
A mechanistic model of fast pyrolysis of hemicellulose†

Abstract
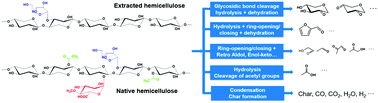
Hemicellulose is one of the major components of lignocellulosic biomass, which is an abundant source of renewable carbon on the Earth and has potential for the production of renewable drop-in transportation fuels and multiple commodity chemicals. In this work, a structure for hemicellulose extracted from corn stover was proposed to capture the experimentally characterized structural properties. A mechanistic model for hemicellulose pyrolysis was constructed based on the reaction family approach that we used for cellulose pyrolysis before. The model described the decomposition of hemicellulose chains, reactions of intermediates, and formation of a range of low molecular weight products (LMWPs) at the mechanistic level and specified rate constants for all the reactions in the network. Overall, 504 reactions of 114 species were included in the mechanistic model for fast pyrolysis of extracted hemicellulose. The mechanistic model closely matched experimental yields of various products with mass yield ≥1 wt%. Modeling results show that both the degree of polymerization and the polydispersity index of hemicellulose have an insignificant effect on the pyrolysis product distribution. Then, the mechanistic model of extracted hemicellulose is further extended to simulate the fast pyrolysis of native hemicellulose. Comparison of the model results showed that fast pyrolysis of native hemicellulose from corn stalk yielded more char, gaseous species, acetol, and much more acetic acid than that of extracted hemicellulose from corn stover, while yielding less 1,2-anhydroxylopyranose, 1,2;3,4-dianhydroxylopyranose and glycolaldehyde.
 Please wait while we load your content...
Please wait while we load your content...

