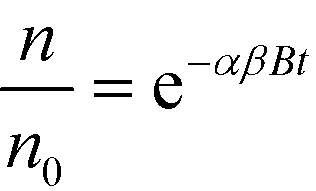 | (2) |
Fig. 3 presents the size distribution of the AuNPs at different concentrations for both Cit and bPEI coating using spICP-MS in the <0.45 μm fraction after 24 h of agitation. To quantify the aggregation rate, an aggregation number (AN) can be determined as described in a previous study21 as the ratio of the mass sum of the aggregates (dimer, trimer, and larger oligimers), maggr, over the mass sum of the monomer (mmono):
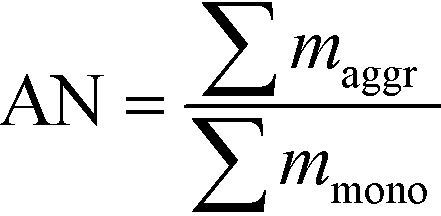 | (3) |
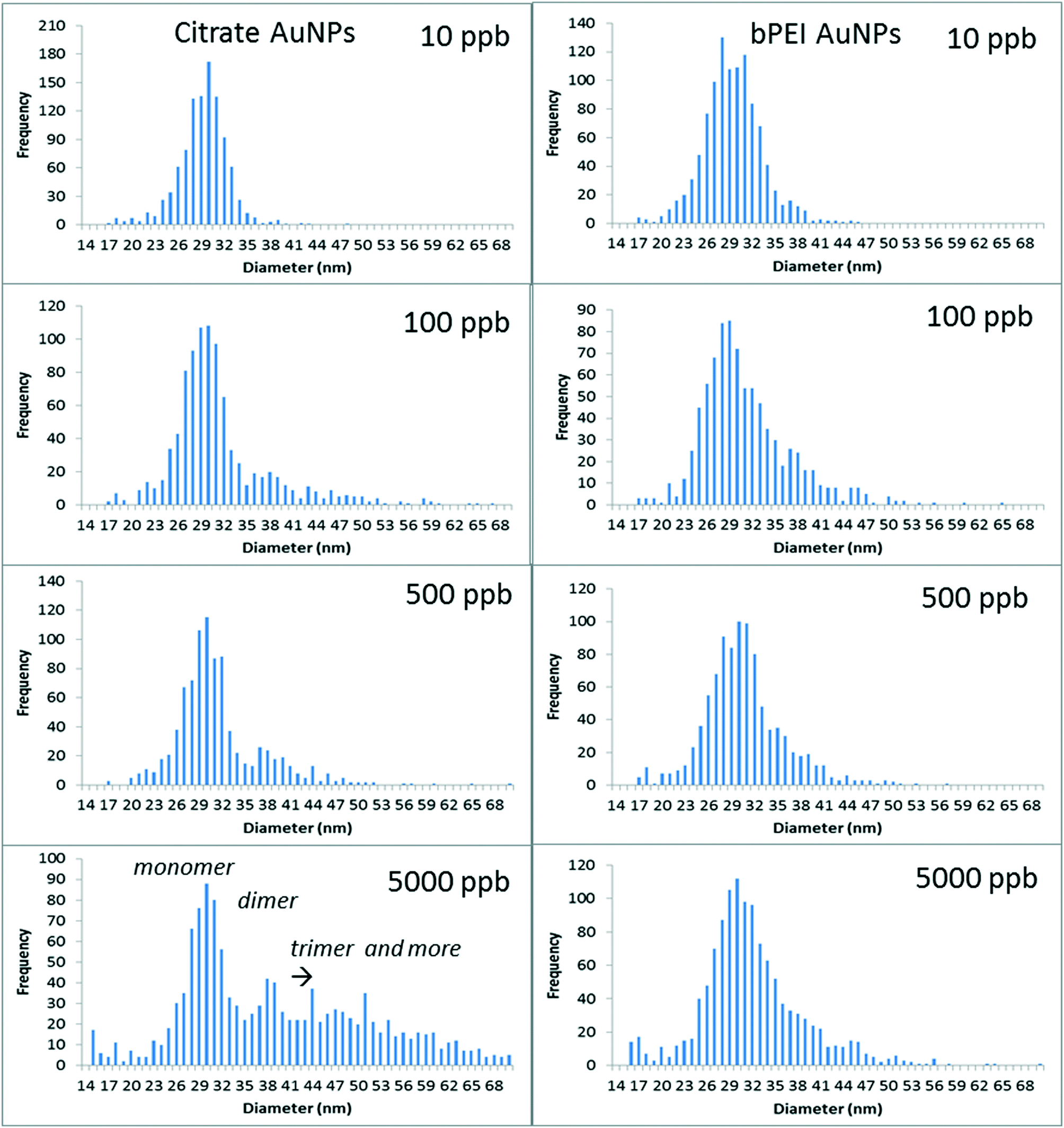 | ||
| Fig. 3 spICP-MS analyses for different AuNP concentrations spiked into SONE-1 soil after 24 h of agitation in the fraction <0.45 μm (for citrate and bPEI coatings). | ||
For citrate coating, the increase of concentration clearly involved the formation of dimers (at around 38 nm) and then trimers (and higher order oligomers). It is important to note that the sample concentrations were adjusted (multiple dilutions) to avoid particle event coincidence during the dwell time. The number of particles is determined in such a way that the particle number is below 300 min−1, at which level coincidence was not observed for the control sample (AuNPs in DI water). For (100 and 500) μg kg−1, AN increased to 0.5, and it was 10 times higher for 5000 μg kg−1 (Table 2). bPEI-AuNPs, to a lesser extent, exhibited aggregates starting at 100 μg kg−1. However, the aggregation rate remained relatively constant up to 5000 μg kg−1, in contrast to Cit-AuNPs. The increase of the AuNP number concentration increased the collision rate for homo-aggregation of AuNPs, and, therefore, the presence of dimers or higher order species in the spICP-MS data. It is important to note that, in the soil extract samples, AuNPs may be homo-aggregated or hetero-aggregated with soil colloids (i.e., single and multiple AuNPs either in a free oligomer or attached to the same colloid can produce similar frequency patterns under certain conditions).
| Sample concentration (μg kg−1) | ANa | |
|---|---|---|
| a For each coating, a control sample was analyzed (Cit- and bPEI-AuNPs diluted directly in DI water) and an ANblank was calculated from the size distribution. Thus, in the AN presented here, the blank was subtracted. The uncertainty was determined as the standard deviation of duplicate samples. | ||
| Cit-AuNPs mixed with SONE-1 | 10 | 0 |
| 100 | 0.50 ± 0.06 | |
| 500 | 0.4 ± 0.2 | |
| 5000 | 5 ± 1 | |
| bPEI-AuNPs mixed with SONE-1 | 10 | 0 |
| 100 | 0.60 ± 0.07 | |
| 500 | 0.25 ± 0.03 | |
| 5000 | 0.5 ± 0.2 | |
To verify hetero-aggregation, AF4-UV-MALS-ICP-MS was performed on samples with a spiked concentration of 5000 μg kg−1. Fig. 4a presents the fractograms (detector signal as a function of tR) for the soil extract samples spiked with Cit-AuNPs. Based on Rg, fractionation is effective from about (50 to 250) nm in the main peak (tR from 15 min to 60 min). The Au ICP-MS trace is present throughout the separation (from tR = 5 min). For comparison, the 30 nm Cit-AuNPs injected alone (no extract) under the same conditions are eluted at tR = 6 min. However, Au is mainly coeluted with the soil colloids, represented in the graph by Al, Fe and Mn for the clay and the (oxy)hydroxides of Fe or Mn present.44 Therefore, these results appear to confirm that hetero-aggregation is dominant. Collections of fractions were performed at three tR values: 5.5 min (F1), 16.5 min (F2) and 45 min (F3) for a 1 min collection time. F1 corresponds to the retention of AuNP monomers injected alone. F2 was collected because of the high Au intensity at this tR (Fig. 4a); moreover, it may correspond to principal aggregates of AuNPs. According to the number of spherical primary particles and the configuration of aggregates (linear, spherical…), the hydrodynamic diameter can vary.45 Finally, F3 was collected at the maximum of the main colloid population. spICP-MS analysis was performed on each fraction and results are presented in Fig. 4b. Aggregation numbers for each fraction were calculated from the spICP-MS size distribution as described in eqn (3) and were as follows: 0.21 ± 0.06, 7 ± 1 and 4.3 ± 0.4, for F1, F2 and F3, respectively. It shows that the higher aggregation rate is associated with F2, where a more important frequency signal (Fig. 4b) between (37 and 80) nm (spherical equivalent diameter) is observed.
 | ||
| Fig. 4 a) AF4-UV-MALS-ICP-MS fractograms of soil extract (<0.45 μm) spiked with 5000 μg kg−1 of Cit-AuNPs and b) spICP-MS size distributions of AuNPs corresponding to the three collected fractions obtained during the AF4 separation. The agitation time was 24h. | ||
The monomer population (single AuNPs), though also present, may simply reflect coelution with soil colloids (i.e., AuNPs attached to colloids). F3 indicates a lower homo-aggregation rate and further confirms that hetero-aggregation is dominant. Additionally, more than one AuNP can be attached to a single colloid. When aggregates are observed in spICP-MS and the tR in AF4 matches the colloid trace (typically for F3), it represents two NPs (if dimers) attached to the same soil particle; these AuNPs might be in contact or not (homo/hetero-aggregation). To help verify which mode is occurring or dominant, one option is to use electron microscopy imaging. Another possible method would be to use fast scanning with microsecond dwell times to assess characteristic changes in peak shape associated with hetero- versus homoaggregation, but this was beyond the scope of the present work and would involve extended studies to validate the approach.
For bPEI-AuNPs, the fractionation profile was similar to Cit-AuNPs. However, for the fractions analyzed by spICP-MS, the rate of homo/hetero-aggregation was lower compared to citrate coating, with AN values of 0.23 ± 0.05, 0.82 ± 0.08 and 0.77 ± 0.07, for F1, F2 and F3, respectively (see ESI,† Fig. S1). As previously shown, F1 corresponds to the monomer and yields a low AN whereas F2 and F3 have the same AN associated with a small amount of homo/hetero-aggregation. It is worth noting that the recovery in the <0.45 μm fraction, for a spiked concentration of 5000 μg kg−1, is higher for citrate (8.3% ± 0.9%) compared to bPEI coating (3.5% ± 0.2%) involving a higher attachment rate for bPEI on larger soil particles. Moreover, in a simple solution (DI water only or MHW) with only AuNPs, Cit-AuNPs tend to homo-aggregate more readily compared to bPEI-AuNPs due to the additional polymer-induced steric stabilization present for bPEI. The difference in recovery can be due to the competition between homo- and hetero-aggregation. Therefore, oligomers of Cit-AuNPs are formed more easily in contrast with bPEI-AuNPs.
For the remainder of this study, a concentration of 500 μg kg−1 was chosen to produce sufficient signal to be detected by the analytical method and to limit the homo-aggregation observed at 5000 μg kg−1. Moreover, according to eqn (1) the influence of concentration up to 500 μg kg−1 is negligible. For real environmental samples a prerequisite step of preconcentration, such as cloud point extraction,21 is often needed to detect ENMs by the standard analytical methods, as environmental concentrations are generally well below the μg kg−1 level.46
 | ||
| Fig. 5 Au recovery for different AuNP coatings in SONE-1 extracts after acid digestion and ICP-MS analysis. The spiked AuNP concentration was 500 μg kg−1. The agitation time was 24 h. | ||
spICP-MS measurements were performed on each soil extract spiked with AuNPs with different coatings (PVP, PEG, SRHA and SONE-HA) (see ESI,† Fig. S2). The size distributions indicate that homoaggregation is not present; the frequency distributions are similar to the initial AuNP samples (diluted stock solution). It should be noted that HA stabilizes AuNPs initially coated with citrate. AF4 fractograms show that AuNPs are coeluted with the soil colloids; similar results were found for the Cit and bPEI samples (data not shown).
 | ||
| Fig. 6 Au recovery for Cit-AuNPs in SONE-1, San Joaquin and estuarine sediment extracts after acid digestion and ICP-MS analysis. The spiked AuNP concentration was 500 μg kg−1. The agitation time was 24 h. | ||
AF4-UV-MALS fractograms for all samples (<0.45 μm fraction) present a broad peak between (20 and 75) min (see ESI,† Fig. S3). The maximum of the UV peak corresponds to a size Rg ≈ 200 nm for the soil samples (SONE-1 and SJ) and ≈180 nm for Sed. To the naked eye, we observed that SJ and Sed were less turbid after centrifugation, compared to SONE-1. Moreover, for the same injected quantity in AF4, the integrated intensity signals (for SJ and Sed) are reduced. The UV signal (at 254 nm) was integrated for each matrix and normalized to the largest area (i.e., that measured for SONE-1). The integrated and normalized UV for SONE-1, SJ and Sed extracts are 1, 0.1 and 0.1, respectively. Similarly, Al, Fe and Mn intensities (determined by ICP-MS) in the extracts (<0.45 μm) are also lower for SJ and Sed (data not shown). The normalization for Al, Fe and Mn contents (combined together) in the different extracts represents 1, 0.45 and 0.06 for SONE-1, SJ and Sed, respectively. These results suggest that Sed and SJ contain a lower fraction of colloids relative to SONE-1. NOM content (as described previously), is higher in SONE-1 and Sed with ≥40 mg C kg−1 compared with about 29 mg C kg−1 for SJ. Thus, the non-negligible Au recovery in Sed extract relative to the number of colloids may be explained by the presence of NOM. NOM can adsorb48 or replace the initial coating and adsorb on other mineral soil particles. Frequency size distributions for AuNPs obtained by spICP-MS show that aggregation (homo or homo/hetero) is most prevalent in the Sed extracts (See ESI,† Fig. S4), where dimer, trimer and higher order oligomer peaks are clearly present. The Sed particles are more negatively charged (Table 1) and therefore could be less of a sink for the negatively charged Cit-AuNPs; this in turn could promote homo-aggregation, but this is speculative without further confirmation.
3.3 Kinetic study
For this set of experiments, the time of contact (i.e., agitation time) between SONE-1 soil and Cit-AuNPs was set between (0 and 48) h with a fixed Au concentration of 500 μg kg−1. Fig. 7 shows that the leachable AuNP content is decreasing rapidly. At t = 0, less than 60% and 40% of total Au is extracted from the fraction <1 and <0.45 μm, respectively. Therefore, attachment on soil particles is rapid. At t = 2 h, less than 20% and 10% of Au is found in the fraction <1 μm and <0.45 μm, respectively. Extracted Au continued to decrease and reached, after a contact time of 48 h, (8.8 ± 0.7) % and (2.0 ± 0.2) %, for <1 μm and <0.45 μm, respectively. Thus, in this system, equilibrium is not reached.28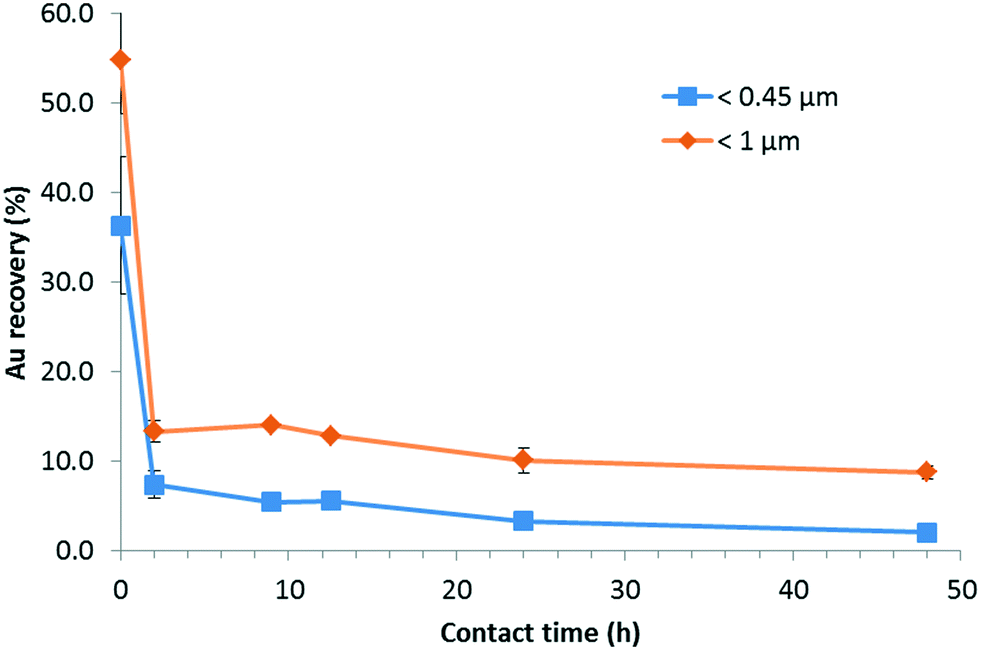 | ||
| Fig. 7 Kinetics of Au loss in SONE-1 soil extracts as a function of the contact (agitation) time. | ||
For each reported contact (agitation) time, AF4-UV-MALS-ICP-MS analysis was performed on the <0.45 μm fraction. Fig. 8 presents the ICP-MS trace (197Au) for the fractograms at five contact times (In Fig. S5† MALS, UV and major elements fractograms are presented). It shows that at t0, Au is eluted principally at 6 min, which corresponds to tR associated with free singlet AuNPs. The Au fraction eluted with the natural colloids between (13 and 75) min is lower. When the contact time increases, it is observed that AuNPs are associated more with the colloidal component. The ratio between the areas of peak 2 (13 min to 75 min) and peak 1 (0 min to 13 min) increase from 0.9 to 7.6, with a linear correlation (y = 0.15x + 0.97, R2 = 0.92). Therefore, larger soil particles (Fig. 7) and colloids (Fig. 8) tend to capture AuNPs over time, removing them from the solution phase.
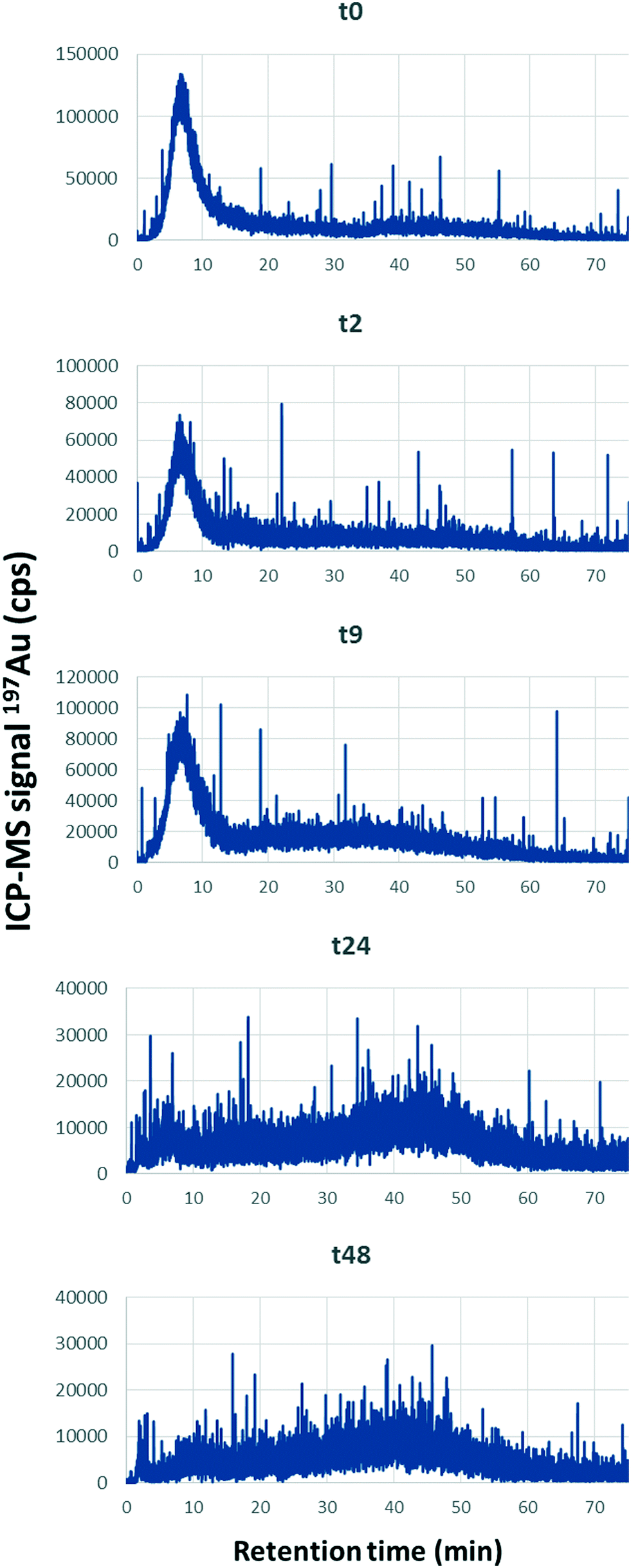 | ||
| Fig. 8 AF4-ICPMS fractograms showing 197Au signal (ICP-MS) at different contact times after extraction of the <0.45 μm fraction. The spiked concentration was 500 μg kg−1 Au. | ||
3.4 UV irradiation
UV exposure was previously observed to transform PEG and PVP coatings on AuNPs.36,49 More specifically, PEG was found to degrade and detach from the AuNP surface, while PVP oxidized but remained attached to the AuNP as a compressed surface coating. Both transformations resulted in diminished stability of the AuNPs against homo-aggregation.36 While UV exposure would likely have a muted impact on soil associated NPs, NPs used in foliar applications or those deposited onto the top layer of soil or associated with disturbed surface layers can be affected. Here, we also employ UV exposure as a well-defined, model approach to induce coating loss.36,49 Other likely routes of coating degradation (e.g., biodegradation) have not yet been so thoroughly characterized. In this study, we exposed the PEG-AuNPs to UV irradiation for 4 days in DI water to induce nearly complete removal of the PEG coating (Fig. S6†) and then evaluated the effect on hetero-aggregation after 2 h of mixing with the SONE-1 soil slurry. Details of the results are reported in the ESI.†Similar percent recovery of spiked Au was obtained in the <0.45 μm fraction regardless of UV exposure: (1.25 ± 0.03)% and (1.39 ± 0.1)%, for the unexposed and UV-exposed PEG-AuNPs, respectively. Likewise, similar percent recovery was also obtained in the <1 μm fraction: (10.6 ± 0.6)% and (11.8 ± 1.1)%, for the unexposed and UV-exposed PEG-AuNPs, respectively. These results are consistent with those from the coating comparison experiments, where similar recoveries of Au in the different size fractions from the soil slurry were obtained regardless of coating type. These results differ notably from our previous study on UV-irradiated PEG-AuNPs, where loss of the coating significantly changed the homo-aggregation rate of the AuNPs.36 The results in the soil slurry imply that, for low concentrations of AuNPs undergoing primarily hetero-aggregation with naturally occurring particles, the presence or absence of the polymeric coating may not be significant with respect to the hetero-aggregation process, despite the steric forces imparted by the dense PEG coating. A likely explanation is that nearly all of the AuNPs have attached to a soil colloid or particle within the mixing time probed here (2 h), because of the high concentration of soil colloids and hence high collision rate. Alternatively, the presence of NOM in the soil slurry may play a role in mitigating differences between coatings. Differences in the attachment efficiency, α, attributable to the presence of the PEG coating may only be measurable at either shorter mixing times (minutes) or lower soil colloid concentrations.
4 Conclusions
In this work, an extraction procedure, based on standard leaching methods and previous studies, was applied to soil and sediment matrices spiked with various AuNP concentrations and surface coatings (as a model ENM). The low percentage of AuNPs extracted (recovered) from soil suspensions after 24 h of contact, suggests that their environmental mobility is greatly reduced. Additionally, the kinetic study has shown that the attachment of AuNPs onto soil particles is rapid (Au loss >80% in first 2 h of contact) and begins as soon as the AuNPs are exposed to soil particles and leachant. Thus, if AuNPs or other ENMs are released into the environment and enter soil or sedimentary compartments, interactions with naturally occurring particles are expected to play a substantial role in mitigating transport and determining fate. The influence of surface coating, including UV degradation of surface coatings, on the attachment of AuNPs to soil particles was not evident. Furthermore, we conclude that NOM plays a substantial role in the fate and transport of ENMs by decreasing the predicted influence of native surface chemistry and functional coatings, much as serum proteins adhere to and mask the intrinsic surface properties of ENMs in biological systems. We have also demonstrated how an approach based on the combination of in situ complementary and hyphenated analytical techniques can effectively be used to interrogate and quantify these complex interactions involving natural matrices. The approach utilized here can be easily extended to other metal containing ENMs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to Stephen Wilson of USGS for kindly providing SONE-1 for this study. SML acknowledges the National Research Council for a postdoctoral fellowship. The authors thank Danielle Gorka, Dave Holbrook and Elijah Petersen of NIST for helpful comments and review of the manuscript.References
- F. Laborda, E. Bolea, G. Cepriá, M. T. Gómez, M. S. Jiménez, J. Pérez-Arantegui and J. R. Castillo, Anal. Chim. Acta, 2016, 904, 10–32 CrossRef CAS PubMed.
- E. J. Petersen, D. X. Flores-Cervantes, T. D. Bucheli, L. C. C. Elliott, J. A. Fagan, A. Gogos, S. Hanna, R. Kägi, E. Mansfield, A. R. M. Bustos, D. L. Plata, V. Reipa, P. Westerhoff and M. R. Winchester, Environ. Sci. Technol., 2016, 50, 4587–4605 CrossRef CAS PubMed.
- S. Slomkowski, V. Alemán José, G. Gilbert Robert, M. Hess, K. Horie, G. Jones Richard, P. Kubisa, I. Meisel, W. Mormann, S. Penczek and F. T. Stepto Robert, Pure Appl. Chem., 2011, 83(12), 2229–2259 CrossRef CAS.
- M. Hassellöv and F. von der Kammer, Elements, 2008, 4, 401–406 CrossRef.
- S. Dubascoux, I. Le Hécho, M. Potin Gautier and G. Lespes, Talanta, 2008, 77, 60–65 CrossRef CAS PubMed.
- H. El Hadri, G. Lespes, P. Chéry and M. Potin-Gautier, Anal. Bioanal. Chem., 2014, 406, 1111–1119 CrossRef CAS PubMed.
- S. Harguindeguy, P. Crançon, F. Pointurier, M. Potin-Gautier and G. Lespes, Chemosphere, 2014, 103, 343–348 CrossRef CAS PubMed.
- S. Serrano, M. A. Gomez-Gonzalez, P. A. O'Day, F. Laborda, E. Bolea and F. Garrido, J. Hazard. Mater., 2015, 286, 30–40 CrossRef CAS PubMed.
- F. He, D. Zhao, J. Liu and C. B. Roberts, Ind. Eng. Chem. Res., 2007, 46, 29–34 CrossRef CAS.
- X.-Q. Li, D. W. Elliott and W.-X. Zhang, Crit. Rev. Solid State Mater. Sci., 2006, 31, 111–122 CrossRef CAS.
- S. J. Klaine, P. J. J. Alvarez, G. E. Batley, T. F. Fernandes, R. D. Handy, D. Y. Lyon, S. Mahendra, M. J. McLaughlin and J. R. Lead, Environ. Toxicol. Chem., 2008, 27, 1825–1851 CrossRef CAS PubMed.
- A. R. Whitley, C. Levard, E. Oostveen, P. M. Bertsch, C. J. Matocha, F. V. D. Kammer and J. M. Unrine, Environ. Pollut., 2013, 182, 141–149 CrossRef CAS PubMed.
- G. Cornelis, L. Pang, C. Doolette, J. K. Kirby and M. J. McLaughlin, Sci. Total Environ., 2013, 463–464, 120–130 CrossRef CAS PubMed.
- K. Tiede, S. P. Tear, H. David and A. B. A. Boxall, Water Res., 2009, 43, 3335–3343 CrossRef CAS PubMed.
- B. Kim, M. Murayama, B. P. Colman and M. F. Hochella, J. Environ. Monit., 2012, 14, 1128–1136 CAS.
- F. Van Koetsem, T. T. Geremew, E. Wallaert, K. Verbeken, P. Van der Meeren and G. Du Laing, Ecol. Eng., 2015, 80, 140–150 CrossRef.
- K. N. M. Mahdi, R. J. B. Peters, E. Klumpp, S. Bohme, M. v. d. Ploeg, C. Ritsema and V. Geissen, Environ. Nanotechnol. Monit. Manage., 2017, 7, 24–33 CrossRef.
- N. Akaighe, R. I. MacCuspie, D. A. Navarro, D. S. Aga, S. Banerjee, M. Sohn and V. K. Sharma, Environ. Sci. Technol., 2011, 45, 3895–3901 CrossRef CAS PubMed.
- E. M. Hotze, T. Phenrat and G. V. Lowry, J. Environ. Qual., 2010, 39, 1909–1924 CrossRef CAS PubMed.
- J. Gigault and V. A. Hackley, Anal. Chim. Acta, 2013, 763, 57–66 CrossRef CAS PubMed.
- H. El Hadri and V. A. Hackley, Environ. Sci.: Nano, 2017, 4, 105–116 RSC.
- B. Collin, E. Oostveen, O. V. Tsyusko and J. M. Unrine, Environ. Sci. Technol., 2014, 48, 1280–1289 CrossRef CAS PubMed.
- V. Merdzan, R. F. Domingos, C. E. Monteiro, M. Hadioui and K. J. Wilkinson, Sci. Total Environ., 2014, 488–489, 316–324 CrossRef CAS PubMed.
- R. F. Domingos, C. Franco and J. P. Pinheiro, Environ. Sci. Pollut. Res., 2015, 22, 2900–2906 CrossRef CAS PubMed.
- EPA, Liquid-Solid partitioning as a function of liquid-to-solid ratio in solid materials using a parallel batch procedure-Method 1316, U.S. Environmental Protection Agency, October, 2012 Search PubMed.
- ISO, Soil quality - Guidance on leaching procedures for subsequent chemical and ecotoxicological testing of soils and soil materials (ISO 18772:2008), International Organisation for Standardisation, Geneva, Switzerland, 2008 Search PubMed.
- A. Praetorius, N. Tufenkji, K.-U. Goss, M. Scheringer, F. von der Kammer and M. Elimelech, Environ. Sci.: Nano, 2014, 1, 317–323 RSC.
- G. Cornelis, Environ. Sci.: Nano, 2015, 2, 19–26 RSC.
- A. L. Dale, G. V. Lowry and E. A. Casman, Environ. Sci.: Nano, 2015, 2, 27–32 RSC.
- NIST, Standard Reference Material 2709a, San Joaquin Soil, Certificate of Analysis, National Institute of Standards and Technology, U.S. Department of Commerce, 2009 Search PubMed.
- NIST, Standard Reference Material 1646a, Estuarine Sediment, Certificate of Analysis, National Institute of Standards and Technology, U.S. Department of Commerce, 2004 Search PubMed.
- EPA, Methods for measuring the acute toxicity of effluents and receiving waters to freshwater and marine organisms, U.S. Environmental Protection Agency, 2002 Search PubMed.
- L. J. Gimbert, P. M. Haygarth, R. Beckett and P. J. Worsfold, Environ. Sci. Technol., 2005, 39, 1731–1735 CrossRef CAS PubMed.
- K. A. Smith, Soil and Environmental Analysis: Physical Methods, Revised, and Expanded, Taylor & Francis, 2000 Search PubMed.
- T. Fischer, J. Alsins and B. Berne, Int. J. Dermatol., 1984, 23, 633–637 CrossRef CAS PubMed.
- S. M. Louie, J. M. Gorham, E. A. McGivney, J. Liu, K. B. Gregory and V. A. Hackley, Environ. Sci.: Nano, 2016, 3, 1090–1102 RSC.
- NIST, Reference Material 8012, Gold Nanoparticles, Nominal 30 nm Diameter, Report of Investigation, National Institute of Standards and Technology, U.S. Department of Commerce, 2015 Search PubMed.
- NIST, Reference Material 8013, Gold Nanoparticles, Nominal 60 nm Diameter, Report of Investigation, National Institute of Standards and Technology, U.S. Department of Commerce, 2015 Search PubMed.
- M. Andersson, B. Wittgren and K.-G. Wahlund, Anal. Chem., 2003, 75, 4279–4291 CrossRef CAS PubMed.
- J. Gigault, J. M. Pettibone, C. Schmitt and V. A. Hackley, Anal. Chim. Acta, 2014, 809, 9–24 CrossRef CAS PubMed.
- N. S. Simon, S. A. Hatcher and C. Demas, Chem. Geol., 1992, 100, 175–189 CrossRef CAS.
- D. B. Smith, W. F. Cannon, L. G. Woodruff, F. Solano, J. E. Kilburn and D. L. Fey, Geochemical and mineralogical data for soils of the conterminous United States, Report 801, Reston, VA, 2013 Search PubMed.
- L. E. Barton, M. Therezien, M. Auffan, J.-Y. Bottero and M. R. Wiesner, Environ. Eng. Sci., 2014, 31, 421–427 CrossRef CAS.
- S. B. Hendricks and L. T. Alexander, Soil Sci., 1939, 48, 257–272 CrossRef CAS.
- D.-H. Tsai, T. J. Cho, F. W. DelRio, J. Taurozzi, M. R. Zachariah and V. A. Hackley, J. Am. Chem. Soc., 2011, 133, 8884–8887 CrossRef CAS PubMed.
- F. Gottschalk, T. Sun and B. Nowack, Environ. Pollut., 2013, 181, 287–300 CrossRef CAS PubMed.
- L. Kvítek, A. Panáček, J. Soukupová, M. Kolář, R. Večeřová, R. Prucek, M. Holecová and R. Zbořil, J. Phys. Chem. C, 2008, 112, 5825–5834 Search PubMed.
- D. P. Stankus, S. E. Lohse, J. E. Hutchison and J. A. Nason, Environ. Sci. Technol., 2010, 45, 3238–3244 CrossRef PubMed.
- S. M. Louie, J. Gorham, J. Tan and V. A. Hackley, Environ. Sci.: Nano, 2017, 4, 1866–1875 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional methods, results and parameters for humic acid extraction, ICP-MS, AF4-UV-MALS-ICP-MS, and spICP-MS, are presented in the electronic supporting information. See DOI: 10.1039/c7en00868f |
| ‡ The identification of any commercial product or trade name does not imply endorsement or recommendation by the National Institute of Standards and Technology. |
| This journal is © The Royal Society of Chemistry 2018 |