
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterisation and thermal properties of Sn(II) pyrrolide complexes†
James D.
Parish
a,
Michael W.
Snook
a,
Andrew L.
Johnson
 *a and
Gabriele
Kociok-Köhn
b
*a and
Gabriele
Kociok-Köhn
b
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: A.L.Johnson@bath.ac.uk
bChemical Characterisation and Analysis Facility, Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 30th April 2018
Abstract
SnO is a rare example of a stable p-type semiconductor material. Here, we describe the synthesis and characterisation of a family of Sn(II) pyrrolide complexes for future application in the MOCVD and ALD of tin containing thin films. Reaction of the Sn(II) amide complex, [{(Me3Si)2N}2Sn], with the N,N-bidentate pyrrole pro-ligand, L1H, forms the hetero- and homoleptic complexes [{L1}Sn{N(SiMe3)2}] (1) and [{L1}2Sn] (2), respectively, bearing the 2-dimethylaminomethyl-pyrrolide ligand (L1). Reaction of [{(Me3Si)2N)}2Sn] with the pyrrole-aldimine pro-ligands, L2H–L7H, results in the exclusive formation of the homoleptic bis-pyrrolide complexes [{L2–7}2Sn] (3–8). All complexes have been characterised by elemental analysis and NMR spectroscopy, and the molecular structures of complexes 1–5 and 8 are determined by single crystal X-ray diffraction. TG analysis and isothermal TG analysis have been used to evaluate the potential utility of these systems as MOCVD and ALD precursors.
Introduction
Transparent semiconducting oxide (TSO) thin films have attracted considerable interest due to their omnipresence in modern technology, finding wide-spread application in solar cells, light emitting diodes, flat panel displays, optical communicators, gas sensors and thin film transistors.1 The majority of commercially available semiconducting oxides are n-type, e.g. ZnO; many potential applications of TSOs are still limited by the scarcity of p-type counterparts.2 The development of high performance p-type TSOs would leverage the inordinate potential of oxides for transparent electronics and optoelectronics by combining them with n-type TSOs in p–n heterojunctions.3 The recent rapid development of both photovoltaics and solar water splitting also calls for p-type electrodes for more efficient hole collection.4 However, the most significant challenge in the realisation of this goal is the paucity of suitable p-type TSOs. To this end, a great deal of experimental work has focused on the development of p-type semiconducting materials such as SnO, Cu2O and N-doped ZnO.2,4,5 Unfortunately, all these metastable materials suffer from an intrinsic instability towards oxidation. Despite this, SnO which possesses a layered PbO-like litharge structure has received considerable attention as a p-type semiconductor.1c,2,5,6 Interest was initially sparked by SnO grown on yttria stabilised ZrO2 (YSZ) reportedly displaying a Hall mobility of 2.4 cm2 V−1 s−1 and with a field effect mobility of 1.3 cm2 V−1 s−1, when used as a p-channel thin film transistor (TFT).7 More recently, Hall mobility values as high as ∼18.71 cm2 V−1 s−1 and field effect mobilities of 6.75 cm2 V−1 s−1 have been reported.8Physical vapour deposition (PVD)8,9 and chemical vapour deposition (CVD)10 have both been used to produce thin films of SnO with varying degrees of success. Since modern devices are topographically diverse structures, a vapor phase technique capable of producing thin films with exceptional conformality is required. Atomic layer deposition (ALD), and to a lesser extent metal–organic chemical vapor deposition (MOCVD), offer such a solution. However, SnO is intrinsically reactive towards oxygen, and formation of both Sn2O3 and SnO2 as phase impurities results in thin films with undesirable properties.10g Therefore, precise control over the oxidation state of the metal is paramount. Whilst a number of Sn-precursor/reactant combinations have been surveyed for the growth of SnO, the majority have focused on the utility of Sn(IV) precursor combinations, e.g. SnCl4/H2O11/H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 SnI4/O2,13 Sn(NMe2)4/H2O/H2O2,14 SnEt4/H2O2/O2/O3-plasma and Bu3SnOEt/O3.15 Of the precursor/reactant combinations investigated, only three have utilised Sn(II) precursors (Fig. 1): in the case of the stannylene complex (A), reaction with H2O failed to produce SnO, and reaction with either H2O216 or NO17 resulted in the formation of a mixed phase of SnO/SnO2 (i.e. SnOx). Similarly, Sn(HMDS)2 (B) has also been used in conjunction with either H2O or O3 in an ALD process to deposit Sn(II) and Sn(IV) oxides and SiO2 mixes between 80 and 250 °C.18 To date, only the Sn(II) aminoalkoxide complex (C) has been found to produce phase pure SnO in an ALD process, with H2O, between 90 °C and 210 °C, with crystallinity occurring above 150 °C.19 This dearth of suitable precursors for SnO production has prompted us, and others,20 to investigate new Sn(II)–ligand combinations. In an attempt to optimise precursor reactivity and thermal stability, we chose to investigate the utility of the amino-pyrrolide (L1) and pyrrolylaldiminato ligands (L2–L6) shown in Scheme 1.21 The ligands (L1–L6) can be tuned by the substitution of the R groups to limit oligomerisation and tune volatility. The bidentate chelating effect should increase the thermal stability of the resulting metal compounds. Whilst numerous metal compounds with pyrrole ligands can be found in the literature, their application as CVD or ALD precursors is limited to selected titanium,22 barium23 and copper(I)24 complexes. To date, Sn(II)–pyrrolide systems are limited to the stannocene complex [Sn{η5-2,5-NC4tBu2H2}2],25 and the carbonyl-substituted pyrrolide complexes, [Sn{κ2−N,O-NC4H3CH(O)R}2] (R = NMe2 or Me).26 Here we describe the details of the synthesis and structure of a series of bidentate pyrrolide based Sn(II) complexes, including investigations into their thermal properties.
12 SnI4/O2,13 Sn(NMe2)4/H2O/H2O2,14 SnEt4/H2O2/O2/O3-plasma and Bu3SnOEt/O3.15 Of the precursor/reactant combinations investigated, only three have utilised Sn(II) precursors (Fig. 1): in the case of the stannylene complex (A), reaction with H2O failed to produce SnO, and reaction with either H2O216 or NO17 resulted in the formation of a mixed phase of SnO/SnO2 (i.e. SnOx). Similarly, Sn(HMDS)2 (B) has also been used in conjunction with either H2O or O3 in an ALD process to deposit Sn(II) and Sn(IV) oxides and SiO2 mixes between 80 and 250 °C.18 To date, only the Sn(II) aminoalkoxide complex (C) has been found to produce phase pure SnO in an ALD process, with H2O, between 90 °C and 210 °C, with crystallinity occurring above 150 °C.19 This dearth of suitable precursors for SnO production has prompted us, and others,20 to investigate new Sn(II)–ligand combinations. In an attempt to optimise precursor reactivity and thermal stability, we chose to investigate the utility of the amino-pyrrolide (L1) and pyrrolylaldiminato ligands (L2–L6) shown in Scheme 1.21 The ligands (L1–L6) can be tuned by the substitution of the R groups to limit oligomerisation and tune volatility. The bidentate chelating effect should increase the thermal stability of the resulting metal compounds. Whilst numerous metal compounds with pyrrole ligands can be found in the literature, their application as CVD or ALD precursors is limited to selected titanium,22 barium23 and copper(I)24 complexes. To date, Sn(II)–pyrrolide systems are limited to the stannocene complex [Sn{η5-2,5-NC4tBu2H2}2],25 and the carbonyl-substituted pyrrolide complexes, [Sn{κ2−N,O-NC4H3CH(O)R}2] (R = NMe2 or Me).26 Here we describe the details of the synthesis and structure of a series of bidentate pyrrolide based Sn(II) complexes, including investigations into their thermal properties.
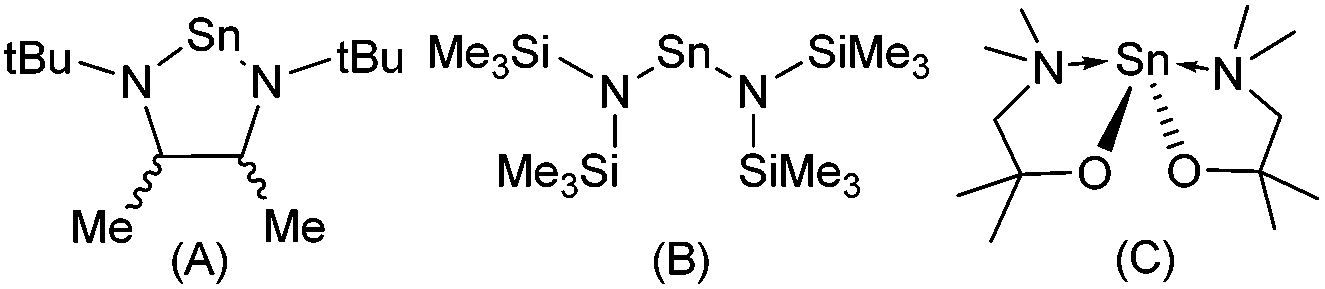 | ||
| Fig. 1 Molecular structures of the three reported Sn(II) ALD precursors which have been used to produce tin oxide thin films. | ||
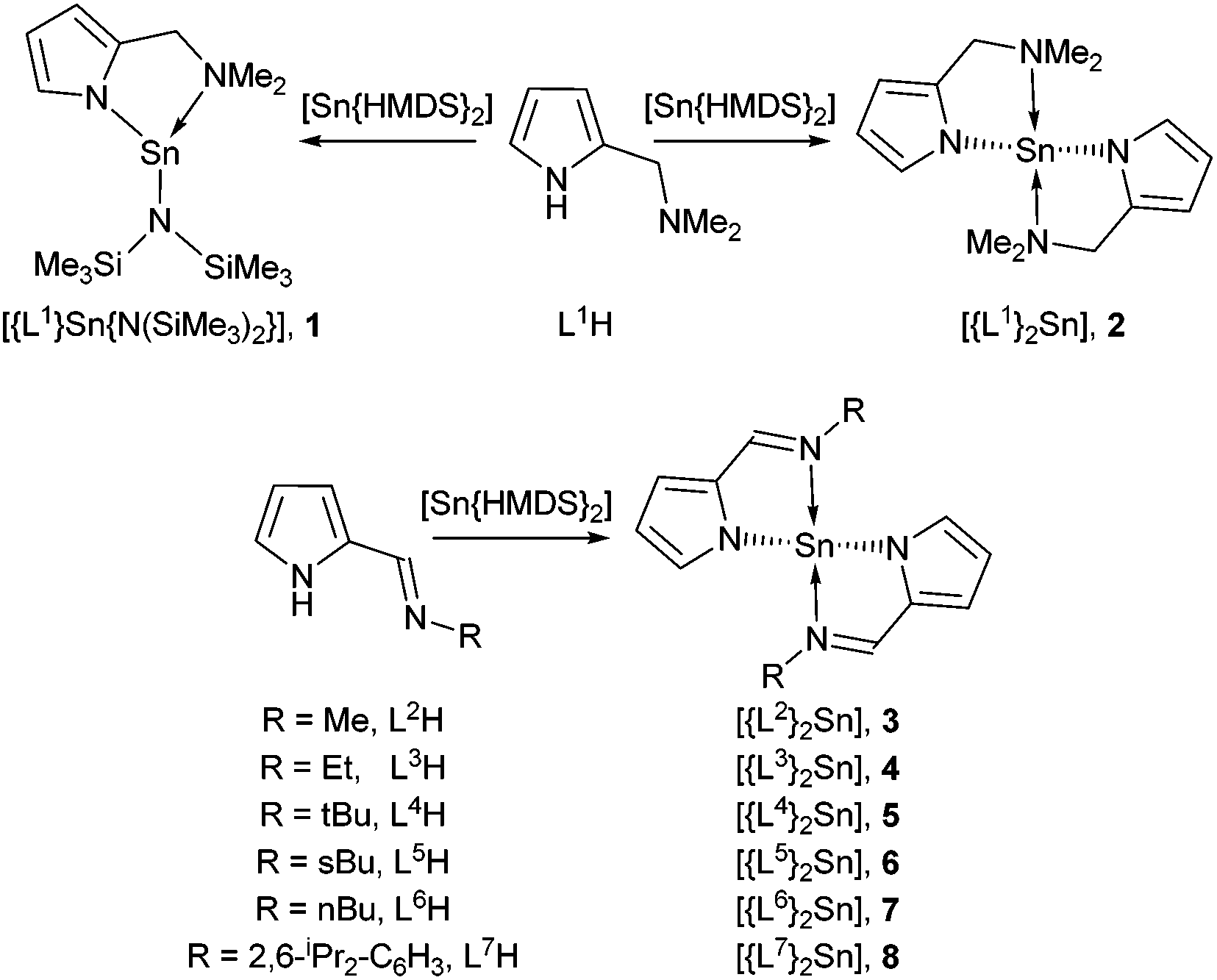 | ||
| Scheme 1 | ||
Results and discussion
Synthesis of tin(II) complexes
In all cases, isolated products were characterised by solution state NMR (1H, 13C and 119Sn) spectroscopy and elemental analysis. Initial attempts to prepare the mono(dimethylamido)Sn(II) compounds by a direct stoichiometric (1:1) reaction of bis(dimethylamido)tin(II) with the pyrrole ligands, L1H–L7H, in both hexane and THF, respectively, were unsuccessful. However, reactions did result in the formation of bis(pyrrolide) compounds 2–8. We attribute this failure to prepare the dimethylamido-tin(II) species to the stronger acidity of the pyrrole {N–H} relative to that of the dimethylamine hydrogen, combined with the enhanced basicity of the dimethylamide group in the monoamide intermediate, [{pyrrolide}Sn-NMe2], relative to [Sn{NMe2}2].
In contrast, the reaction of the bulkier and less basic amide system [Sn{N(SiMe3)2}2] with the ligand L1H in a stoichiometric 1:1 reaction results in the formation, and isolation after recrystallisation, of the mono-pyrrolide complex 1. The 1H NMR spectrum of 1 clearly shows the presence of a singlet resonance at δ = 0.25 ppm representative of the presence of a {HMDS} group, in an 18H:3H ratio with the dimethylamine group, {NMe2}, associated with {L1} (δ = 1.76 ppm), indicative of the presence of the {L1} and {HMDS} ligands in a 1:1 ratio. A comparable reaction of [Sn{HMDS}2] in an equimolar reaction with L2H–L8H results in the formation of the bis-pyrrolide complexes, 2–8, in yields <50%, suggestive of a Schlenk equilibrium in which the putative mono-amide intermediates are unstable with respect to disproportionation, and formation of the bis-pyrrole complex. The 1H NMR spectra of 2–8 clearly show the absence of resonances associated with the {HMDS} ligands and are consistent with the formation of the bis-pyrrole complexes. In the case of the sec-Bu derivative complex, 6, a racemic (±)sec-butyl amine was used for the synthesis of the proligand L5H, resulting in the 1H and 13C NMR spectra containing two sets of resonances corresponding to the presence of the associated (R,R/S,S and R,S/S,R) stereoisomers in solution. Elemental analysis confirms the formation of the bis-pyrrole complexes. The intrinsic C2 symmetry of complexes 2–7 is negated somewhat in solution by a rapid, so-called, “flip-flop” equilibrium process in which the N→Sn coordination bonds repeatedly open and close. In compound 8, however, the methyl and methine groups of the isopropyl substituents display a series of convoluted multiplets (δ = 0.89–1.36 ppm, 12H), alongside two broad resonances respectively (δ = 3.00 ppm, 1H; and 3.43 ppm, 1H) suggestive of a slow rotation, on the NMR timescale, about the N–C(phenyl) bond. The stoichiometric reaction (2:1) of the pro-ligands, L1H–L7H, with [Sn{HMDS}2] produces the expected complexes cleanly in moderate to high yields (64–87%).
Solid state structures
X-ray diffraction studies of single crystals of complexes 1, 2, 3, 4, 5 and 8 unambiguously established their solid state structures. The structures of the heteroleptic and the homoleptic complexes [Sn{κ2-N,N-NC4H3CH2NMe2}{N(SiMe3)2}] (1) and [Sn{κ2-N,N-NC4H3CH2NMe2}2] (2) are shown in Fig. 2. While compounds 1 and 2 are both chiral, the enantiomers co-crystallise in noncentrosymmetric space groups.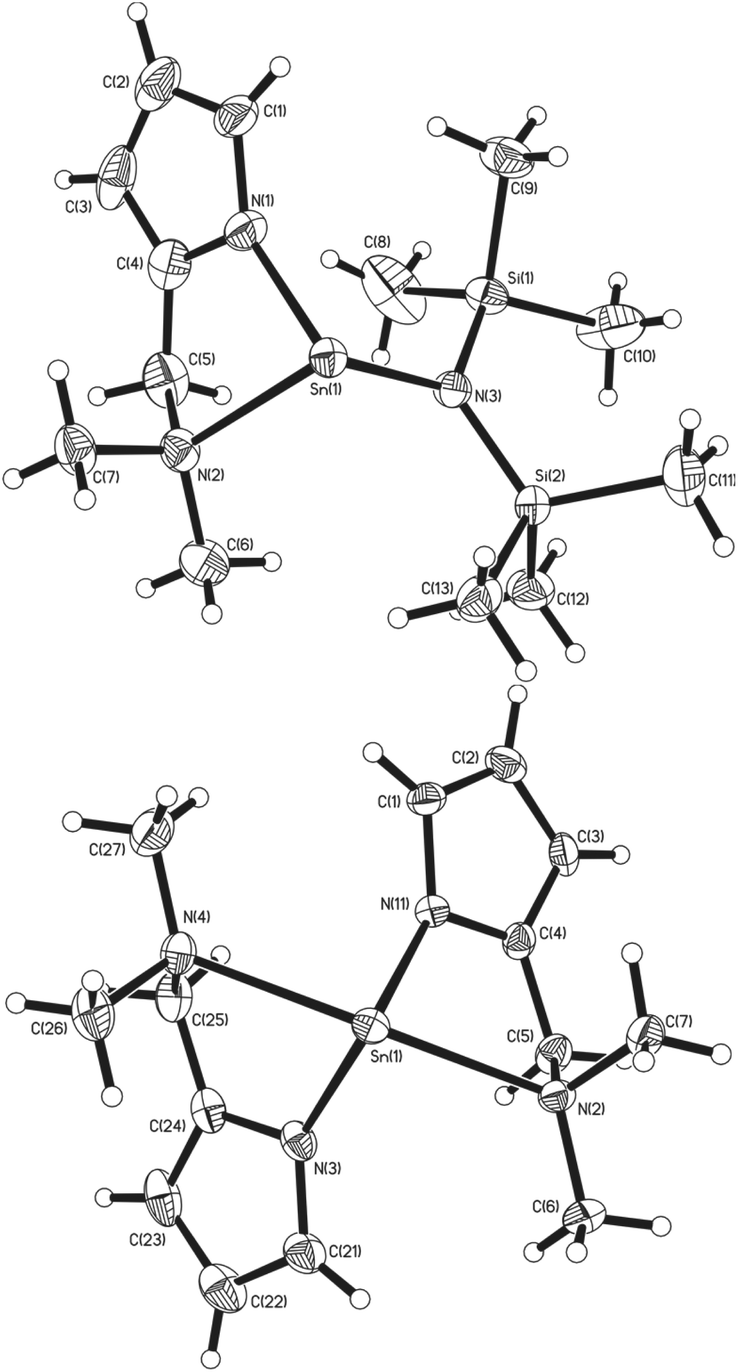 | ||
| Fig. 2 The molecular structures of complex 1 (top) and 2 (bottom) (50% probability ellipsoids). | ||
In the solid state, 1 crystallises in the monoclinic space group P21/c. The asymmetric unit cell contains a single monomeric complex with a three coordinate, pseudo-trigonal-pyramidal geometry about the Sn(II) centre, with the {L1}− ligand coordinated in a κ2 fashion via the pyrrolide nitrogen and the pendant {NMe2} group, as well as the nitrogen of the {HMDS} ligand, in a terminal bonding mode (Fig. 2).
The Sn(1)–N(1) (2.152) and Sn(1)–N(3) (2.127) bond lengths are comparable to those already reported for Sn-amide compounds,27 whereas the dative Sn(II)←NMe2 bond [Sn(1)–N(2) (2.418)] is expectedly longer. Despite a constrained bite angle for the {L1}− ligand [N(1)–Sn(1)–N(2) (74.52°)], the N–Sn–N bond angles in 1 [N(1)–Sn(1)–N(3) (96.36°) and N(2)–Sn(1)–N(3) (96.84°)] suggest the absence of sp-hybridisation at the Sn(II) centre and that the tin–ligand bonds almost exclusively involve the p-orbitals; the nature of the electron lone pair in compound 1 can therefore be considered as essentially a 5s2 configuration and therefore non-directional.
Complex 2, which has intrinsic C2 symmetry, crystallises in the monoclinic space group P21/c and is shown in Fig. 2. Here the asymmetric unit cell contains a single molecule of the complex with a four coordinate, pseudo-trigonal bipyramidal geometry (τ = 0.83)28 in which the two {L1}− ligands are coordinated in the same κ2 fashion observed in 1, with the N-atoms of the pyrrolide ligands occupying two equatorial, and the pendant {NMe2} groups occupying the axial positions. A cursory analysis of the bond angles about the Sn(II) centre in 2 [N(1)–Sn(1)–N(3) (97.44°) & N(2)–Sn(1)–N(3) (147.21°)] again suggests that the tin–ligand bonds almost exclusively involve the p-orbitals on Sn, and that the lone pair of electrons in 2 is therefore again essentially 5s2 based. The Sn–Npy [Sn(1)–N(1) (2.179°) & Sn(1)–N(3) (2.165°)] and Sn←NMe2 [Sn(1)–N(2) (2.516°) & Sn(1)–N(4) (2.528°)] bond lengths in 2 are commensurate with 1 and comparable complexes.
For the imine complexes 3, 4 and 5, which are structurally related to 2, the molecular structures are shown in Fig. 3. For complexes 3 and 5, which crystallise in the centrosymmetric space group P21/n, the asymmetric unit cell contains one full molecule of the bis-(pyrrolylaldiminate)Sn(II) complex. Complexes 3 and 5 are disordered such that all ligand atoms, with the exception of Sn(1), N(1) and N(3), exhibit 67:33 and 80:20 disorder, respectively, via a pseudo-mirror plane containing the three non-affected atoms. Complex 4 crystallises in the polar space group P21 with only one enantiomer of the chiral complex in the crystals, while in all three cases, the central Sn(II) atoms are four-coordinate; analysis of the bond angles about the tin centre suggests a trigonal bipyramidal geometry28 [3: τ = 0.92, 4: τ = 0.82, 5: τ = 1.06] with the imine nitrogen atoms occupying the axial coordination sites and the pyrrole nitrogen atoms the equatorial positions. While the N(imine)–Sn–N(imine) bond angles increase from ∼143° to 152° as the imine substituent changes from methyl to ethyl and t-butyl, respectively, the Npy–Sn–Npy angles [3: 92.17(10)°, 4: 95.64(19)°, 5: 88.33(15)°] are all around 90° suggesting that the Sn–Npy bonds involve mostly the Sn(II) p-orbitals. The Sn–Npy and Sn←NR bond lengths (shown in Table 1) are all similar, ibid.
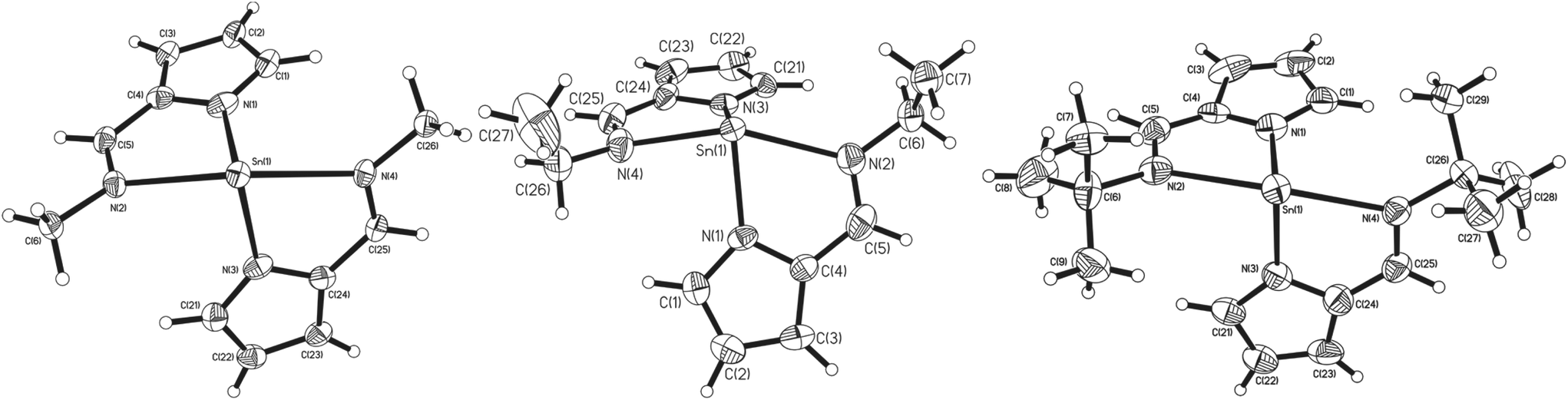 | ||
| Fig. 3 The molecular structures of complex 3 (40% probability ellipsoids), 4 (50% probability ellipsoids) and 5 (50% probability ellipsoids). Complexes 3 and 5 are disordered such that all ligand atoms, with the exception of Sn(1), N(1) and N(3), exhibited 67:33 and 80:20 disorder, respectively, via a pseudo-mirror plane containing the three non-affected atoms. The major component only is depicted for clarity. | ||
| 1 | 2 | 3 | 4 | 5 | 8 | |
|---|---|---|---|---|---|---|
|
a Complexes 3 and 5 are disordered such that all ligand atoms, with the exception of Sn(1), N(1) and N(3), exhibited 67:33 and 80:20 disorder respectively, via a pseudo-mirror plane containing the three non-affected atoms. Only the major component is shown here for clarity.
|
||||||
| Selected bond lengths (Å) | ||||||
| Sn(1)–N(1) | 2.152(2) | 2.179(2) | 2.181(3) | 2.171(5) | 2.185(4) | 2.3138(16) |
| Sn(1)–N(2) | 2.418(2) | 2.516(2) | 2.409(3) | 2.470(8) | 2.462(5) | 2.3308(16) |
| Sn(1)–N(3) | 2.127(2) | 2.165(2) | 2.186(3) | 2.167(5) | 2.165(4) | 2.2871(16) |
| Sn(1)–N(4) | — | 2.528(2) | 2.439(3) | 2.375(7) | 2.442(5) | 2.3127(15) |
N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
— | — | 1.294(6) | 1.279(12) | 1.300(10) | 1.297(2) |
| 1.280(5) | 1.302(11) | 1.273(10) | 1.304(2) | |||
| Selected bond angles (°) | ||||||
| N(1)–Sn(1)–N(2) | 74.52(8) | 72.14(8) | 70.13(11) | 70.0(2) | 69.99(18) | 71.26(6) |
| N(2)–Sn(1)–N(3) | 96.84(8) | 86.71(8) | 84.73(10) | 86.3(2) | 88.31(17) | 81.34(5) |
| N(3)–Sn(1)–N(4) | — | 72.16(8) | 70.26(11) | 72.1(2) | 73.54(17) | 71.84(6) |
| N(1)–Sn(1)–N(3) | 96.36(8) | 97.44(8) | 92.17(10) | 95.64(19) | 88.33(15) | 127.03(6) |
| N(2)–Sn(1)–N(4) | — | 147.21(7) | 142.69(11) | 144.9(2) | 151.8(2) | 117.62(5) |
Similarly to complexes 2–5, compound 8 is chiral (Fig. 4), possessing molecular C2 symmetry; the other enantiomer is also formed in the product, with 8 crystallising in the centrosymmetric monoclinic space group P21/n. Exhibiting a 4-coordinate Sn(II) centre, the geometry about the Sn(II) atom is best described as square based pyramidal [τ = 0.15]. Interestingly, the Sn–Npy bonds in 8 [Sn(1)–N(1) = 2.3138(16) Å, Sn(1)–N(3) = 2.2871(16) Å] are significantly longer than those reported for 1–5. Similarly, the Sn←NR bonds are also significantly longer [Sn(1)–N(2) = 2.3308(16) Å, Sn(1)–N(4) = 2.3127(15) Å] than those observed in 3–5. Consistent with this observation, the Npy–Sn–Npy and N(imine)–Sn–N(imine) angles observed in 8 are both close to 120° [127.03(6)° and 117.62(5)° respectively], suggesting that the tin–ligand bonds almost exclusively involve sp2 hybridised orbitals on the tin, with the lone pair in 8 considered to be essentially based in a directional sp2 orbital.
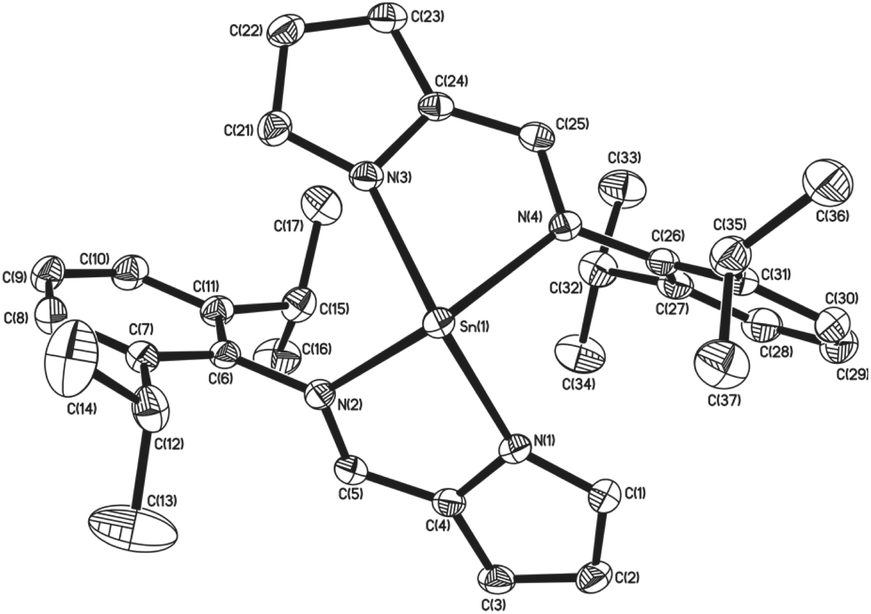 | ||
| Fig. 4 The molecular structures of complex 8 (50% probability ellipsoids), hydrogen atoms have been omitted for clarity. | ||
Thermal profiles
Two of the main precursor requirements for MOCVD and ALD applications are the need for volatility and thermal stability.29 As the primary goal of synthesising compounds 1–8 was driven by our interest in their application as precursors for the MOCVD and ALD of Sn(II) oxide films, melting point analysis, thermogravimetric analysis (TGA) and isothermal studies were employed to investigate the volatility and thermal stability of complexes 1–8. The melting points and analysis of compounds 1–8 were recorded with instruments housed in an argon filled glove-box in order to minimise reaction with atmospheric moisture/air. For the amino-pyrrolide complexes 1 and 2, results suggest that these materials are unsuitable for application as ALD precursors. Table 2 shows the melting and decomposition points for these complexes. Complex 1 displays a rather low decomposition temperature (100 °C) quite close to its melting point (95 °C). Similarly for 2, a relatively low decomposition (50 °C) was observed before any phase transition could be detected, suggesting the possible lack of utility of these systems as ALD precursors. Despite this observation, the TGA of Sn(II)bis-(pyrrolide) 2–8 was performed in order to gain greater insight into the relative volatilities and thermal stabilities of the compounds.| Compound R= | Melting point (°C) | Decompn point (°C) | Evaporation ratea (μg min−1 cm−2) |
|---|---|---|---|
| a Isothermal TGA recorded at 130 °C. b Isothermal TGA recorded at 160 °C. | |||
| (1) | 95 | ∼100 | — |
| (2) | — | ∼50 | — |
| -Me (3) | — | 140 | 6.409(8) |
| -Et (4) | 130 | 137 | 19.894(1) |
| -tBu (5) | — | 141 | 19.974(7) |
| -nBu (6) | 85 | 100 | 14.085(8) |
| -sBu (7) | Viscous oil | 94 | 5.611(6) |
| -Dipp (8) | 153 | 290 | 2.357(5) |
| -Dipp (8)b | 12.653(9) | ||
As seen in Fig. 5, compounds 3–8 exhibit very similar thermal behaviour, consistent with single step evaporation. For all precursors, the onset of volatilisation (∼100 °C) and the temperature at which the evaporation is completed (between 220 and 255 °C for 3–7 and by 286 °C for 8) are similar.
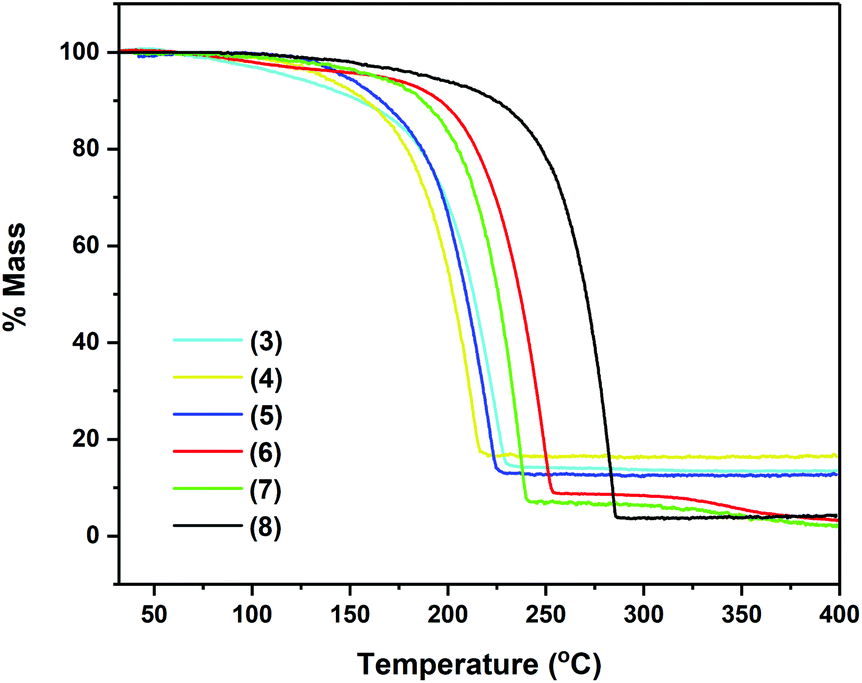 | ||
| Fig. 5 Thermogravimetric analysis data for complexes 3–8. | ||
Table 3 shows germane data, relating to the TG analysis of compounds 3–8, i.e. % residual mass and wt% of Sn in complexes. Fig. 4 clearly shows that compounds 3–8 exhibit very similar thermal behaviour, undergoing a clear, single mass loss event over a small temperature window to yield stable residues of between 4 and 16.5%, consistent with a single step evaporation process. In the case of complexes 6–8, the final mass residues are considerably lower than the expected mass residue for the production of the Sn metal, strongly suggestive of a high degree of volatility within these systems. For complexes 3–5, the mass residues are proportionately higher although still below the % mass residue expected if decomposition resulted in the formation of the Sn metal. Compound 2, which was also analysed, showed a complicated and shallow decomposition profile with mass loss starting at 36 °C. At 400 °C, the residual mass is ∼67%, indicative of a non-volatile material with incomplete thermal decomposition (Fig. S1, ESI†).
Given the nature of the ligand systems involved in 3–8, it is unlikely that the TGA residues contain oxide products (i.e. SnOx), and instead are more likely to be metallic Sn (with possible carbon impurities). This is consistent with the observation of metallic deposits (of Sn) in the TGA crucibles after decomposition studies, suggesting the possible application of these systems in the deposition of metallic tin under a non-oxidative atmosphere.
While the TGA data provide an indication of the volatility of the complexes, decomposition characteristics are less easy to discern for complexes with significant volatility. However, no stepwise decomposition processes are observed in the TGA profiles of 3–8, corresponding to the systematic breakdown of the pyrrolide ring systems, as postulated in other studies.30 More relevant investigations have suggested that pyrrolide complexes are susceptible to β-hydride elimination processes,22 in these cases most likely arising from hydride abstraction from the aldimine substituents. This is in contrast to complex 2, which possesses pendant {CH2NMe2} groups, and as such does not share the same electronic delocalisation observed for the aldimine systems 3–8. Consequently, the thermal analysis of 2 (ESI†) shows a stepwise decomposition over a broad temperature range, consistent with the aforementioned decomposition pathway.
Remarkably, no discernible trends are observed between pyrrolide-aldimine substituents and volatilities/stabilities, with the ethyl (3) and t-butyl (4) substituted complexes showing the highest volatility followed by the 2,6-diisopropyl-phenyl complex (8). However, it is noteworthy that the aryl containing system, 8, displays a strikingly high thermal stability, in contrast to the other systems investigated here.
We suggest that this high degree of thermal stability is in part due to the absence of a suitable hydride abstraction process, as discussed previously. This observation is the focus of further studies to enhance the thermal stability of selected precursor systems, and to expand the ALD window of selected compounds, whilst inhibiting CVD processes.
The thermal behaviour of complexes 3–8 was further investigated using isothermal TGA studies (Fig. 6). At a fixed temperature of 130 °C, the mass loss for each compound was measured over a period of 120 min (2 h). In all measurements, an approximate linear weight loss was observed, which could be indicative of sublimation, with limited signs of decomposition. However, for complexes 6 and 7, visual (m.p. studies) decomposition appears to begin at below 100 °C (Table 2). From the gradient of the corresponding plots, the evaporation rates at a set temperature of 130 °C were determined (Table 2). The evaporation rates were found to be in the range of 2.4–20 μg min−1 cm−2. From the thermal studies, one can conclude that among the Sn(II) pyrrolide complexes reported here, the ethyl and tert-butyl substituted complexes 4 and 5 show greatest promise for MOCVD and ALD applications with comparable evaporation rates of ca. 20 μg min−1 cm−2. These values are within a range previously determined to be suitable for a vapour based deposition process. The difference in the evaporation rate between the sec-Bu, n-Bu and t-Bu derivatives 5, 6 and 7, respectively, is noticeable, and indicates clearly that not only the molecular mass of a compound but also the factors such as steric crowding, electronic saturation, crystal packing, intermolecular interactions, and so forth, strongly influence thermal behaviour, highlighting the importance of precursor screening. In the case of compound 8, which produced the lowest % residue in the TG analysis (4%) and demonstrated an unusually high thermal stability cf.1–7 in this series, isothermal analyses were performed at both 130 °C and 160 °C, respectively, where a significantly higher volatility was observed above the recorded melting point of 153 °C.
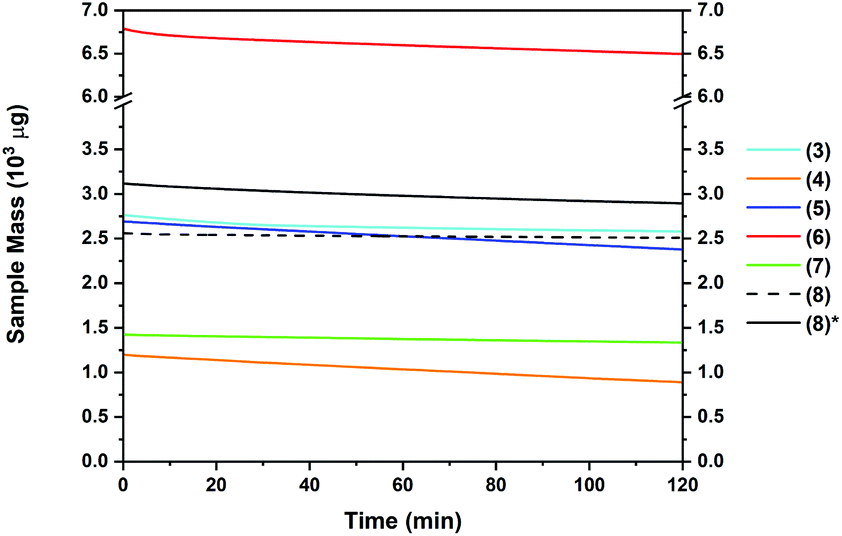 | ||
| Fig. 6 Mass loss (mg) for complexes 3–8 over 120 min at 120 °C and 160 °C*. | ||
It should be noted that these differences would not have been recognised from a standard plot of % mass loss versus time commonly found elsewhere in the literature, which fails to take into account the differences in mass between the samples, and instead reports the change in mass as a % of sample size, which is rarely consistent on scales such as these.
Conclusions
With the use of amine and aldimine substituted pyrroles as chelating ligands, a series of novel homoleptic Sn(II) complexes have been developed. All the compounds are monomeric and volatile, showing variable sublimation behaviour. Given the limited choice of precursors available for MOCVD and ALD of Sn(II) oxide thin films, the ethyl, t-butyl and 2,6-di-isopropylphenyl complexes of 4, 5 and 8, reported here, are promising precursor candidates for vapour deposition processes.The work presented here primarily concerns precursor development and molecular characterisation. Detailed studies on the MOCVD and ALD of Sn(II) oxides using these precursors, and subsequent thin film characterisation, will be published separately.
Experimental
General procedures
Elemental analyses were performed using an Exeter Analytical CE 440 analyser. 1H, 13C and 119Sn NMR spectra were recorded on Bruker Advance 300 or 500 MHz FT-NMR spectrometers, as appropriate, in saturated solutions at room temperature. Chemical shifts are expressed in ppm with respect to Me4Si (1H and 13C). TGA and PXRD analyses were performed using a PerkinElmer TGA7 and Bruker D8 instrument (Cu-Kα radiation), respectively.All reactions were carried out under an inert atmosphere using standard Schlenk techniques. Solvents were dried and degassed under an argon atmosphere over activated alumina columns using an Innovative Technology solvent purification system (SPS). The Sn(II) amides, [Sn{NMe2}2] and [Sn{N(SiMe3)2}2], were prepared by literature methods.27e,31 The pro-ligands L1H–L7H were synthesized using literature methods.21
4: Storage at −28 °C afforded colourless crystals. Yield: 1.16 g, 64%. Elemental analysis for C14H18N4Sn (expected): C 46.46 (46.58)%, H 4.87 (5.03)%, N 15.61 (15.52)%. 1H NMR (500 MHz, C6D6): 7.49–7.56 (m, 1H, PyrCHNEt), 7.04–7.07 (m, 1H, Pyr, C4–H), 6.73–6.77(m, 1H, Pyr, C3–H), 6.50–6.53 (m, 1H, Pyr, C2–H), 3.18–3.25 (s, 2H, CH2), 1.03 (t, 3H, CH3). 13C NMR (75.5 MHz, C6D6): 156.9 (1C, PyrCHNEt), 137.3 (1C, Pyr, C1), 133.1 (1C, Pyr, C4), 117.4 (1C, Pyr, C3), 112.5 (1C, Pyr, C2), 51.2 (1C, CH2), 17.3 (1C, CH3). 119Sn NMR (111.8 MHz, C6D6): −402.3.
5: Storage at −28 °C afforded colourless crystals. Yield: 1.69 g, 81%. Elemental analysis for C18H26N4Sn (expected): C 51.93 (51.83)%, H 6.15 (6.28)%, N 13.86 (13.43)%. 1H NMR (500 MHz, C6D6): 7.97–8.02 (m, 1H, PyrrCHNtBu), 7.19–7.21 (m, 1H, Pyrr, C4–H), 6.76–6.79 (m, 1H, Pyrr, C3–H), 6.46–6.48 (m, 1H, Pyrr, C2–H), 1.19 (s, 9H, CH3). 13C NMR (75.5 MHz, C6D6): 154.0 (1C, PyrrCHNtBu), 138.5 (1C, Pyrr, C1), 133.7 (1C, Pyrr, C4), 118.3 (1C, Pyrr, C3), 112.6 (1C, Pyrr, C2), 57.5 (1C, C(CH3)3), 31.3 (3C, C(CH3)3). 119Sn NMR (111.8 MHz, C6D6): −384.1.
6: Storage at −28 °C afforded colourless crystals. Yield: 1.50 g, 72%. Elemental analysis for C18H26N4Sn (expected): C 52.13 (51.83)%, H 6.37 (6.28)%, N 13.28 (13.43)%. 1H NMR (500 MHz, C6D6): 7.70–7.77 (m, 1H, PyrrCHNsBu), 7.15–7.19 (m, 1H, Pyrr, C4–H), 6.77–6.80 (m, 1H, Pyrr, C3–H), 6.47–6.50 (m, 1H, Pyrr, C2–H), 3.10–3.20 (m, 1H, –NCH(CH3)CH2CH3), 1.54–1.66 (m, 1H, NCH(CH3)CH2CH3), 1.36–1.47 (m, 1H, NCH(CH3)CH2CH3), 1.14–1.19 (m, 3H, NCH(CH3)CH2CH3), 0.70–0.75 (m, 3H, NCH(CH3)CH2CH3). 13C NMR (125.7 MHz, C6D6): 156.0 (1C, PyrrCHNsBu), 137.6 (1C, Pyrr, C1), 133.7 (1C, Pyrr, C4), 118.2 (1C, Pyrr, C3), 112.7 (1C, Pyrr, C2), 64.3 (br, d, 1C, NC(CH3)CH2CH3), 32.3 (d, 1C, NC(CH3)CH2CH3), 22.6 (d, 1C, NC(CH3)CH2CH3), 11.5 (d, 1C, NC(CH3)CH2CH3). 119Sn NMR (186.4 MHz, C6D6): −385.7.
7: Storage at −28 °C afforded colourless crystals. Yield: 1.56 g, 75%. Elemental analysis for C18H26N4Sn (expected): C 51.55 (51.83)%, H 6.20 (6.28)%, N 13.12 (13.43)%. 1H NMR (500 MHz, C6D6): 7.55–7.61 (m, 1H, PyrrCHNnBu), 7.09–7.12 (m, 1H, Pyrr, C4–H), 6.76–6.79 (m, 1H, Pyrr, C3–H), 6.53–6.55 (m, 1H, Pyrr, C2–H), 3.26–3.31 (t, J = 6.85 Hz, 2H, NCH2CH2CH2CH3), 1.47–1.54 (m, 2H, PyrrCHNCH2CH2CH2CH3), 1.12–1.20 (m, 2H, –NCH2CH2CH2CH3), 0.79 (t, J = 7.34 Hz 3H, NCH2CH2CH2CH3). 13C NMR (125.7 MHz, C6D6): 157.82 (s, 1C, PyrrCHNnBu), 137.6 (1C, Pyrr, C1), 133.5 (1C, Pyrr, C4), 117.8 (1C, Pyrr, C3), 112.9 (1C, Pyrr, C2), 57.4 (1C, NCH2CH2CH2CH3), 34.6 (1C, NCH2CH2CH2CH3), 21.1 (1C, NCH2CH2CH2CH3), 14.3 (1C, NCH2CH2CH2CH3). 119Sn NMR (186.4 MHz, C6D6): −401.5.
8: Storage at −28 °C afforded colourless crystals. Yield: 2.63 g, 84%. Elemental analysis for C34H42N4Sn (expected): C 65.37 (65.29)%, H 6.83 (6.77)%, N 8.91 (8.96)%. 1H NMR (500 MHz, C6D6): 7.85–7.86 (m, 1H, PyrrCHNDipp), 7.13–7.23 (m, 3H, ortho, meta-Dipp), 6.84–6.86 (m, 1H, Pyrr, C4–H), 6.62–6.64 (m, 1H, Pyrr, C3–H), 6.35–6.37 (m, 1H, Pyrr, C2–H), 3.43 (br s, 1H, CHMe2), 3.00 (br s, 1H, CHMe2), 0.89–1.36 (br, m, 12H, CHMe2). 13C NMR (125.7 MHz, C6D6): 158.6 (1C, PyrrCHNDipp), 149.8 (1C, ipso-Dipp), 145.6 (1C, Pyrr, C1), 142.8 (1C, ortho-Dipp), 142.3 (1C, ortho-Dipp), 137.0 (s, 1C, Pyrr, C3), 126.7 (br, 2C, meta-Dipp), 124.6 (br, 1C, para-Dipp), 121.0 (1C, Pyrr, C4), 114.5 (1C, Pyrr, C2), 29.1 (br, 1C, CHMe2), 28.7 (br, 1C, CHMe2), 26.4 (br, CHMe2), 24.9 (br, CHMe2), 24.6 (br, CHMe2), 23.1 (br, CHMe2). 119Sn NMR (111.8 MHz, C6D6): −419.0.
Single crystal X-ray diffraction
Experimental details related to the single-crystal X-ray crystallographic studies of compounds 1–5 and 8 are summarised in Table 4. All crystallographic data were collected at 150(2) K either on a SuperNova (Dual, EosS2) diffractometer using radiation Cu-Kα (λ = 1.54184 Å) or Mo-Kα (λ = 0.71073 Å). All structures were solved by direct methods followed by full-matrix least squares refinement on F2 using the WINGX-2014 suite of programs32 or OLEX2.33 All hydrogen atoms were included in idealised positions and refined using the riding model. Crystals were isolated from an argon filled Schlenk flask and immersed in oil before being mounted onto the diffractometer.| Compound number | 1 | 2 | 3 | 4 | 5 | 8 |
|---|---|---|---|---|---|---|
| Chemical formula | C13H29N3Si2Sn | C14H22N4Sn | C12H14N4Sn | C14H18N4Sn | C18H26N4Sn | C34H42N4Sn |
| Formula mass | 402.26 | 365.04 | 332.96 | 361.01 | 417.12 | 625.40 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P121/c1 | P121/c1 | P21/n | P21 | P21/n | P121/n1 |
| a/Å | 11.8512(4) | 9.3277(3) | 13.7265(4) | 8.5561(3) | 12.1057(3) | 15.0740(1) |
| b/Å | 10.4036(3) | 11.3766(4) | 5.93000(10) | 8.8998(3) | 9.7711(2) | 11.8067(1) |
| c/Å | 15.6557(5) | 14.9148(6) | 16.1287(4) | 10.0193(3) | 16.0995(4) | 18.2296(2) |
| α/° | 90 | 90 | 90 | 90 | 90 | 90 |
| β/° | 96.431(3) | 98.662(4) | 104.195(3) | 93.866(3) | 95.487(2) | 106.666(1) |
| γ/° | 90 | 90 | 90 | 90 | 90 | 90 |
| Unit cell volume/Å3 | 1918.13(11) | 1564.67(10) | 1272.76(6) | 761.21(4) | 1895.62(8) | 3108.11(5) |
| Crystal size (mm) | 0.403 × 0.37 × 0.293 | 0.516 × 0.378 × 0.266 | 0.372 × 0.040 × 0.028 | 0.320 × 0.150 × 0.030 | 0.389 × 0.251 × 0.037 | 0.277 × 0.238 × 0.196 |
| Temperature/K | 150.00(10) | 150.00(10) | 150(2) | 150(2) | 150(2) | 150.00(10) |
| Z | 4 | 4 | 4 | 2 | 4 | 4 |
| Radiation type | Mo-Kα | Mo-Kα | Cu-Kα | Cu-Kα | Cu-Kα | Cu-Kα |
| Theta range (°) | 6.54 to 54.958 | 6.586 to 54.958 | 3.797 to 72.307 | 4.423 to 73.010 | 4.375 to 73.438 | 6.732 to 145.674 |
| Absorption coefficient, μ/mm−1 | 1.451 | 1.627 | 15.833 | 13.285 | 10.746 | 6.741 |
| No. of reflections measured | 15655 |
13218 |
13015 |
5956 | 12882 |
26377 |
| No. of independent reflections | 4329 | 3586 | 2487 | 2288 | 3772 | 6142 |
| R int | 0.0354 | 0.0361 | 0.0405 | 0.0279 | 0.0598 | 0.0393 |
| Final R1 values (I > 2σ(I)) | 0.0295 | 0.0291 | 0.0260 | 0.0336 | 0.0495 | 0.0258 |
| Final wR(F2) values (I > 2σ(I)) | 0.0561 | 0.0563 | 0.0631 | 0.0894 | 0.1407 | 0.0697 |
| Final R1 values (all data) | 0.0372 | 0.0381 | 0.0291 | 0.0338 | 0.0536 | 0.0265 |
| Final wR(F2) values (all data) | 0.0596 | 0.0607 | 0.0646 | 0.0896 | 0.1453 | 0.0702 |
| Goodness of fit on F2 | 1.068 | 1.061 | 1.128 | 1.077 | 1.090 | 1.062 |
| Largest diff. peak and hole (e Å−3) | 0.42 and −0.35 | 0.33 and −0.51 | 1.008 and −0.401 | 1.203 and −0.788 | 1.728 and −0.965 | 0.73 and −0.70 |
| CCDC number | 1820592 | 1820591 | 1820595 | 1820596 | 1820594 | 1820593 |
The asymmetric unit cell of 3 comprises one molecule of the complex in which all ligand atoms, with the exception of Sn1, N3 and N1, exhibited 80:20 disorder via a pseudo-mirror plane containing the three non-affected atoms. Bond length restraints were included (for chemically equivalent bonds in both the major/minor components), in addition to ADP restraints.
Complex 5 suffers from similar disorder to that observed in 3, i.e. the asymmetric unit cell comprises one molecule of the complex in which all ligand atoms, with the exception of N3 and N1, exhibited 67:33 disorder via a pseudo-mirror plane containing the three non-affected atoms. Distance-similarity restraints were included (for chemically equivalent bonds in both the major/minor components), in addition to ADP restraints, to assist convergence. The Sn centre is disordered over two sites in the 9:1 ratio.
Thermogravimetric analysis (TGA)
TGA was performed using a TGA 4000 PerkinElmer system, housed in an argon filled glovebox. Samples were prepared air sensitively, and TGAs were performed under a flow of Ar at 20 ml min−1 and heated from 30 °C to 400 °C at a ramp rate of 5 °C min−1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the University of Bath (PhD studentship to J. D. P.) and the Department of Chemistry (Masters studentship to M. W. S.).References
-
(a) P. P. Edwards, A. Porch, M. O. Jones, D. V. Morgan and R. M. Perks, Dalton Trans., 2004, 2995 RSC
; (b) J. Robertson, R. Gillen and S. J. Clark, Thin Solid Films, 2012, 520, 3714–3720 CrossRef
- K. H. L. Zhang, K. Xi, M. G. Blamire and R. G. Egdell, J. Phys.: Condens. Matter, 2016, 28, 383002 CrossRef PubMed
- A. N. Banerjee and K. K. Chattopadhyay, Prog. Cryst. Growth Charact. Mater., 2005, 50, 52–105 CrossRef
- L. Chen, J. Yang, S. Klaus, L. J. Lee, R. Woods-Robinson, J. Ma, Y. Lum, J. K. Cooper, F. M. Toma, L.-W. Wang, I. D. Sharp, A. T. Bell and J. W. Ager, J. Am. Chem. Soc., 2015, 137, 9595–9603 CrossRef PubMed
- Z. Wang, P. K. Nayak, J. A. Caraveo-Frescas and H. N. Alshareef, Adv. Mater., 2016, 28, 3831–3892 CrossRef PubMed
- K. J. Saji, K. Tian, M. Snure and A. Tiwari, Adv. Electron. Mater., 2016, 2, 1500453 CrossRef
- Y. Ogo, H. Hiramatsu, K. Nomura, H. Yanagi, T. Kamiya, M. Kimura, M. Hirano and H. Hosono, Phys. Status Solidi A, 2009, 206, 2187–2191 CrossRef
- J. A. Caraveo-Frescas, P. K. Nayak, H. A. Al-Jawhari, D. B. Granato, U. Schwingenschlogl and H. N. Alshareef, ACS Nano, 2013, 7, 5160–5167 CrossRef PubMed
-
(a) D. Das and R. Banerjee, Thin Solid Films, 1987, 147, 321–331 CrossRef
-
(a) N. Hollingsworth, G. A. Horley, M. Mazhar, M. F. Mahon, K. C. Molloy, P. W. Haycock, C. P. Myers and G. W. Critchlow, Appl. Organomet. Chem., 2006, 20, 687–695 CrossRef
- H. Viirola and L. Niinistö, Thin Solid Films, 1994, 249, 144–149 CrossRef
-
(a) X. Du, Y. Du and S. M. George, J. Phys. Chem. A, 2008, 112, 9211–9219 CrossRef PubMed
- J. Sundqvist, J. Lu, M. Ottosson and A. Hårsta, Thin Solid Films, 2006, 514, 63–68 CrossRef
-
(a) H.-E. Cheng, D.-C. Tian and K.-C. Huang, Procedia Eng., 2012, 36, 510–515 CrossRef
- C. E. Nanayakkara, G. Liu, A. Vega, C. L. Dezelah, R. K. Kanjolia and Y. J. Chabal, Langmuir, 2017, 33, 5998–6004 CrossRef PubMed
-
(a) J. Heo, A. S. Hock and R. G. Gordon, Chem. Mater., 2010, 22, 4964–4973 CrossRef
- J. Heo, S. B. Kim and R. G. Gordon, J. Mater. Chem., 2012, 22, 4599 RSC
- J. Tupala, M. Kemell, M. Mattinen, K. Meinander, S. Seppälä, T. Hatanpää, J. Räisänen, M. Ritala and M. Leskelä, J. Vac. Sci. Technol., A, 2017, 35, 041506 Search PubMed
- J. H. Han, Y. J. Chung, B. K. Park, S. K. Kim, H.-S. Kim, C. G. Kim and T.-M. Chung, Chem. Mater., 2014, 26, 6088–6091 CrossRef
- S. M. George, J. H. Nam, G. Y. Lee, J. H. Han, B. K. Park, C. G. Kim, D. J. Jeon and T.-M. Chung, Eur. J. Inorg. Chem., 2016, 2016, 5539–5546 CrossRef
-
(a) Y. Yang, D. Cui and X. Chen, Dalton Trans., 2010, 39, 3959 RSC
- K. Black, A. C. Jones, J. Bacsa, P. R. Chalker, P. A. Marshall, H. O. Davies, P. N. Heys, P. O'Brien, M. Afzaal, J. Raftery and G. W. Critchlow, Chem. Vap. Deposition, 2010, 16, 93–99 CrossRef
- S. Acharya, J. Torgersen, Y. Kim, J. Park, P. Schindler, A. L. Dadlani, M. Winterkorn, S. Xu, S. P. Walch, T. Usui, C. Schildknecht and F. B. Prinz, J. Mater. Chem. C, 2016, 4, 1945–1952 RSC
-
(a) B. Vidjayacoumar, D. J. H. Emslie, J. M. Blackwell, S. B. Clendenning and J. F. Britten, Chem. Mater., 2010, 22, 4854–4866 CrossRef
- N. Kuhn, G. Henkel and S. Stubenrauch, J. Chem. Soc., Chem. Commun., 1992, 760 RSC
- K. Hubler and U. Hubler, Z. Anorg. Allg. Chem., 2000, 626, 1224–1236 CrossRef
-
(a) G. L. Fondong, E. Y. Njua, A. Steiner, C. F. Campana and L. Stahl, Polyhedron, 2011, 30, 2856–2862 CrossRef
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
-
(a) A. Devi, Coord. Chem. Rev., 2013, 257, 3332–3384 CrossRef
- G. B. Bacskay, M. Martoprawiro and J. C. Mackie, Chem. Phys. Lett., 1999, 300, 321–330 CrossRef
-
(a) P. Foley and M. Zeldin, Inorg. Chem., 2002, 14, 2264–2267 CrossRef
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1820591–1820596. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt00490k |
| This journal is © The Royal Society of Chemistry 2018 |