
Constructing canopy-shaped molecular architectures to create local Pt surface sites with high tolerance to H2S and CO for hydrogen electrooxidation†
Tao
Wang‡
a,
Zhi-Xin
Chen‡
a,
Song
Yu
a,
Tian
Sheng
 b,
Hai-Bin
Ma
a,
Lu-Ning
Chen
a,
Muhammad
Rauf
a,
Hai-Ping
Xia
a,
Zhi-You
Zhou
*a and
Shi-Gang
Sun
a
b,
Hai-Bin
Ma
a,
Lu-Ning
Chen
a,
Muhammad
Rauf
a,
Hai-Ping
Xia
a,
Zhi-You
Zhou
*a and
Shi-Gang
Sun
a
aCollaborative Innovation Center of Chemistry for Energy Materials, State Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: zhouzy@xmu.edu.cn
bCollege of Chemistry and Materials Science, Anhui Normal University, Wuhu, 241000, P. R. China
First published on 30th November 2017
Abstract
Rational design and construction of the local environment of active sites on noble metal surfaces is a promising, but challenging, approach for developing high-selectivity catalysts. This study presents an effective approach, via engineering local active sites, aiming to solve the critical problem of H2S and CO poisoning of Pt catalysts for H2 electrooxidation, the anode reaction of polymer electrolyte membrane fuel cells. A canopy-shaped molecular architecture was constructed by immobilizing an organic molecule, 2,6-diacetylpyridine (DAcPy), on Pt surface, which exhibits high H2S (1 ppm) and CO (100 ppm) tolerance. Through electrochemical, spectroscopic, and DFT studies, as well as comparative investigation of analogous structure molecules, it was revealed that DAcPy can be strongly adsorbed on Pt surface through tridentate coordination (two Pt–C and one Pt–N bonds), allowing it to compete with CO and H2S adsorption. The pyridine ring of DAcPy is in a tilted orientation, providing some protection underneath the ring for Pt atoms. Such a height-limited space is just accessible for small-sized H2, but not for relatively large H2S and CO. This study demonstrates that regulating steric hindrance to protect active sites is a promising approach for designing highly selective electrocatalysts.
Broader contextPolymer electrolyte membrane fuel cell (PEMFC) is an efficient and clean power source for electric vehicles. The fuel of PEMFC is H2, which is mainly supplied by steam reforming from fossil fuels cost-efficiently. Unfortunately, reformed H2 usually contains some impurities like CO and H2S, which can poison severely Pt catalysts in fuel cells. Classic approach to enhance CO tolerance relies on combining Pt with another metal (e.g., PtRu) or metallic oxide to promote CO oxidation. Although this approach has been developed for several decades, it is yet unable to meet the challenge requirements. Herein, we developed a new strategy to enhance CO and H2S tolerance for H2 oxidation on Pt by constructing active sites protected by molecular architectures. We found that a pyridine derivative, 2,6-diacetylpyridine, can be strongly adsorbed on Pt surface, and creates size-selective wedge-shaped spaces, which are accessible for small-sized H2, but not for relatively large H2S and CO. This study indicates that regulating steric hindrance on metal surface active sites is a promising approach for designing high selective electrocatalysts. |
Introduction
Precise construction of local active sites to regulate the reaction environment can result in superior selectivity of enzyme towards different substrates. Analogously, in heterogeneous catalytic systems, rational design and precise construction of local active sties could be used in development of advanced catalytic systems, which remains a great challenge.1–5 Pt-based catalysts are widely used in many catalytic processes; however, they show a weakness in selectivity. In particular, some small-size molecules (such as CO and H2S), which can strongly adsorb on Pt, will block the active sites and finally deactivate the catalysts. This disadvantage severely limits the performance of Pt-based catalysts in some practical applications.6–12 For instance, Pt is highly active for the hydrogen oxidation reaction (HOR), and is the essential catalyst for the anode reaction of polymer electrolyte membrane fuel cells (PEMFCs). In industry, H2 is mainly supplied by reforming fossil fuels because of the low cost of this process. However, the H2 produced contains some impurities such as CO and H2S, which can poison anode Pt catalysts.13–17 Therefore, there is an urgent need to design a Pt-based catalyst system that can tolerate these poisonous species. Taking inspiration from bio-systems, in which local active sites are constructed by surface modification to regulate the mass transfer process, could offer a potentially valid approach for solving this challenging problem.Chemical modification of electrodes (CME) can be an effective way to introduce new physical and (electro)chemical properties to the electrodes.18 Substrate electrodes can be chemically modified using, for example, a molecular monolayer, polymer thin film, metal–organic supramolecule, etc.19 Recent developments in CME have yielded some design principles for constructing functional molecular architectures, contributing to regulation of the electronic state and the distribution of active sites on metal surfaces.7,20–35 For example, Markovic and co-workers demonstrated that modification of the Pt surface with cyanide can result in some isolated Pt atoms, which avoid any poisoning by non-covalent adsorbates such as PO43− and SO42− in the oxygen reduction reaction.36 However, to the best of the authors’ knowledge, as yet, there are no reports of CME that can prevent poisoning of Pt catalysts by strongly adsorbed CO or H2S. The challenges for designing such a functional catalytic system mainly originate from two aspects: (1) the strongly covalent interaction between CO/H2S and the Pt surface, giving them the ability to ‘kick out’ the modified functional molecules.37 Thus, the interaction between molecular architectures and surface needs to be strong enough to compete with CO/H2S. In coordination chemistry, a chelate that forms through a central metal ion with multidentate ligands is much more stable than a common complex with monodentate ligands. Hence, a multidentate-ligand approach could be promising in building stable molecular architectures on metal surfaces; (2) unlike large hydrated PO43− and SO42− ions, the diameters of CO and H2S molecules go down to 2–5 Å, allowing them to adsorb on isolated metal atoms created by molecular architectures. For example, previous studies in CME were mainly concerned with building a highly ordered 2D pattern to separate atoms on the metal surfaces and generate isolated active sites.23,38–41 The constructed steric hindrance of this approach is in the planar dimension or parallel to the surface. The vertical direction of the exposed active sites is free of hindrance, so that CO and H2S can still adsorb on such sites. To address this limitation, it is necessary to design molecular architectures from two dimensions to three dimensions, that is to construct steric hindrance in the vertical direction of the exposed active sites.
Herein, a new approach is proposed to engineer local Pt active sites for H2 oxidation with high CO and H2S tolerance by constructing an organic molecular architecture. In practice, a kind of pyridine derivative with two carbonyl groups, named 2,6-diacetylpridine (DAcPy) is reduced and strongly adsorbed on Pt surface through tridentate coordination (two Pt–C and one Pt–N bonds). This molecular architecture acts as a canopy, under which Pt atoms can be accessible for small-sized H2, but not for relatively large CO and H2S. Thus, DAcPy-modified Pt exhibits even higher CO tolerance than most frequently used PtRu alloy catalysts.
Results and discussion
To evaluate the proposed approach, HOR catalyzed by commercial Pt/C was carried out in 0.5 M H2SO4 solution with Na2S (existing form is H2S in acidic solution) as a poison reagent. Bare Pt/C is very susceptible to H2S poisoning. Fig. 1a depicts HOR polarization curves of the bare Pt/C in 0.5 M H2SO4 solution at different concentrations of Na2S. In the 10 μM Na2S solution, only 69% of HOR current at 0.1 V was maintained. When the Na2S concentration was increased to 100 μM, the bare Pt/C was poisoned completely by H2S, and no considerable HOR current could be observed.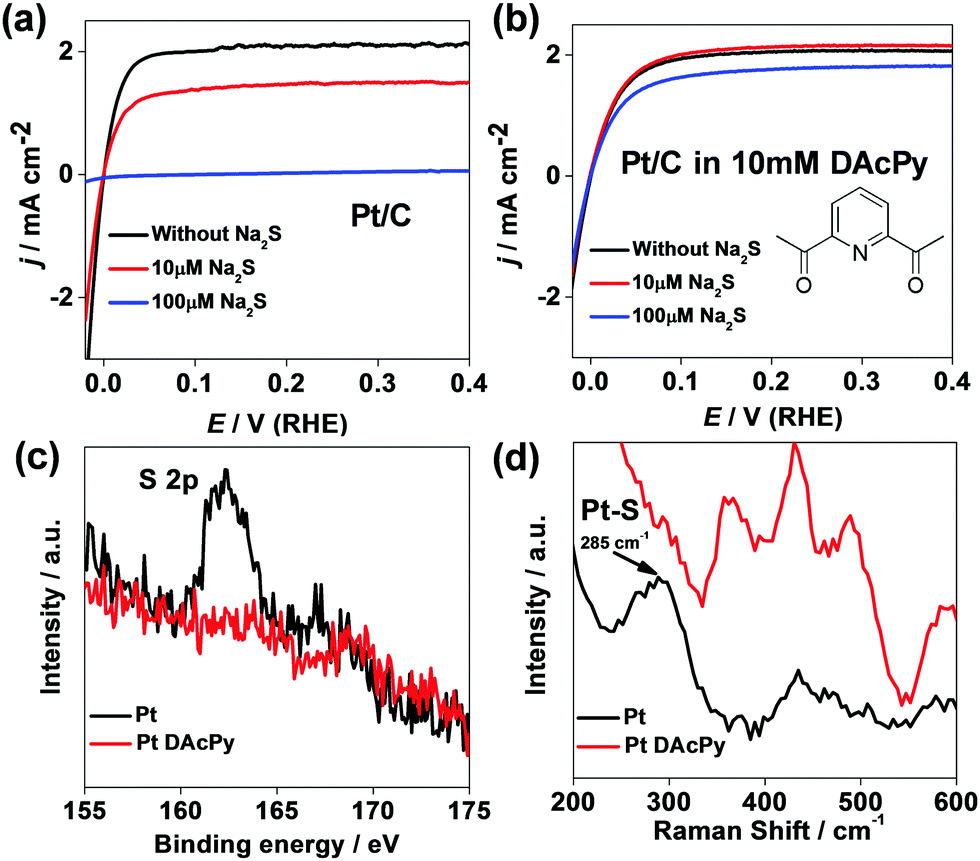 | ||
| Fig. 1 HOR polarization curves of commercial Pt/C in 0.5 M H2SO4 (a) and 0.5 M H2SO4 + 10 mM DAcPy (b) at different concentrations of Na2S. Scan rate: 10 mV s−1; rotating rate: 900 rpm. (c) XPS of Pt foils poisoned by 10 μM Na2S with (red) and without (black) DAcPy modification. (d) Raman spectra of Au@Pt core–shell nanoparticles collected in 10 μM Na2S + 0.5 M H2SO4 with (red) and without (black) 10 mM DAcPy. | ||
Interestingly, the addition of DAcPy to the solution could enhance significantly the H2S tolerance for the Pt catalyst. Fig. 1b shows HOR polarization curves of the Pt/C in 10 mM DAcPy + 0.5 M H2SO4 solution. DAcPy can spontaneously adsorb on the Pt surface in this solution. Clearly, the HOR catalytic current changed little after adding 10 μM Na2S. When the Na2S concentration was increased to 100 μM, the Pt/C modified with DAcPy still maintained 84% of initial activity at 0.1 V.
To confirm the success of blocking sulfur poisoning by DAcPy, the surface characterization was carried out using X-ray photoelectron spectroscopy (XPS) and in situ electrochemical surface-enhanced Raman spectroscopy (EC-SERS).
The commercial Pt/C contains high-surface-area carbon black, allowing physical adsorption of sulfur-containing species including H2S. To avoid such interference, a smooth Pt foil was used instead of the Pt/C for the XPS test. Fig. 1c shows the XPS of Pt foil with and without DAcPy modification after immersion in 10 μM Na2S and then washing. Clearly, there is no detectable S element signal on DAcPy-modified Pt surface; in contrast, a remarkable peak at 162 eV, which belongs to S2−, is observed in the spectrum of the Pt foil without DAcPy modification.
Commercial Pt/C has no surface-enhanced Raman effect. In the EC-SERS test, Au@Pt core–shell nanoparticles were used, which can provide Raman enhancement from the Au core. Fig. 1d shows Raman spectra of Au@Pt core–shell nanoparticles collected in 10 μM Na2S + 0.5 M H2SO4 with and without 10 mM DAcPy. When bare Au@Pt nanoparticles were poisoned by 10 μM Na2S, a peak at 285 cm−1 was observed clearly, attributed to Pt–S bonding.42 This peak disappears from the spectrum of Au@Pt modified with DAcPy under the same conditions. This SERS result confirms that modification of Pt with DAcPy can help to prevent H2S adsorption on Pt surface.
For fuel cell applications, it is better to use DAcPy-coated Pt/C rather than adding DAcPy into the solution, as the additive can be easily leached out by water, a product of the oxygen reduction reaction. For this goal, Pt/C coated with DAcPy was prepared. In brief, the Pt/C and DAcPy were dispersed in isopropanol, and heated at 80 °C for 12 h. It was found that DAcPy can adsorb on Pt surface more firmly using this approach rather than simply immersing in 10 mM DAcPy aqueous solution. The obtained Pt/C coated with DAcPy was separated by centrifugation and washing. The size (3.4 nm) of Pt nanoparticles changed little after the DAcPy modification, as shown by transmission electron microscopy (TEM) (Fig. S1, ESI†). Fig. 2 shows the 5 h stability tests of two types of DAcPy-modified Pt to resist H2S and CO during HOR. When the H2 containing 1 ppm H2S was fed, only 37% of initial current remained for the bare Pt/C after the stability test, compared with 83% for the Pt/C in 0.5 M H2SO4 + 10 mM DAcPy, and 90% for DAcPy-coated Pt/C in blank 0.5 M H2SO4. Results were similar for 100 ppm CO: DAcPy-coated Pt/C has better CO tolerance than Pt/C in DAcPy solution, with remaining activity f 87% versus 78% after the stability test. In contrast, the bare Pt/C only maintained 24% of initial activity. These results demonstrate the good prospects of DAcPy-coated Pt/C for fuel cell applications.
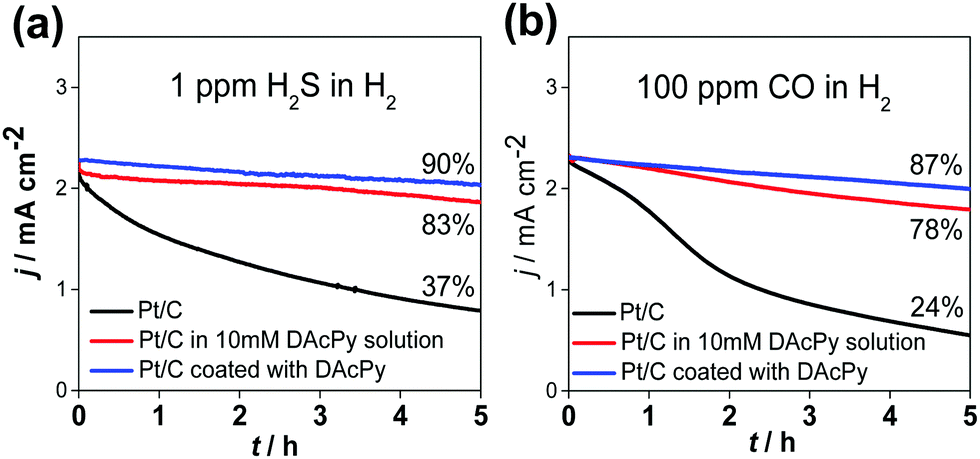 | ||
| Fig. 2 H2S and CO tolerance test of commercial Pt/C (black), Pt/C in 10 mM DAcPy solution (red), and Pt/C coated with DAcPy (blue) in 0.5 M H2SO4 solution saturated with H2/1 ppm H2S balance gas (a) and H2/100 ppm CO balance gas (b). The potential was held at 0.1 V (RHE). Rotating rate: 1600 rpm. | ||
PtRu alloy is well-known as a CO-tolerant electrocatalyst for HOR. PtRu catalyst and PtRu modified with DAcPy were tested for tolerance to 100 ppm CO. The I–t curves in Fig. 3a show that DAcPy was also able to enhance CO tolerance on Pt–Ru alloyed surface. A comparison of the CO tolerance of Pt/C, PtRu and DAcPy modified catalysts is given in Fig. 3b. The time (t−20%) to lose 20% current density was selected to assess the ability to resist CO. The result shows that DAcPy-modified Pt/C catalyst exhibited remarkable improvement in CO tolerance, and was even better than PtRu. Moreover, DAcPy modified PtRu exhibited the best CO tolerance among these five catalysts. This indicates that an approach including DAcPy modification is also valid for Pt-based alloy catalysts.
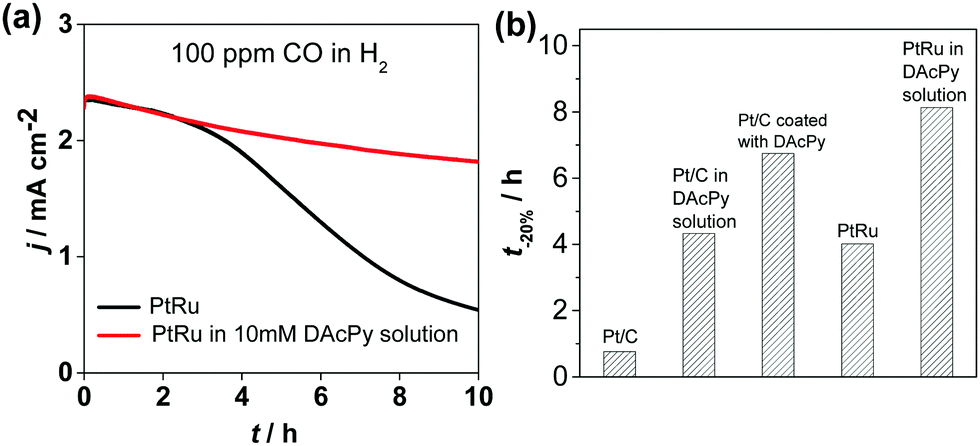 | ||
| Fig. 3 (a) CO tolerance test of commercial PtRu in 0.5 M H2SO4 solution with (red line) and without (green line) 10 mM DAcPy. The solution was purged with H2/100 ppm CO balance gas. Rotating rate: 1600 rpm. (b) Comparison of CO tolerance of Pt/C, PtRu, Pt/C with DAcPy, and PtRu with DAcPy. The time for decline of 20% current density was used to evaluate the stability. | ||
DAcPy is a relatively complex organic molecule with two acetyl groups and one pyridine cycle. To identify any effects of molecular structure, the sulfur tolerance was tested of three analogous compounds: 2-acetylpyridine (AcPy) with only one acetyl group, pyridine (Py) without acetyl group, and 1,3-diacetylbenzene (DAcPh) with two acetyl groups, but with a benzene ring rather than a pyridine ring. All these molecules can be adsorbed on Pt surface, resulting in considerable suppression of hydrogen adsorption/desorption on Pt (Fig. S2, ESI†). Fig. 4a–c shows HOR polarization curves of Pt/C modified with AcPy, Py, and DAcPh, in H2-saturated 0.5 M H2SO4 solution with different concentrations of Na2S. Clearly, unlike DAcPy, all of these molecules are unable to enhance the sulfur tolerance for Pt/C. The current densities remaining at 0.1 V were used to evaluate the sulfur tolerance (Fig. 4d). When 10 μM Na2S was added to the solution, the current densities at 0.1 V were decreased by 25%, 40%, and 58% for AcPy, Py, and DAcPh, respectively. When the Na2S concentration was increased to 100 μM, the Pt/C modified with Py was deactivated completely by sulfur as bare Pt/C, and Pt/C modified with AcPy and DAcPh only retained 5–15% of the initial activity. The much higher sulfur tolerance of DAcPy than AcPy, Py and DAcPh indicates that the combination of pyridinic N with two acetyl groups is essential for high sulfur tolerance. Both the pyridinic N and acetyl group may be bonded on Pt surface. It is predicted that more coordination sites on a ligand will result in stronger interaction between adsorbed molecules and Pt surface.
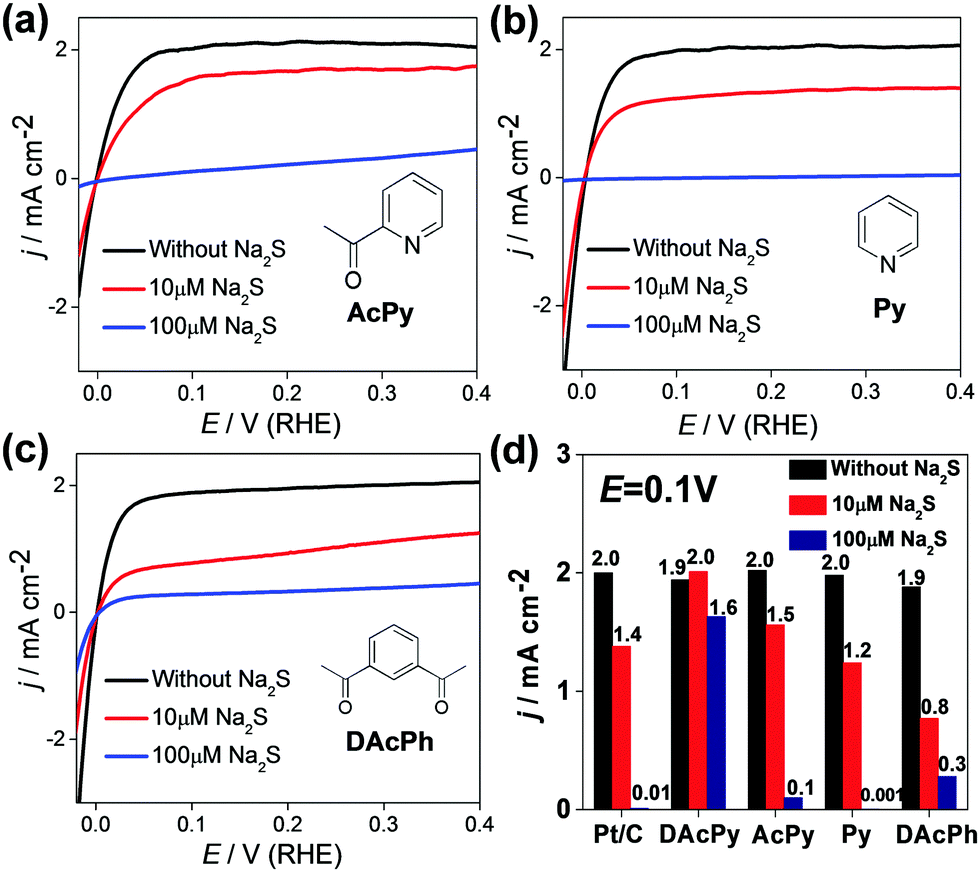 | ||
| Fig. 4 Polarization curves of Pt/C with AcPy (a), Py (b) and DAcPh (c) in H2-saturated 0.5 M H2SO4 solution with 10 μM Na2S (red line), 100 μM Na2S (blue line) and without Na2S (black line). The concentration of AcPy and Py was 10 mM, and about 3.5 mM for DAcPh (saturation solution). All polarization curves were recorded at 10 mV s−1. (d) Comparison of sulfur tolerance of Pt/C modified with different organic molecules. The current density at 0.1 V was used to evaluate the sulfur tolerance. | ||
To investigate the adsorption structure of DAcPy on Pt, electrochemical and spectroscopic characterization was carried out. Fig. 5a depicts cyclic voltammograms (CVs) of Pt/C in Ar-saturated 0.5 M H2SO4 with and without 10 mM DAcPy. Clearly, the adsorption of DAcPy significantly suppressed underpotential deposited hydrogen (HUPD, 0.05–0.35 V) and hydroxyl species (>0.80 V) on the Pt surface. The remaining surface accessible for HUPD was measured to be about 19% of the initial surface. Moreover, considerable current reduction at potential lower than 0.1 V could be observed in the first cycle in DAcPy solution. This reduction process declined quickly in the following cycles, but the HUPD current changed little (Fig. S3, ESI†). This result indicates that the cathodic current mainly came from the reduction reaction of DAcPy adsorbed on Pt. More information about adsorbed DAcPy was obtained through EC-SERS, with spectra collected on the Au@Pt core–shell nanoparticles at different potentials depicted in Fig. 5b. The bands at 994 and 1024 cm−1 were assigned to ring breathing mode and symmetric ring deformation mode of pyridine ring, respectively (Table S1, ESI†).43,44 The band at 1316 cm−1 was assigned to C–C stretching between the pyridine ring and carbonyl group; the band at 1560 cm−1 was assigned to pyridine ring C–C stretching mode.43 Besides, the band at around 1690 cm−1 was assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carbonyl group. As the potential decreased from 0.5 to 0.2 V, the band center of the carbonyl group shifted negatively from 1694 to 1681 cm−1. When the potential was further lowered to 0.1 V (this potential is close to the reduction potential in CVs in Fig. 5a), the band intensity of carbonyl group decreased significantly, and a new band appeared at around 370 cm−1. According to reference results,45–47 this new band could be attributed to Pt–C bond. The EC-SERS results indicate that adsorbed DAcPy on Pt can be converted into a reduced form through Pt–C coordination at the low potential.
O stretching of carbonyl group. As the potential decreased from 0.5 to 0.2 V, the band center of the carbonyl group shifted negatively from 1694 to 1681 cm−1. When the potential was further lowered to 0.1 V (this potential is close to the reduction potential in CVs in Fig. 5a), the band intensity of carbonyl group decreased significantly, and a new band appeared at around 370 cm−1. According to reference results,45–47 this new band could be attributed to Pt–C bond. The EC-SERS results indicate that adsorbed DAcPy on Pt can be converted into a reduced form through Pt–C coordination at the low potential.
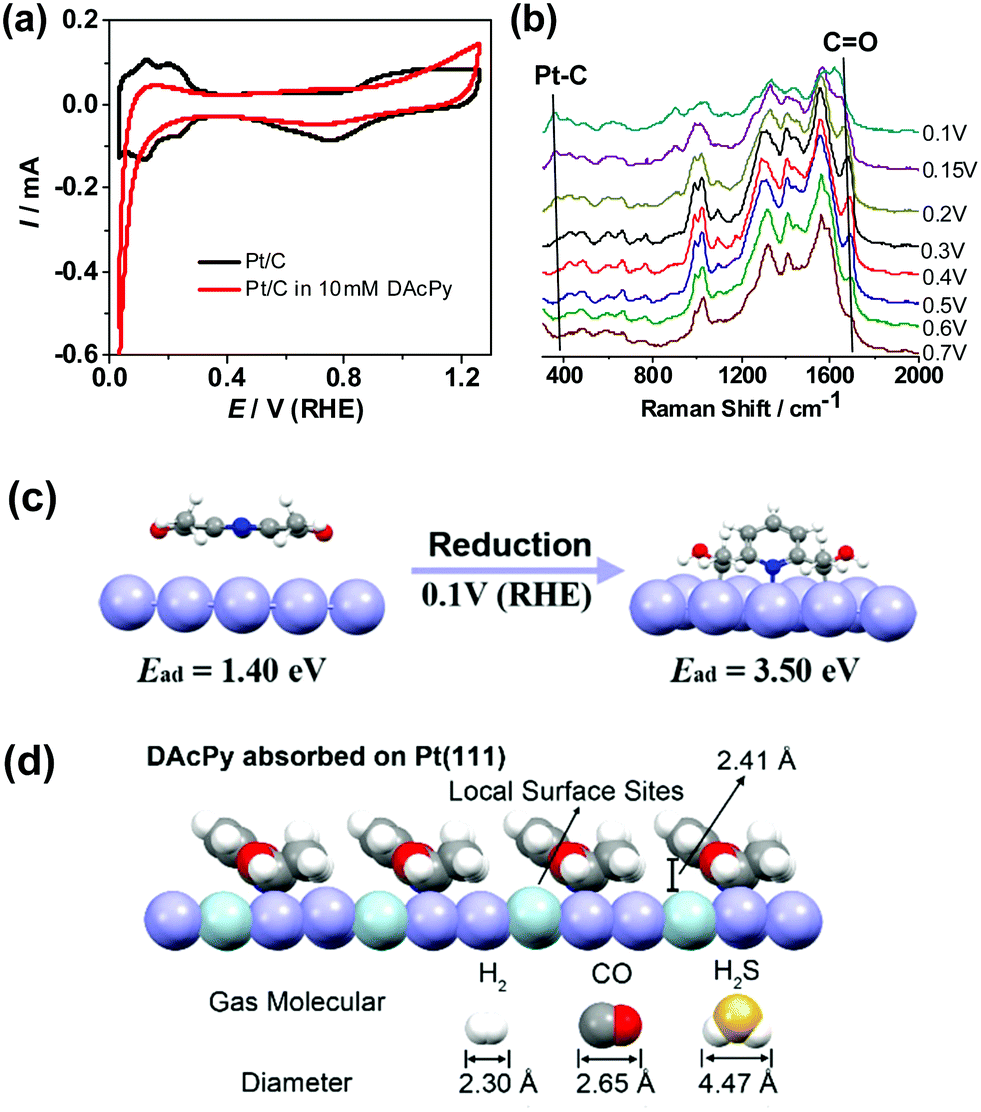 | ||
| Fig. 5 (a) Cyclic voltammograms of Pt/C in Ar-saturated 0.5 M H2SO4 without and with 10 mM DAcPy. (b) Electrochemical Raman spectra of Au@Pt core–shell nanoparticles collected in 0.5 M H2SO4 + 10 mM DAcPy at different potentials. (c) Adsorption structure of DAcPy on Pt(111) calculated from DFT. After reduction, DAcPy can be strongly adsorbed on Pt through tridentate coordination (two Pt–C and one Pt–N bonds) with the pyridine ring in tilted orientation. (d) The proposed model for reduced-form DAcPy adsorbed on Pt(111) from the side view. The height between pyridine ring and Pt atoms (light green spheres) under DAcPy is 2.41 Å. According to the diameters of H2, CO, and H2S, only H2 can be adsorbed on such narrow spaces. | ||
To better understand the adsorption mode of DAcPy on Pt, DFT calculations were performed (see ESI,† for detail). Pristine DAcPy prefers an adsorption configuration parallel to the surface through van der Waals forces because of large steric hindrance from the two acetyl groups. The adsorption energy of DAcPy by this mode is only 1.40 eV, which is even lower than that of Py (1.61 eV) through N atom adsorption (Fig. S4 and Table S2, ESI†). Clearly, this result cannot interpret the much higher sulfur tolerance of DAcPy than Py. However, when adsorption of DAcPy was reductive, the adsorption energy dramatically increased to 3.50 eV, much higher than that of CO (1.91 eV for CO adsorbed on fcc hollow sites) and H2S (1.12 eV), seen in Fig. S5 (ESI†). That is, DAcPy accepts two couples of proton and electron, and two carbonyl groups are reduced to hydroxyl groups and carbon atoms bonding with the hydroxyl group interact with Pt to form Pt–C bond (i.e., Pt–C–OH). This process is thermodynamically favorable, and the reaction energy can be as high as −2.10 eV. In this configuration, the adsorption of reduced DAcPy occurs via three coordination sites (two Pt–C bonds and one Pt–N bond) with the pyridine ring in tilted orientation (Fig. 5c). The very strong interaction between the reduced DAcPy and Pt enables this molecule to compete with H2S and CO adsorption. The adsorption energies of AcPy and its reduced form were also calculated, being 1.22 and 2.51 eV, respectively. This result is in agreement with the better sulfur tolerance of DAcPy than AcPy.
The coverage of DAcPy on the Pt surface is also an important parameter for blocking H2S and CO adsorption. Thermogravimetric analysis (TGA) and element analysis were used to estimate the coverage of DAcPy. To avoid the physical adsorption of DAcPy on carbon support, unsupported Pt black was used instead of Pt/C. The mass loss of adsorbed DAcPy on Pt black was 1.3%, and mainly occurred at around 150 °C (Fig. S6, ESI†). CHN element analysis indicated that the weight contents of C, N, and H were 1.158%, 0.162%, and 0.158%, respectively, after background correction (Table S2, ESI†). The DAcPy content calculated from the C element is 1.74%, slightly higher than that measured by TGA (1.3%). The average weight content of DAcPy was 1.5% according to the TGA and element analysis. The area occupied per DAcPy molecule was then calculated to be 37 Å2 (eqn (1)), corresponding to 5.6 Pt atoms on Pt(111).
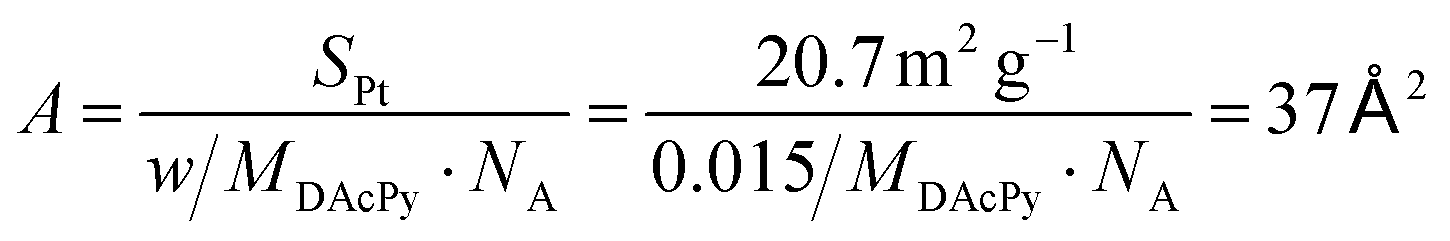 | (1) |
Each DAcPy will bind to three Pt atoms (two Pt–C and one Pt–N bonds). On average, 2.6 out of 5.6 Pt atoms (or 46% of surface atoms) are unoccupied. The CV result (Fig. 5a) indicates that only 19% of the Pt surface is free for HUPD. So, about 40% of unoccupied Pt atoms are accessible for HUPD for DAcPy-modified Pt surface.
On the basis of the adsorption model predicted by DFT calculation and the coverage information, a model of adsorbed DAcPy to block CO and H2S as shown in Fig. 5d is proposed. The DAcPy is adsorbed on the Pt surface with tilted-orientation pyridine ring. Under the ring, there exists a wedge-shaped space, where the height between the Pt and pyridine ring is 2.41 Å. When HOR proceeds, small-sized (2.30 Å) H2 can approach the isolated Pt atoms (light green spheres in Fig. 5d) under the pyridine ring, while larger CO (2.65 Å) and H2S (4.47 Å) will be hindered by the pyridine ring. That is, adsorbed DAcPy acts as a canopy to provide a height-limited space, which can block large-sized CO/H2S, but is still accessible for small-sized H2. To the authors’ knowledge, this is the first study to develop molecular architectures for Pt-based catalysts to resist CO and H2S by regulating the steric hindrance of local active sites. Such an approach does not rely on highly ordered and compact 2D molecular patterns, and could be a universal method for HOR to resist other poisoning species beyond CO and H2S.
Conclusions
An organic molecular architecture was constructed on a Pt surface to enhance H2S and CO tolerance for H2 electrooxidation. 2,6-Diacetylpyridine was selected, which can be strongly anchored on Pt surface through tridentate coordination with tilted-orientation pyridine ring. This molecule acts as a canopy, under which some isolated Pt atoms are accessible for small-sized H2, but not for larger CO and H2S. This approach was universally valid for different surfaces such as Pt nanoparticles and alloy surfaces. Moreover, this study also showed that constructing the local environment of active sites is a promising approach for developing new generation catalysts with high selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by grants from the Major State Basic Research Development Program of China (2015CB932303), National Science Foundation of China (91645121, 21373175 and 21621091).Notes and references
- P. Liu, R. Qin, G. Fu and N. Zheng, J. Am. Chem. Soc., 2017, 139, 2122–2131 CrossRef CAS PubMed.
- Y. J. Kang, P. D. Yang, N. M. Markovic and V. R. Stamenkovic, Nano Today, 2016, 11, 587–600 CrossRef CAS.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef PubMed.
- A. Cuesta, ChemPhysChem, 2011, 12, 2375–2385 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, S. Kuhl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- O. A. Baturina, A. Epshteyn, P. A. Northrup and K. E. Swider-Lyons, J. Electrochem. Soc., 2011, 158, B1198–B1205 CrossRef CAS.
- D. Y. Chung, M. J. Lee, M. Kim, H. Shin, M. J. Kim, J. M. Yoo, S. Park and Y. E. Sung, Catal. Today, 2017, 293, 2–7 CrossRef.
- Q. G. He, B. Shyam, M. Nishijima, D. Ramaker and S. Mukerjee, J. Phys. Chem. C, 2013, 117, 4877–4887 CAS.
- Y. C. Hsieh, Y. Zhang, D. Su, V. Volkov, R. Si, L. Wu, Y. Zhu, W. An, P. Liu, P. He, S. Ye, R. R. Adzic and J. X. Wang, Nat. Commun., 2013, 4, 2466 Search PubMed.
- A. Verdaguer-Casadevall, P. Hernandez-Fernandez, I. E. L. Stephens, I. Chorkendorff and S. Dahl, J. Power Sources, 2012, 220, 205–210 CrossRef CAS.
- C. Hulteberg, Int. J. Hydrogen Energy, 2012, 37, 3978–3992 CrossRef CAS.
- A. U. Nilekar, K. Sasaki, C. A. Farberow, R. R. Adzic and M. Mavrikakis, J. Am. Chem. Soc., 2011, 133, 18574–18576 CrossRef CAS PubMed.
- S. M. M. Ehteshami and S. H. Chan, Electrochim. Acta, 2013, 93, 334–345 CrossRef CAS.
- S. J. Lee, S. Mukerjee, E. A. Ticianelli and J. McBreen, Electrochim. Acta, 1999, 44, 3283–3293 CrossRef CAS.
- A. U. Nilekar, K. Sasaki, C. A. Farberow, R. R. Adzic and M. Mavrikakis, J. Am. Chem. Soc., 2011, 133, 18574–18576 CrossRef CAS PubMed.
- T. C. Deivaraj and J. Y. Lee, J. Power Sources, 2005, 142, 43–49 CrossRef CAS.
- Z. L. Liu, X. Y. Ling, X. D. Su and J. Y. Lee, J. Phys. Chem. B, 2004, 108, 8234–8240 CrossRef CAS.
- R. W. Murray, Acc. Chem. Res., 1980, 13, 135–141 CrossRef CAS.
- R. Alkire, D. Kolb and J. Lipkowski, Chemically modified electrodes, Wiley-VCH, Weinheim, Germany, 2009 Search PubMed.
- R. Gutzler, S. Stepanow, D. Grumelli, M. Lingenfelder and K. Kern, Acc. Chem. Res., 2015, 48, 2132–2139 CrossRef CAS PubMed.
- Y. H. Chung, D. Y. Chung, N. Jung and Y. E. Sung, J. Phys. Chem. Lett., 2013, 4, 1304–1309 CrossRef CAS PubMed.
- Y. H. Chung, S. J. Kim, D. Y. Chung, H. Y. Park, Y. E. Sung, S. J. Yoo and J. H. Jang, Chem. Commun., 2015, 51, 2968–2971 RSC.
- E. G. Ciapina, P. P. Lopes, R. Subbaraman, E. A. Ticianelli, V. Stamenkovic, D. Strrncnik and N. M. Markovic, Electrochem. Commun., 2015, 60, 30–33 CrossRef CAS.
- G. De Leener, F. Evoung-Evoung, A. Lascaux, J. Mertens, A. G. Porras-Gutierrez, N. Le Poul, C. Lagrost, D. Over, Y. R. Leroux, F. Reniers, P. Hapiot, Y. Le Mest, I. Jabin and O. Reinaud, J. Am. Chem. Soc., 2016, 138, 12841–12853 CrossRef CAS PubMed.
- G. Fu, X. Jiang, L. Tao, Y. Chen, J. Lin, Y. Zhou, Y. Tang and T. Lu, Langmuir, 2013, 29, 4413–4420 CrossRef CAS PubMed.
- N. Jung, H. Shin, M. Kim, I. Jang, H. J. Kim, J. H. Jang, H. Kim and S. J. Yoo, Nano Energy, 2015, 17, 152–159 CrossRef CAS.
- K. Miyabayashi, H. Nishihara and M. Miyake, Langmuir, 2014, 30, 2936–2942 CrossRef CAS PubMed.
- E. Morsbach, M. Nesselberger, J. Warneke, P. Harz, M. Arenz, M. Baumer and S. Kunz, New J. Chem., 2015, 39, 2557–2564 RSC.
- J. E. Newton, J. A. Preece, N. V. Rees and S. L. Horswell, Phys. Chem. Chem. Phys., 2014, 16, 11435–11446 RSC.
- Y. J. Tong, Chem. Soc. Rev., 2012, 41, 8195–8209 RSC.
- E. C. Tse, C. J. Barile, N. A. Kirchschlager, Y. Li, J. P. Gewargis, S. C. Zimmerman, A. Hosseini and A. A. Gewirth, Nat. Mater., 2016, 15, 754–759 CrossRef CAS PubMed.
- V. Viswanathan and H. A. Hansen, Top. Catal., 2013, 57, 215–221 CrossRef.
- G. R. Xu, J. Rai, L. Yao, Q. Xue, J. X. Jiang, J. H. Zeng, Y. Chen and J. M. Lee, ACS Catalysis, 2017, 7, 452–458 Search PubMed.
- G. R. Zhang, M. Munoz and B. J. Etzold, Angew. Chem., Int. Ed. Engl., 2016, 55, 2257–2261 CrossRef CAS PubMed.
- Z. Y. Zhou, X. W. Kang, Y. Song and S. W. Chen, J. Phys. Chem. C, 2012, 116, 10592–10598 CAS.
- D. Strmcnik, M. Escudero-Escribano, K. Kodama, V. R. Stamenkovic, A. Cuesta and N. M. Markovic, Nat. Chem., 2010, 2, 880–885 CrossRef CAS PubMed.
- J. Monzo, M. T. Koper and P. Rodriguez, ChemPhysChem, 2012, 13, 709–715 CrossRef CAS PubMed.
- M. Escudero-Escribano, M. E. Michoff, E. P. Leiva, N. M. Markovic, C. Gutierrez and A. Cuesta, ChemPhysChem, 2011, 12, 2230–2234 CrossRef CAS PubMed.
- M. Escudero-Escribano, G. J. Soldano, P. Quaino, M. E. Z. Michoff, E. P. M. Leiva, W. Schmickler and A. Cuesta, Electrochim. Acta, 2012, 82, 524–533 CrossRef CAS.
- L. F. Lu, R. H. Li, K. Fujiwara, X. Q. Yan, H. Kobayashi, W. Z. Yi and J. Fan, J. Phys. Chem. C, 2016, 120, 11572–11580 CAS.
- B. Genorio, D. Strmcnik, R. Subbaraman, D. Tripkovic, G. Karapetrov, V. R. Stamenkovic, S. Pejovnik and N. M. Markovic, Nat. Mater., 2010, 9, 998–1003 CrossRef CAS PubMed.
- Z. Q. Tian, B. Ren and B. W. Mao, J. Phys. Chem. B, 1997, 101, 1338–1346 CrossRef CAS.
- P. Sett, S. Chattopadhyay and P. K. Mallick, Vib. Spectrosc., 2009, 49, 84–95 CrossRef CAS.
- D. Y. Wu, X. M. Liu, S. Duan, X. Xu, B. Ren, S. H. Lin and Z. Q. Tian, J. Phys. Chem. C, 2008, 112, 4195–4204 CAS.
- N. H. Kim and K. Kim, J. Phys. Chem. B, 2006, 110, 1837–1842 CrossRef CAS PubMed.
- W. Krasser and A. J. Renouprez, Solid State Commun., 1982, 41, 231–235 CrossRef CAS.
- B. Ren, X. Q. Li, D. Y. Wu, J. L. Yao, Y. Xie and Z. Q. Tian, Chem. Phys. Lett., 2000, 322, 561–566 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ee02641b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |