
Dissolution, migration, and deposition of transition metal ions in Li-ion batteries exemplified by Mn-based cathodes – a critical review
 *a
and
Khalil
Amine
*a
*a
and
Khalil
Amine
*a
Abstract
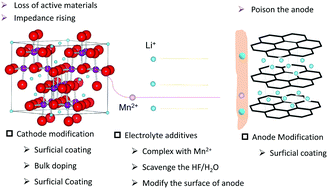
Unlike the revolutionary advances in the anodes of lithium-ion batteries from Li intercalation materials to Li alloy and/or conversion reaction materials, the development of the cathode is still dominated by the Li intercalation compounds. Transition metal ions are essential in these cathodes as the rapid redox reaction centers, and one of the biggest challenges for the TM-based cathodes is the capacity and power fading especially at an elevated temperature, which is directly associated with the dissolution–migration–deposition (DMD) process of TMs from the cathode materials. This process not only alters the surface structure of the cathode materials, but more importantly, changes the SEI composition at the anode side. There is no doubt that the TM-DMD issue should be addressed thoroughly to unlock the potential of these compounds to enable a prolonged battery lifetime. This review article mainly focuses on research activities with regard to the DMD process in TM-based cathode materials. In the first four sections, we choose Mn-based cathodes as an example to discuss how Mn DMD relates to the capacity fade of the cell, and what possible approaches might suppress the DMD process by modification of the electrode or electrolyte. In the fifth section, we discuss the TM DMD process in Ni-, Co-, Fe- and V-containing cathode materials. This article reviews the frontier electrochemical research on TM-based cathodes and summarizes the progress and challenges, thereby helping to advance future R&D of LIBs.
- This article is part of the themed collection: 2018 Energy and Environmental Science HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

