
Construction of hierarchical Ni–Co–P hollow nanobricks with oriented nanosheets for efficient overall water splitting†

Abstract
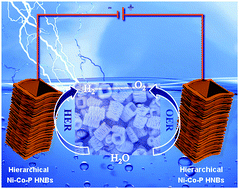
Complex nano-architectures with ordered two-dimensional (2D) building blocks are a class of promising electrocatalysts for different electrochemical technologies. In this work, a novel template-engaged strategy followed by sequential etching and phosphorization treatments is demonstrated to fabricate open and hierarchical Ni–Co–P hollow nanobricks (HNBs) via the assembly of oriented 2D nanosheets. Benefiting from the unique nano-architectures with large electrolyte-accessible surface and abundant mass diffusion pathways, the as-prepared Ni–Co–P HNBs exhibit high electrocatalytic activity, which affords the current density of 10 mA cm−2 at low overpotentials of 270 mV and 107 mV for oxygen and hydrogen evolution reactions respectively, and excellent stability in an alkaline medium. Remarkably, when used as both the anode and cathode, a low cell voltage of 1.62 V is required to reach the current density of 10 mA cm−2, making the Ni–Co–P HNBs an efficient bifunctional electrocatalyst for overall water splitting.
- This article is part of the themed collection: 2018 Energy and Environmental Science HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

