
A novel low-thermal-budget approach for the co-production of ethylene and hydrogen via the electrochemical non-oxidative deprotonation of ethane†

Abstract
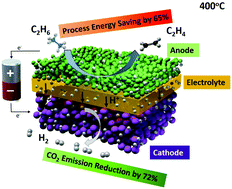
The oversupply of ethane, a major component of natural gas liquids, has stimulated the wide applications of ethylene since the shale gas revolution. However, ethylene production is energy-intensive and represents the most energy-consuming single process in the chemical industry. In this communication, we report, for the first time, a novel low-thermal-budget process for the co-production of ethylene and pure hydrogen using a proton-conducting electrochemical deprotonation cell. At a constant current density of 1 A cm−2, corresponding to a hydrogen production rate of 0.448 mol cm−2 per day, and 400 °C, a close to 100% ethylene selectivity was achieved under an electrochemical overpotential of 140 mV. Compared to an industrial ethane steam cracker, the electrochemical deprotonation process can achieve a 65% saving in process energy and reduce the carbon footprint by as much as 72% or even more if renewable electricity and heat are used. If the heating value of produced hydrogen is taken into account, the electrochemical deprotonation process actually has a net gain in processing energy. The electrochemical deprotonation process at reduced temperatures in the present study provides a disruptive approach for petrochemical manufacturing, shifting the paradigm from thermal chemical practice to a clean energy regime.
 Please wait while we load your content...
Please wait while we load your content...

