
Carbon dioxide in the cage: manganese metal–organic frameworks for high performance CO2 electrodes in Li–CO2 batteries†
 *a
Bo
Wang
*a
*a
Bo
Wang
*a
Abstract
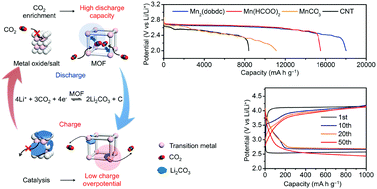
Improving the reversibility and energy efficiency of Li–CO2 electrochemistry would help in developing practical Li–air batteries capable of providing stable power supply in the presence of CO2. However, it is hard for most existing electrodes to convert CO2 effectively (high discharge capacity) and efficiently (low charge potential). Herein, we have, for the first time, identified the potential of metal–organic frameworks (MOFs) as porous catalysts in CO2 electrodes, taking advantage of their high capability for CO2 capture and monodispersed active metal sites for Li2CO3 decomposition. In particular, eight porous MOFs (Mn2(dobdc), Co2(dobdc), Ni2(dobdc), Mn(bdc), Fe(bdc), Cu(bdc), Mn(C2H2N3)2, and Mn(HCOO)2) and two nonporous materials (MnCO3 and MnO) were investigated. Among them, Mn2(dobdc) achieves a remarkable discharge capacity of 18 022 mA h g−1 at 50 mA g−1, while Mn(HCOO)2 retains a low charge potential of ∼4.0 V even at 200 mA g−1 for over 50 cycles. The Li–CO2 electrochemistry on MOF electrodes was investigated with an arsenal of characterization techniques, including X-ray diffraction, scanning electron microscopy, electrochemical impedance spectroscopy, Raman spectroscopy and in situ differential electrochemical mass spectrometry. The findings provide useful design principles for improving the reversibility and energy efficiency of Li–CO2 electrochemistry and will herald the advent of practical technologies that enable energy-efficient utilization of CO2 for electrochemical energy storage and supply.
- This article is part of the themed collection: 2018 Energy and Environmental Science HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

