
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Group 3 metal trihalide complexes with neutral N-donor ligands – exploring their affinity towards fluoride†
Emily
Curnock
a,
William
Levason
 a,
Mark E.
Light
a,
Sajinder K.
Luthra
b,
Graeme
McRobbie
b,
Francesco M.
Monzittu
a,
Gillian
Reid
*a and
R. Nathan
Williams
a
a,
Mark E.
Light
a,
Sajinder K.
Luthra
b,
Graeme
McRobbie
b,
Francesco M.
Monzittu
a,
Gillian
Reid
*a and
R. Nathan
Williams
a
aChemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
bGE Healthcare, The Grove Centre, White Lion Road, Amersham HP7 9LL, UK
First published on 17th April 2018
Abstract
Fluorination of [ScCl3(Me3-tacn)] (Me3-tacn = 1,4,7-trimethyl-1,4,7-triazacyclononane) and [ScCl3(BnMe2-tacn)] (BnMe2-tacn = 1,4-dimethyl-7-benzyl-1,4,7-triazacyclononane) by Cl/F exchange with 3 mol. equiv. of anhydrous [NMe4]F in CH3CN solution yields the corresponding [ScF3(R3-tacn)] (R3 = Me3 or BnMe2). These are the first examples of scandium fluoride complexes containing neutral co-ligands. The fluorination occurs stepwise, and using a deficit of [NMe4]F produced [ScF2Cl(Me3-tacn)]. Attempts to fluorinate [YCl3(Me3-tacn)], [YI3(Me3-tacn)], [LaCl3(Me3-tacn)(OH2)] or [MCl3(terpy)] (M = Sc, Y or La; terpy = 2,2′:6′2′′-terpyridyl) using a similar method were unsuccessful, due to the Cl/F exchange being accompanied by loss of the neutral ligand from the metal centre. Fluorination of [ScCl3(Me3-tacn)] or [ScCl3(terpy)] with Me3SnF was also successful. The products were identified as the very unusual heterobimetallic [Sc(Me3-tacn)F2(μ-F)SnMe3Cl] and [Sc(terpy)F(μ-F)2(SnMe3Cl)2], in which the Me3SnCl formed in the reaction behaves as a weak Lewis acid towards the scandium fluoride complex, linked by Sc–F–Sn bridges. [Sc(terpy)F(μ-F)2(SnMe3Cl)2] decomposes irreversibly in solution but, whilst multinuclear NMR data show that [Sc(Me3-tacn)F2(μ-F)SnMe3Cl] is dissociated into the [ScF3(Me3-tacn)] and Me3SnCl in CH3CN solution, the bimetallic complex reforms upon evaporation of the solvent. The new scandium fluoride complexes and the chloride precursors have been characterised by microanalysis, IR and multinuclear NMR (1H, 19F, 45Sc) spectroscopy as appropriate. X-ray crystal structures provide unambiguous evidence for the identities of [Sc(Me3-tacn)F2(μ-F)SnMe3Cl], [ScF2Cl(Me3-tacn)], [YI3(Me3-tacn)], [{YI2(Me3-tacn)}2(μ-O)], [ScCl3(terpy)], [YCl3(terpy)(OH2)], and [{La(terpy)(OH2)Cl2}2(μ-Cl)2]. Once formed, the [ScF3(R3-tacn)] complexes are stable in water and unaffected by a ten-fold excess of Cl− or MeCO2−, although they are immediately decomposed by excess F−. The potential use of [ScF3(R3-tacn)] type complexes as platforms for 18F PET (positron emission tomography) radiopharmaceuticals is briefly discussed. Attempts to use the Group 3 fluoride “hydrates”, MF3·xH2O, as precursors were unsuccessful; no reaction with R3-tacn or terpy occurred either on reflux in CH3CN or under hydrothermal conditions (H2O, 180° C, 15 h). PXRD data showed that these “hydrates” actually contain the anhydrous metal trifluorides with small amounts of surface or interstitial water.
Introduction
The coordination chemistries of scandium and yttrium have been explored much less than those of the other 3d and 4d metals. The presence of colourless metal ions, in a single (3+) oxidation state and with closed shell configurations (hence no magnetic or d–d spectroscopic fingerprints), coupled with their limited availability, low purity and high cost, restricted early work.1,2 Often their chemistry was included in studies of the lanthanide elements, which tended to see similarities rather than explore differences.2 More recent work3 has shown that there are significant differences, especially for scandium, and the structural chemistry of scandium is surprisingly diverse.4 A rich, but synthetically challenging, organometallic chemistry of both metals has been explored in recent years,5 and C–H bond activation, ethene, styrene and α-olefin polymerisation, and aromatic C–F bond activation have all been observed in appropriate systems.5–9 Scandium fluoride has attracted considerable interest due to its negative thermal expansion,10 and nanocrystals of ScF3 doped with lanthanide ions exhibit tuneable luminescent properties.11 As expected for oxophilic metal ions, the coordination chemistry with neutral ligands has been dominated by oxoanion salts – nitrate, triflate, carboxylates, etc., with much less work on the metal halides.3 Apart from a range of fluoro-anions mostly made by solid state syntheses,2,12 complexes containing Sc–F or Y–F bonds are very rare and contain charged N- or C-donor co-ligands,4e.g. [ScL(μ-F)2(SnMe3Br)2] (L− = N,N′′-(1,3-dimethyl-1,3-propanediylidine)bis(N′,N′-diethyl-1,2-ethanediamine)) made from [ScLBr2] and Me3SnF,13 [Sc{MeC(N(2,6-iPrC6H3))CHC(Me)(NCH2CH2NMe)NH(2,6-iPrC6H3)}F],6 [Sc2(μ-F)2{Fe(C5H4NSitBuMe2)2}],9 [{Cp2Y(μ-F)(thf)2}2],8 and [{Cp2ScF}3].14 Attempts to prepare complexes of ScF3 or YF3 with neutral donor ligands,15 such as R3PO,16 have failed, whilst treatment of [ScI3{o-C6H4(PMe2)2}2] with [NMe4]F in CH2Cl2 resulted in liberation of the diphosphine and precipitation of ScF3.17In an effort to develop new PET (positron emission tomography) imaging agents, the ability of Group 13 metal (Al, Ga or In) aza-macrocycle complexes to undergo Cl/F exchange, including in some cases with radiofluorine, 18F, under mild conditions and in aqueous solution has been demonstrated.18 These include both the neutral trifluoride complexes, [MF3(R3-tacn)] (M = Al, Ga; R3-tacn = 1,4,7-trimethyl-1,4,7-triazacyclononane, 1,4-dimethyl-7-benzyl-1,4,7-triazacyclononane)19 and anionic complexes, [M(nota)F] (H2-R-nota = 1-R-1,4,7-triazacyclononane-4,7-dicarboxylic acid; R = benzyl or a peptide conjugate).20,21 The ideal requirements for PET imaging agents are that the metal complex precursor can be rapidly radiofluorinated in a single step, with minimal subsequent purification needed, and that the resulting complex is stable in water, to reaction with other competitive anions and at near neutral pH. The present work sought to establish whether Group 3 metal centres (Sc, Y or La) could offer prospects as alternative reagents for radiofluorination as potential future PET platforms and therefore explores the synthesis, properties and stability of the metal trifluoride complexes with tridentate neutral N3-donor ligands, R3-tacn and 2,2′:6′2′′-terpyridyl.
Experimental
All complex syntheses were carried out using standard Schlenk and vacuum line techniques. Samples were handled and stored in a glove box under a dry dinitrogen atmosphere to exclude moisture, which decomposes many of the samples. [ScCl3(thf)3] and [YCl2(thf)5][YCl4(thf)2] were prepared by the literature methods.22,23 2,2′:6′2′′-terpyridyl was obtained from Sigma-Aldrich and dried in vacuo prior to use. 1,4,7-Trimethyl-1,4,7-triazacyclononane and 1,4-dimethyl-7-benzyl-1,4,7-triazacyclononane were prepared as described previously.24 Anhydrous [NMe4]F was obtained by recrystallising the commercial sample (Aldrich) from iPrOH as described.25 Infrared spectra were recorded as Nujol mulls between CsI plates using a PerkinElmer Spectrum 100 spectrometer over the range 4000–200 cm−1. 1H, 19F{1H} and 45Sc NMR spectra were recorded from CH2Cl2/CD2Cl2 or CH3CN/CD3CN solutions using a Bruker AV400 spectrometer and referenced to TMS via the residual solvent resonance, CFCl3 and [Sc(H2O)7]3+ in water at pH = 1. Microanalyses were undertaken by London Metropolitan University. n-Hexane was dried by distillation from sodium and CH2Cl2 and CH3CN from CaH2.Metal trifluoride “hydrates”
Method 2: Y2(SO4)3·8H2O (3.0 g, 4.92 mmol) was suspended in hot water (80 °C) until most of the solid dissolved. The liquid was decanted off from any residue and a solution of 40% HF(aq) (3 mL) was added to the solution. A white solid precipitated immediately. The reaction was left stirring for 1.5 h and then the solid was left to settle overnight. The solution was decanted off and the solid dried overnight in a desiccator (1.24 g, 86%).
Metal complexes
X-ray experimental
Crystals of the complexes were grown as reported in the Experimental section. Data collections used a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ SuperBright molybdenum (λ = 0.71073 Å) rotating anode generator with VHF Varimax optics (70 micron focus) with the crystal held at 100 K. Structure solution and refinement were performed using SHELX(S/L)97, SHELX-2013 or SHELX-2014/7.27 H atoms bonded to C were placed in calculated positions using the default C–H distance, and refined using a riding model. Details of the crystallographic parameters are given in Table S1.† CCDC reference numbers in cif format are [ScCl3(terpy)]: 1590538, [YCl3(terpy)(OH2)]: 1590539, [ScF2Cl(Me3-tacn)]: 1590540, [YI3(Me3-tacn)]·CH3CN: 1590541, [{YI2(Me3-tacn)}2(μ-O)]·CH3CN: 1590542, [{La(terpy)(OH2)Cl2}2 (μ-Cl)2]: 1590543, [Sc(Me3-tacn)F2(μ-F)SnMe3Cl]: 1590544, [LuCl3(terpy)(OH2)]: 1590545.† Powder X-ray diffraction data were collected on a Bruker D2 diffractometer using Cu Kα X-rays and refined using the GSAS software.28Results and discussion
Three possible routes to Group 3 metal trifluoride complexes of R3-tacn and terpy (L) were considered:(1) Direct reaction with the “hydrated” Group 3 metal trifluorides with the neutral ligands;
(2) Chloride(iodide)/fluoride exchange by reaction of the pre-formed Group 3 metal trichloride or triiodide complexes, [MX3(L)] (X = Cl or I), with anhydrous [NMe4]F;
(3) Chloride(iodide)/fluoride exchange by reaction of the pre-formed Group 3 metal trichloride or triiodide complexes, [MX3(L)] (X = Cl or I), with Me3SnF.
Attempted synthesis from MF3·xH2O (M = Sc, Y, La)
The anhydrous Group 3 metal fluorides, MF3, are inert, polymeric solids and unreactive towards neutral ligands.1,15 Whilst the anhydrous Group 13 metal fluorides, MF3 (M = Al, Ga, In) are similarly inert, the hydrates MF3·3H2O are more reactive; they can be reacted with neutral ligands to form metal trifluoride ligand complexes, [MF3(L)]·xH2O, most easily under hydrothermal conditions (180 °C).19,29,30 Therefore, we postulated that it may be possible to use the Group 3 metal fluoride hydrates [MF3·xH2O] (M = Sc, Y, La) to form analogous metal trifluoride ligand complexes. Literature data on the hydrates is sparse and the amount of water present apparently variable, although “x” typically appears to be less than one.The addition of a fluoride source, either KF or aqueous HF, to aqueous solutions of yttrium or lanthanum salts, gave immediate white precipitates, MF3·xH2O. Using scandium salts and aqueous HF also gave ScF3·xH2O, but using alkali metal fluorides gave mixtures, and in one case pure KSc2F7, which was identified by its PXRD pattern (see ESI†). The PXRD patterns obtained from MF3·xH2O sometimes showed rather broad reflections, but corresponded to the patterns reported for anhydrous MF3 (Fig. 1 and ESI†).
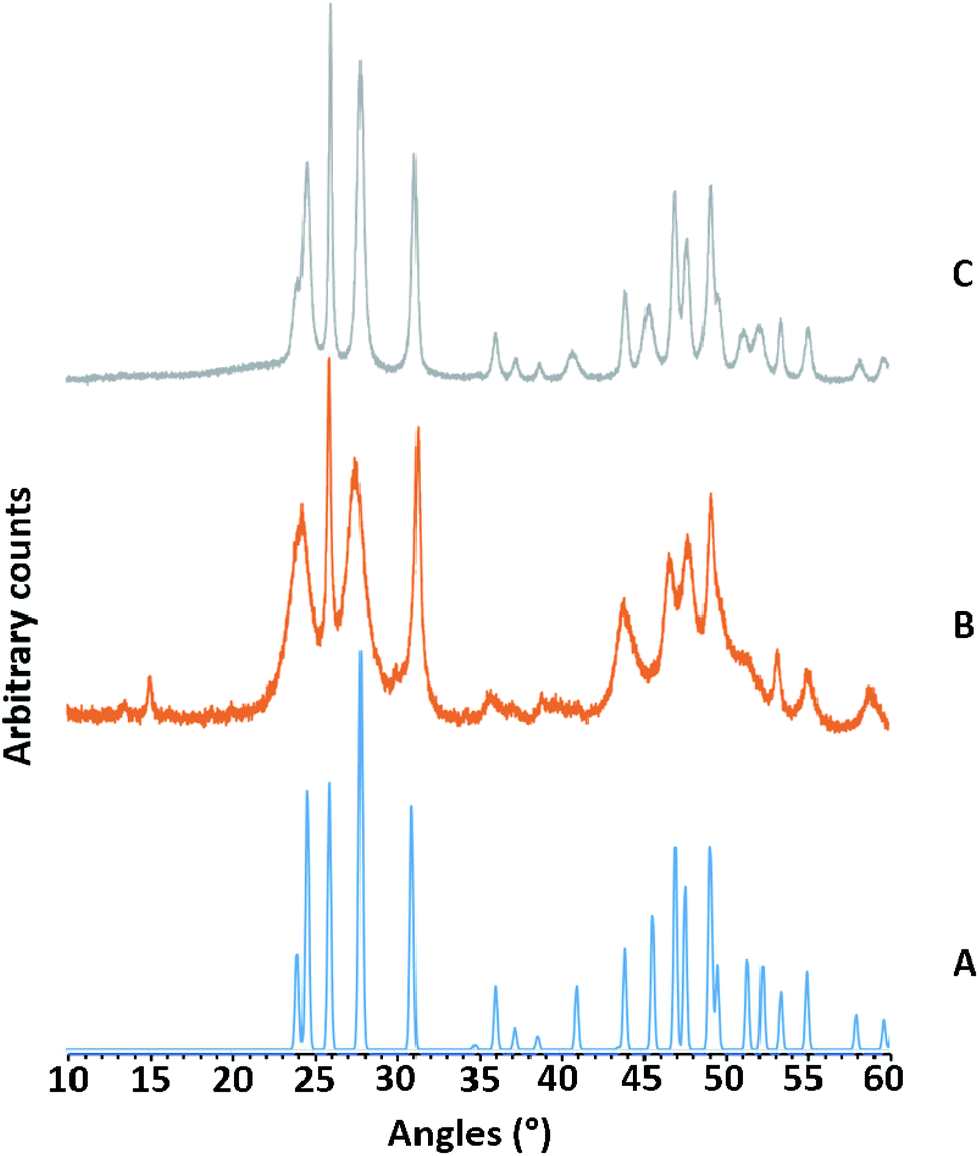 | ||
| Fig. 1 PXRD patterns obtained from the attempted synthesis of yttrium fluoride hydrate. (A) PXRD of anhydrous YF3, A. K. Cheetham and N. Norman, Acta Chem. Scand., Ser. A, 1974, 28, 55; (B) YF3·xH2O (this work); (C) YF3·xH2O after heating under hydrothermal conditions. | ||
Thus, we conclude that the MF3·xH2O actually comprise of the “anhydrous” MF3 polymer, with water of crystallisation on the surface or occupying voids in the crystal lattice, rather than coordinated to the metal ion. This also explains the range of values of x in MF3·xH2O reported in the limited literature available.1,3 Attempts to react the MF3·xH2O with terpy or Me3-tacn under hydrothermal conditions (180 °C, 15 h) were unsuccessful, with the MF3·xH2O being recovered, although with noticeably increased crystallinity (sharper PXRD patterns).
These results explain why ‘hydrated’ Group 3 fluorides are not a viable entry into the coordination chemistry of these fluorides with neutral ligands. This contrasts sharply with the Group 13 fluoride hydrates,19,29,30 and the Group 4 compounds, [MF4(OH2)2] (M = Zr, Hf),31 whose crystal structures show the water is coordinated to the metal, from which it can be displaced by neutral ligands. These are effective synthons for wider coordination chemistry. We reported previously that the f-block tetrafluorides, [MF4·xH2O] (M = Ce, Th) have very limited coordination chemistry (CeF4·xH2O dissolves only very slowly in refluxing dmso to form [CeF4(dmso)2]). They too are likely to contain only lattice/surface water.32 The wider implications of these results in metal fluoride coordination chemistry suggest that for other metals, only those ‘hydrated’ fluorides that contain water within the metal coordination sphere, are likely to be viable synthons for neutral ligand complexes.
Chloride/iodide precursor complexes
Many trichloride complexes of Sc(III), Y(III) and La(III) with neutral ligands are highly moisture sensitive and must be synthesised and handled in anhydrous systems. In work with other early d-block systems, we noted that the corresponding metal iodides and iodo-complexes were often more soluble in weakly coordinating solvents, probably due to lower lattice energy, although the gain in solubility comes at the cost of even greater moisture sensitivity.17,33,34 In the present study we synthesised complexes with Me3-tacn and terpy, which give examples of pseudo-octahedral complexes with fac and mer geometries for scandium, respectively, although for Y and La higher coordination numbers were often produced. The known26 [MCl3(Me3-tacn)] (M = Y, Sc) were made from [ScCl3(thf)3] or [YCl2(thf)5][YCl4(thf)2] and the triaza macrocycle in anhydrous acetonitrile. The corresponding [ScCl3(BnMe2-tacn)] was prepared similarly, whilst reaction of LaCl3·7H2O with Me3-tacn in EtOH yielded [LaCl3(Me3-tacn)(OH2)] as a white powdered solid. All complexes have been characterised spectroscopically (IR, 1H, 45Sc NMR, Table 1, as appropriate) and by microanalysis.| Complex | δ(45Sc)/ppm | δ(19F{1H})/ppm |
|---|---|---|
| a CH3CN/CD3CN except. b CH2Cl2/CD2Cl2. | ||
| [ScCl3(Me3-tacn) | +300 | — |
| [ScCl3(BnMe2-tacn) | +302 | — |
| [ScFCl2(Me3-tacn)] | +219 | 77.2 |
| [ScF2Cl(Me3-tacn)] | +155 | 40.1 |
| [ScF3(Me3-tacn)] | 104 (q, 1JSc–F = 219 Hz) | 7.7 |
| [ScF3(BnMe2-tacn) | 104 | 10.1 |
| [ScF3(terpy)] | +64 | −37.7 [2F], −53.3 [F] |
| [ScCl3(terpy)]b | +254 | — |
Treatment of YI3 with one molar equivalent of Me3-tacn in anhydrous acetonitrile under a dinitrogen atmosphere formed a light yellow, extremely moisture sensitive solid, identified as [YI3(Me3-tacn)]·1.5CH3CN. The 1H NMR spectrum of the product displayed multiplets at 2.81 and 2.73 ppm, corresponding to the ligand CH2 groups, as well as a singlet at 2.58 ppm, due to the ligand methyl groups, consistent with facial complexation of the Me3-tacn. Crystallisation from CH3CN solution yielded two different crystal morphologies, both of which were analysed by single crystal X-ray diffraction and shown to be [YI3(Me3-tacn)]·CH3CN (Fig. 2), with the expected tridentate Me3-tacn and three mutually facial iodides completing the distorted octahedral environment, and the partial hydrolysis product, [{YI2(Me3tacn)}2(μ-O)] (Fig. 3), formed via reaction with trace water. The Y–N bond distances in [YI3(Me3-tacn)] are similar to those reported in functionalised tacn complexes, e.g. [{iPr2-tacn(CH2)2NtBu}Y(CH2SiMe3)2].35
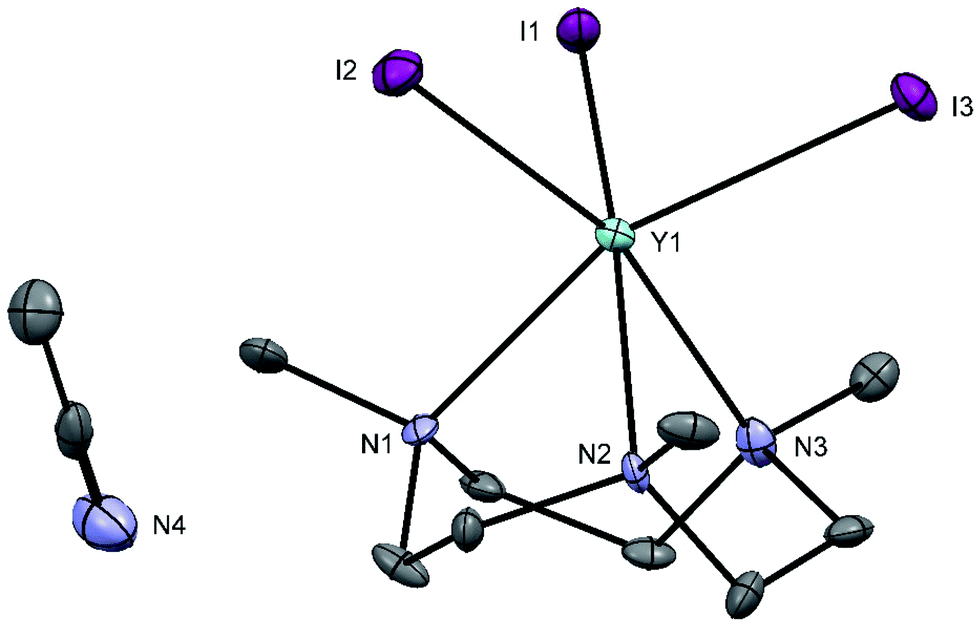 | ||
| Fig. 2 Structure of [YI3(Me3-tacn)]·CH3CN with atom numbering scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Select bond lengths (Å) and angles (°): Y1–I1 = 2.9671(8), Y2–I2 = 2.9509(9), Y1–I3 = 2.9460(8), Y1–N1 = 2.468(6), Y1–N2 = 2.480(5), Y1–N3 = 2.467(7), I3–Y1–N1 = 160.9(1), I2–Y1–N3 = 162.3(2), I1–Y1–N2 = 161.6(1), I1–Y1–I2 = 99.59(2), I3–Y1–I2 = 98.87(2), I3–Y1–N2 = 94.5(1), N1–Y1–N2 = 72.0(2). | ||
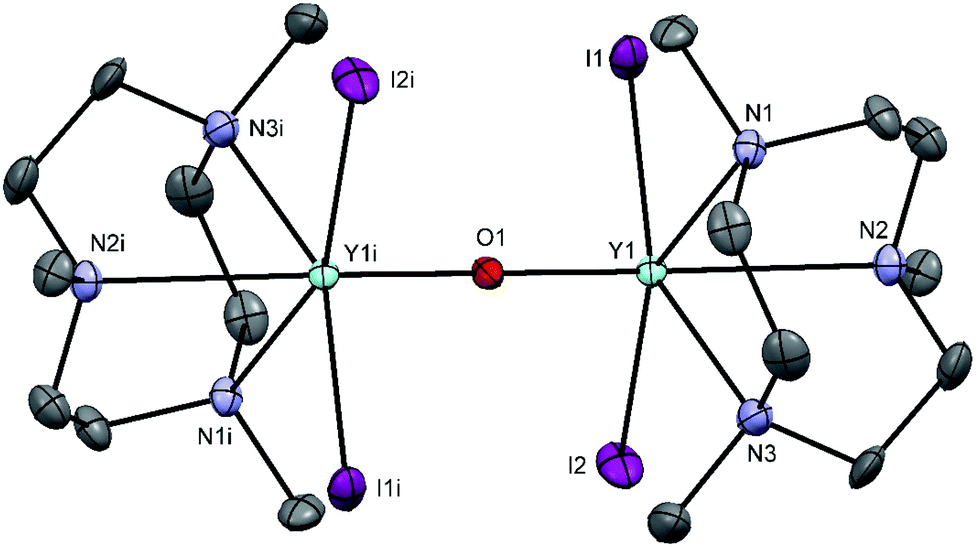 | ||
| Fig. 3 The structure of [{YI2(Me3tacn)}2(μ-O)]·CH3CN with atom numbering scheme. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Y1–I1 = 3.0213(6), Y1–I2 = 2.9969(7), Y1–O1 = 2.0332(5), Y1–N2 = 2.580(5), Y1–N1 = 2.473(5), Y1–N3 = 2.495(5), Y1–O1–Y1 = 180.00(3), I2–Y1–I1 = 93.991(19), O1–Y1–I1 = 104.69(2), O1–Y1–I2 = 104.12(2), O1–Y1–N2 = 156.16(11), O1–Y1–N1 = 90.72(11), O1–Y1–N3 = 93.79(11). | ||
The reaction of [ScCl3(thf)3] with terpy in anhydrous CH3CN gave mer-[ScCl3(terpy)] (Fig. 4).
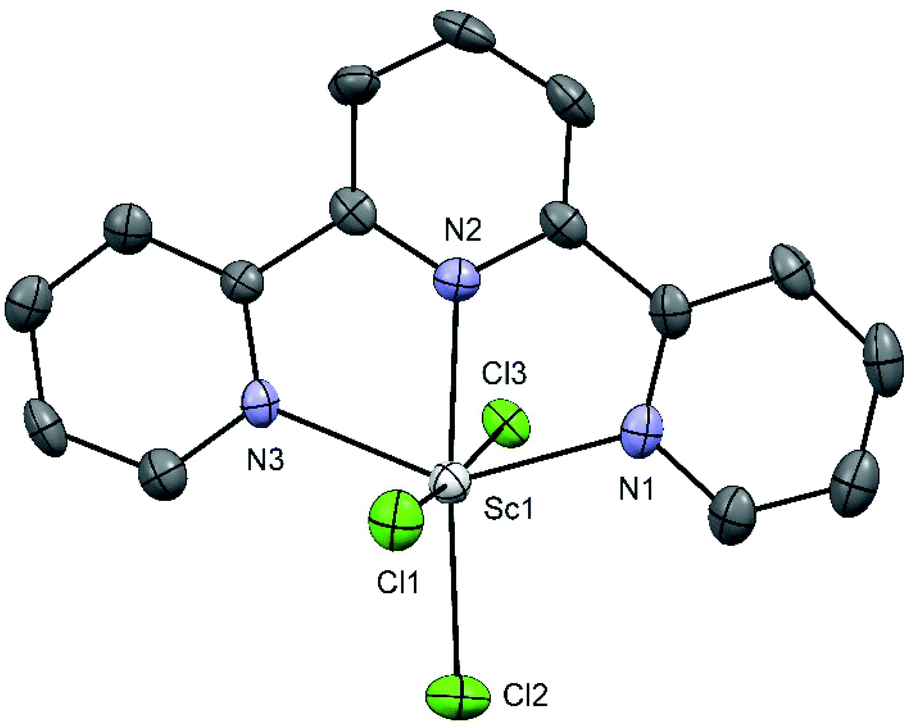 | ||
| Fig. 4 Structure of mer-[ScCl3(terpy)] with ellipsoids drawn at 50% probability level. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Sc1–Cl1 = 2.414(2), Sc1–Cl2 = 2.396(2), Sc1–Cl3 = 2.451(2), Sc1–N1 = 2.229(4), Sc1–N2 = 2.231(5), Sc1–N3 = 2.248(4), Cl1–Sc1–Cl3 = 174.51(7), Cl2–Sc1–N2 = 174.2(1), N1–Sc1–N3 = 142.3(2), N1–Sc1–N2 = 71.3(2), N2–Sc1–N3 = 71.1(2), N2–Sc1–Cl3 83.7(1), Cl3–Sc1–Cl2 91.22(6), Cl2–Sc1–N1 = 105.9(1). | ||
The complex has a distorted octahedral coordination around the metal centre conferred by the rigid terpy ligand, the angles involving the ligand are significantly less than the 180/90° expected for a regular octahedron, with N1–Sc1–N3 = 142.3°. The extended crystal structure of [ScCl3(terpy)] shows π-stacking interactions (3.82 Å) between the aromatic ring of the terpy ligand of the adjacent molecule, connecting them into 1D zig-zag chains (see ESI Fig. S4†).
Reaction of [YCl2(thf)5][YCl4(thf)2] with terpy in CH3CN yields the colourless complex [YCl3(terpy)(OH2)] incorporating adventitious water and characterised spectroscopically and by microanalysis. Confirmation of the seven-coordinate geometry follows from a crystal structure determination (Fig. 5).
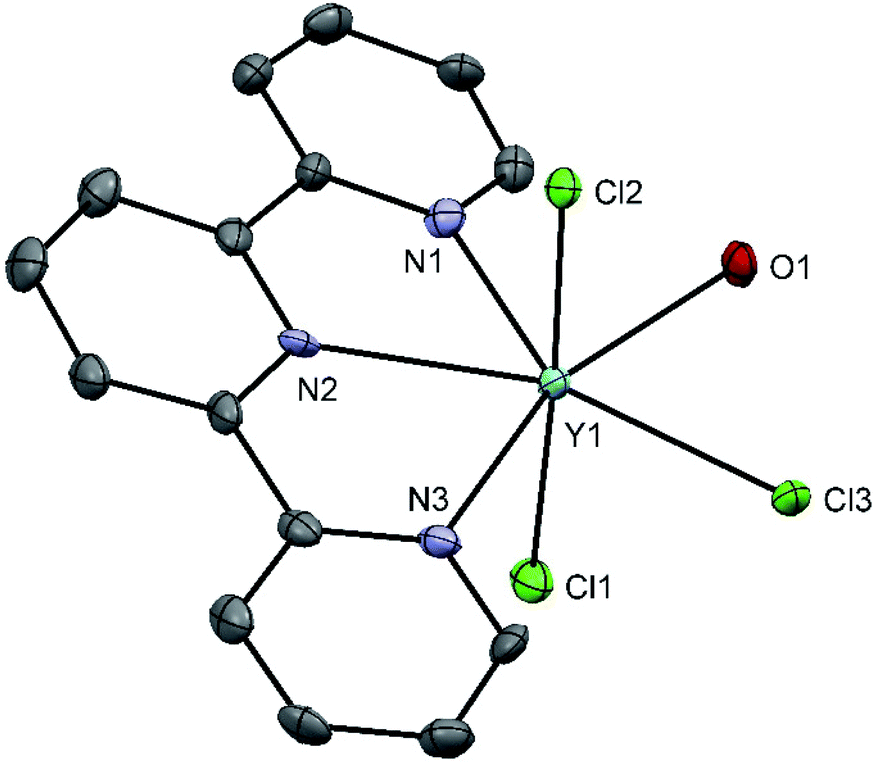 | ||
| Fig. 5 Crystal structure of [YCl3(terpy)(OH2)] with ellipsoid drawn at 50% probability level. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Y1–Cl1 = 2.572(2), Y1–Cl2 = 2.611(2), Y1–Cl3 = 2.629(1), Y1–N1 = 2.507(5), Y1–N2 = 2.480(4), Y1–N3 = 2.514(5), Y1–O1 2.348(3), Cl2–Y1–Cl1 176.49(5), Cl3–Y1–Cl1 90.77(5), Cl1–Y1–O1 96.6(1), Cl1–Y1–N2 96.3(1), Cl2–Y1–N2 80.8(1), N2–Y1–N1 64.9(2). | ||
The crystal structure (Fig. 5) shows a pentagonal-bipyramidal coordination around the metal centre with the Y–Cl bond lengths in the axial positions shorter than that in the equatorial plane. The angle between the yttrium centre and the nitrogen atoms are less than the 72° value expected for the perfect pentagonal–bipyramidal conformation, due to the rigid terpy ligand and the equatorial plane is puckered. Furthermore, the packing in the crystal structure shows both H-bonding (Cl⋯HOH) between adjacent molecules to form associated dimers, and weak π-stacking (4.04 Å) linking the dimers into zig-zag chains (Fig. S5†). The bond lengths are generally shorter than in the eight-coordinate [YCl(terpy)(OH2)4]Cl2·2H2O.36
Using the heavier f-block ions, La(III) and Lu(III), [LaCl3(terpy)(OH2)]·4H2O and [LuCl3(terpy)(OH2)] were obtained via reaction of LaCl3·7H2O or LuCl3·6H2O, respectively, with one mol. equiv. of terpy in ethanol. X-ray crystallographic analyses show that the La(III) complex exists as a chloro-bridged dimer, [{La(terpy)(OH2)Cl2}2(μ-Cl)2] (Fig. 6) involving eight-coordinate La(III) with adjacent molecules linked into chains via H-bonding interactions between the coordinated water molecule on one La(III) centre and the Cl ligands on adjacent molecules (Fig. S6†). On the other hand, [LuCl3(terpy)(OH2)] (Fig. S7†) is a seven-coordinate monomer, isostructural with the Y(III) analogue above (and hence also displaying the same H-bonding and π-stacking interactions in the solid state).
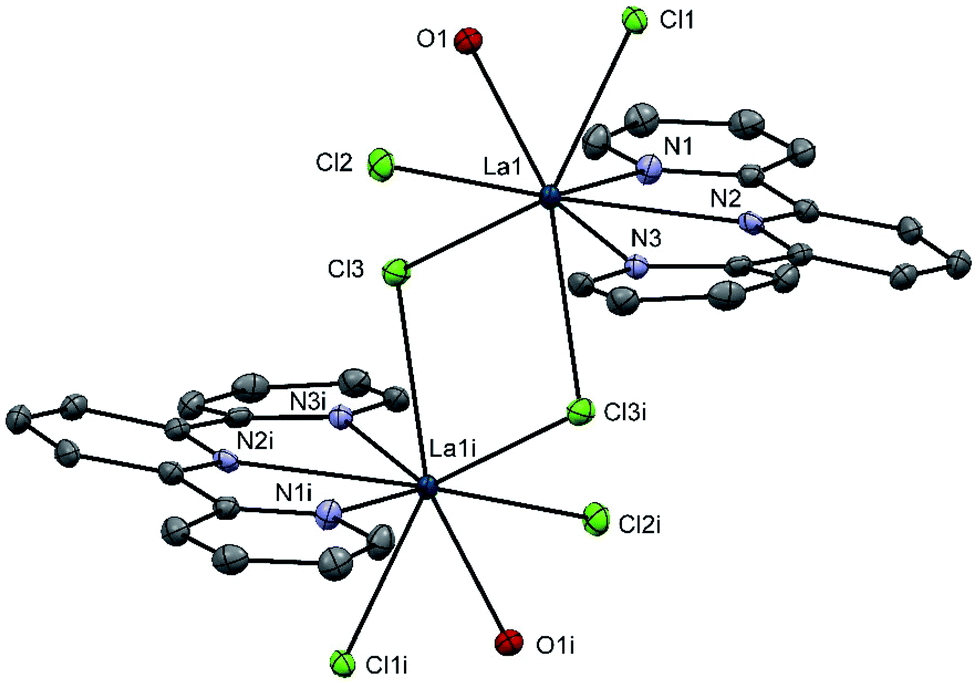 | ||
| Fig. 6 Crystal structure of [{La(terpy)(OH2)Cl2}2(μ-Cl)2] with ellipsoids drawn at 50% probability level. H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): La1–Cl1 = 2.8385(9), La1–Cl2 = 2.8014(8), La1–Cl3 = 2.8895(9), La1–N1 = 2.638(3), La1–N2 = 2.678(3), La1–N3 = 2.658(3), La1–O1 = 2.576(3), La1–Cl3i = 2.9209(9), Cl1–La1–Cl3 = 144.65(2), Cl1–La1–Cl2 = 86.82(2), Cl1–La1–O1 = 72.14(7), Cl2–La1–O1 = 78.77(7), Cl3–La1–O1 = 140.69(7), N3–La1–N1 123.52(9), N3–La1–O1 145.03(9), N3–La1–N2 = 62.21(9). | ||
Chloride(iodide)/fluoride exchange reactions using [NMe4]F
Addition of three mol. equiv. of anhydrous [NMe4]F to a CH3CN solution of [ScCl3(Me3-tacn)] gave a colourless solution whose 19F{1H} and 45Sc NMR spectra37 each showed three broadened resonances at 77.2, 40.1 and 7.7 ppm (19F{1H}) and 219, 155 and 104 ppm (45Sc) (Fig. 7, Table 1), corresponding to [ScFCl2(Me3-tacn)] (very minor), [ScF2Cl(Me3-tacn)] and [ScF3(Me3-tacn)], respectively. The modest quadrupole moment of 45Sc (I = 7/2) means that resonances are observed in many systems, but couplings to other nuclei are often lost in the line broadening, unless the scandium is in a high symmetry environment.17,34,38,39 Since these complexes are the first examples of scandium fluoride species with neutral ligands, there are no comparable literature data, but the chemical shifts of the chloro-species are reasonable compared to data on other ScCl3 adducts.34,38,39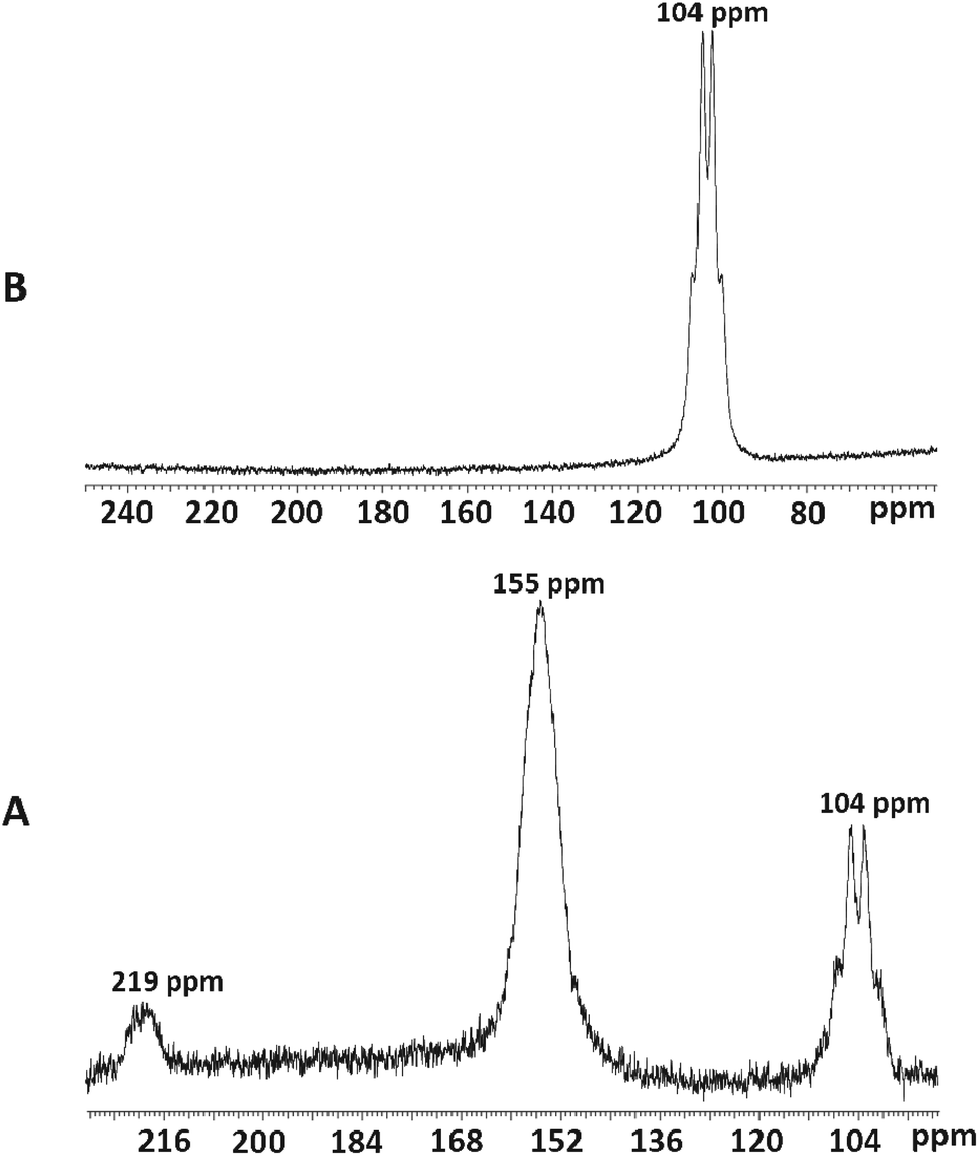 | ||
| Fig. 7 Fluorination of [ScCl3(Me3tacn)]: A: 45Sc NMR showing the species [ScFCl2(Me3tacn)] (219 ppm), [ScF2Cl(Me3tacn)] (155 ppm) and [ScF3(Me3tacn)] (104 ppm) when a deficit of [NMe4]F is used; B: 45Sc NMR of [ScF3(Me3tacn)] showing the quartet at 104 ppm (1JSc–F = 219 Hz). | ||
Adding further small aliquots of [NMe4]F in CH3CN initially led to depletion of the resonances assigned to the mixed chloro/fluoro complexes, and enhancement of the broadened quartet at δ = 104 ppm, attributed to [ScF3(Me3-tacn)] (Fig. 7). The broad quartet shows coupling to three equivalent fluorides with 1JScF = 219 Hz (since the efg is small). However, excess fluoride caused complete loss of all the 45Sc and 19F{1H} resonances from the tacn complexes (see below).
The 19F{1H} and 45Sc NMR data for the fluorination reaction are also strongly indicative of the fluorination of [ScCl3(Me3-tacn)] occurring in a stepwise manner. Further confirmation of this follows from a single crystal X-ray structure determination on [ScF2Cl(Me3-tacn)], a few crystals of which were grown by concentrating the NMR solution in acetonitrile (Fig. 8).
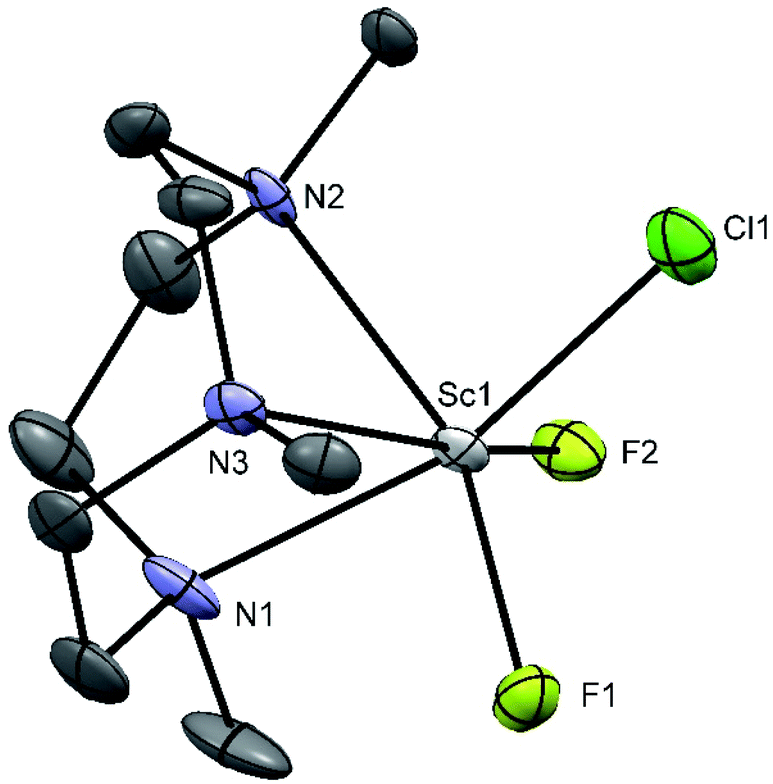 | ||
| Fig. 8 Crystal structure of [ScF2Cl(Me3-tacn)] with ellipsoids drawn at the 50% probability level. H atoms are omitted for clarity. Select bond lengths (Å) and angles (°): Sc1–F1 = 1.947(3), Sc1–F2 = 1.940(3), Sc1–Cl1 = 2.444(2), Sc1–N1 = 2.339(5), Sc1–N2 = 2.364(4), Sc–N3 = 2.334(4), Cl1–Sc1–F1 = 101.7(1), Cl1–Sc1–N1 = 162.4(1), Cl1–Sc1–N2 = 89.9(1), F1–Sc1–F2 = 103.3(1), N2–Sc1–N3 = 74.6(1), F1–Sc1–N3 = 158.8(1). | ||
The complex is six-coordinate with the three nitrogen atoms of the ligand coordinated to the metal in a facial conformation, while the three other positions are occupied by two fluorine atoms and one chlorine, with no evidence for disorder being present. As expected, the Sc–F bonds are significantly shorter than the Sc–Cl bond (1.94 against 2.44 Å). The analogous [ScCl3(BnMe2-tacn)] shows similar behaviour with [NMe4]F, with [ScF3(BnMe2-tacn)] showing a broad 45Sc NMR resonance at 104 ppm, with δ(19F{1H}) = 10.1 ppm.
Attempts to react [ScCl3(terpy)] with dry [NMe4]F in CH3CN on an NMR scale caused complete loss of the resonances from the trichloro complex, without the appearance of any new resonances in either the 45Sc or 19F spectra.
The reaction of [YCl3(Me3-tacn)] with three equivalents of [NMe4]F in CH3CN resulted in a white precipitate that was insoluble in CH3CN or CH2Cl2 and the 1H NMR spectrum of the solution showed liberation of Me3-tacn, whilst the 19F NMR spectrum showed only small amounts of free fluoride. Similar results were obtained using [YI3(Me3-tacn)], and it was concluded that Cl(I)/F exchange resulted in decomposition and loss of the Me3-tacn from the yttrium. Similar decomposition occurred using [LaCl3(Me3-tacn)(OH2)].
Chloride/fluoride exchange reactions using Me3SnF
Me3SnF is a useful fluorinating agent, its polymeric structure makes it insoluble in most solvents,40,41 but it dissolves as the Cl/F exchange reaction proceeds, and usually, the Me3SnCl (which contains tbp tin centres weakly chlorine-bridged into polymeric chains)42 formed is easily removed from the products by washing with hexane. The reagent does not provide free fluoride ions so an excess can be used without the risk of the decomposition observed using [NMe4]F. The fac-octahedral trifluoro complexes [ScF3(R3-tacn)] were readily obtained by treatment of the trichloro species with three mol. equiv. of Me3SnF (and adding excess Me3SnF has no further effect) (Fig. 9). The broad “doublets” observed in the 19F{1H} NMR spectra result from partial collapse of the couplings to 45Sc (I = 7/2) in the low symmetry environments.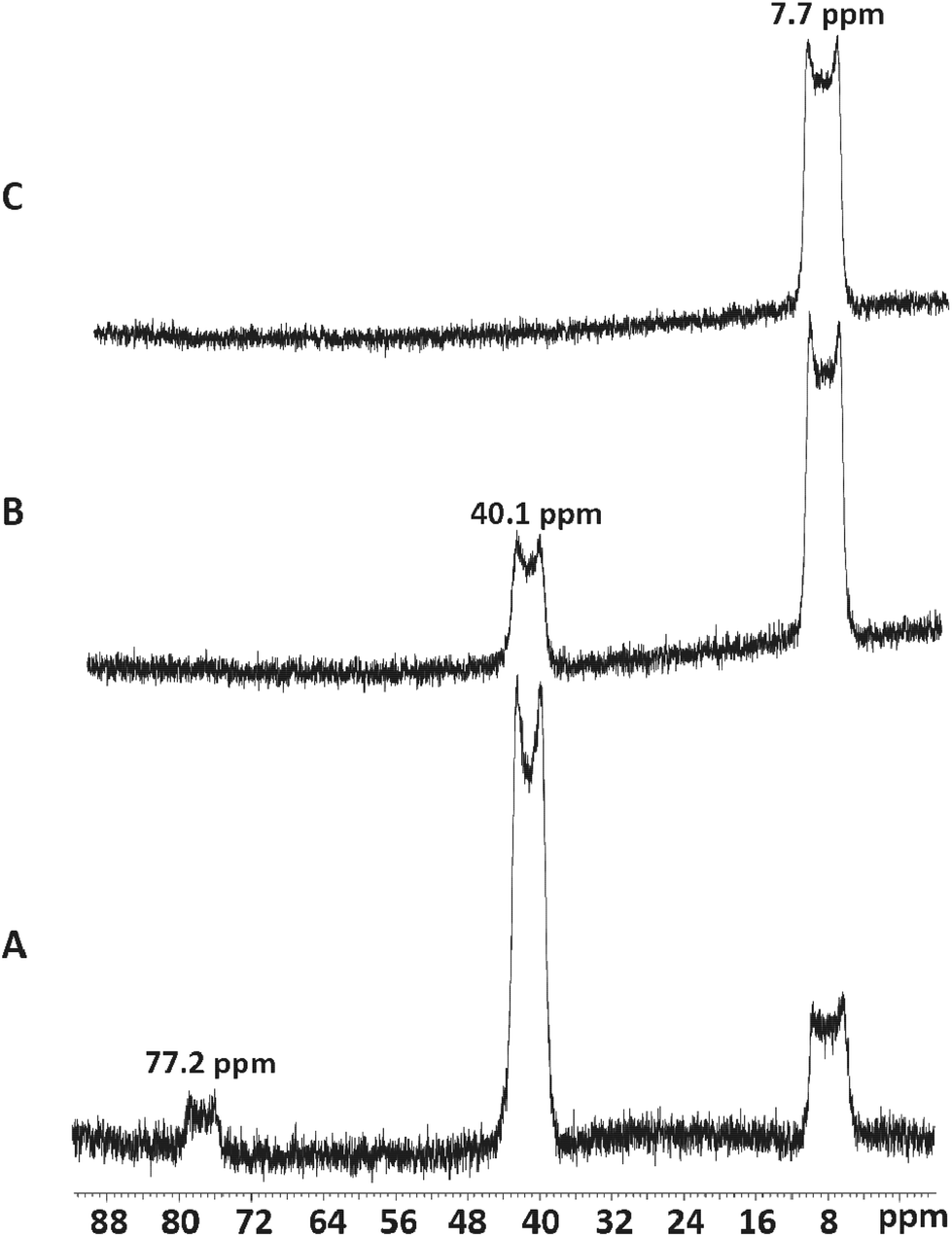 | ||
| Fig. 9 19F{1H} NMR spectra of [ScFCl2(Me3tacn)] (77.2 ppm), [ScF2Cl(Me3tacn)] (40.1 ppm) and [ScF3(Me3tacn)] (7.7 ppm). A: ∼ 2.5 mol. equiv. of Me3SnF were added to a CD3CN solution of [ScCl3(Me3tacn)]; B: <3 mol. equiv. of Me3SnF were added to a CD3CN solution of [ScCl3(Me3tacn)]; C: >3 mol. equiv. of Me3SnF were added to a CD3CN solution of [ScCl3(Me3tacn)]. | ||
However, the product obtained had a microanalysis corresponding to [Sc(Me3-tacn)F2(μ-F)SnMe3Cl]; note that whilst three equivalents of Me3SnCl are produced in the reaction, only one is retained in the scandium complex.
Crystals of [Sc(Me3-tacn)F2(μ-F)SnMe3Cl] were obtained by slow evaporation from a solution of the product in CH3CN. The X-ray crystal structure confirms the presence of three mutually facial fluoride ligands, with the tridentate triaza macrocycle completing the distorted octahedral geometry at Sc with Sc–F = 1.980(6), 1.92(2) and Sc–N = 2.334(6), 2.351(4) Å. The [ScF3(Me3-tacn)] complex acts as a neutral Lewis base towards the Me3SnCl Lewis acid through one bridging fluoride ligand, Sc–F1 = 1.980(6), Å, Sn1–F1 = 2.307(6) Å (Fig. 10). This results in a trigonal bipyramidal geometry at Sn, with the Cl and F ligands in the axial positions and a near linear F–Sn–Cl unit (178.7(2)°). The Sc–N and Sc–Fterminal bonds are similar to those in [ScF2Cl(Me3tacn)], but the Sc–F(Sn) bond is significantly longer. The Sn–F bond is longer than that in Me3SnF (2.1620(7) Å),41 whilst the Sn–Cl is also longer than that in Me3SnCl polymer (2.430(2) Å).42 The Lewis basic properties of fac-[MF3(R3-tacn)] (M = Al, Ga, Fe, Cr, etc.) complexes has been noted previously, and exploited to form mixed metal species with alkali metal43 and lanthanide cations, the latter with applications as molecular magnets.44
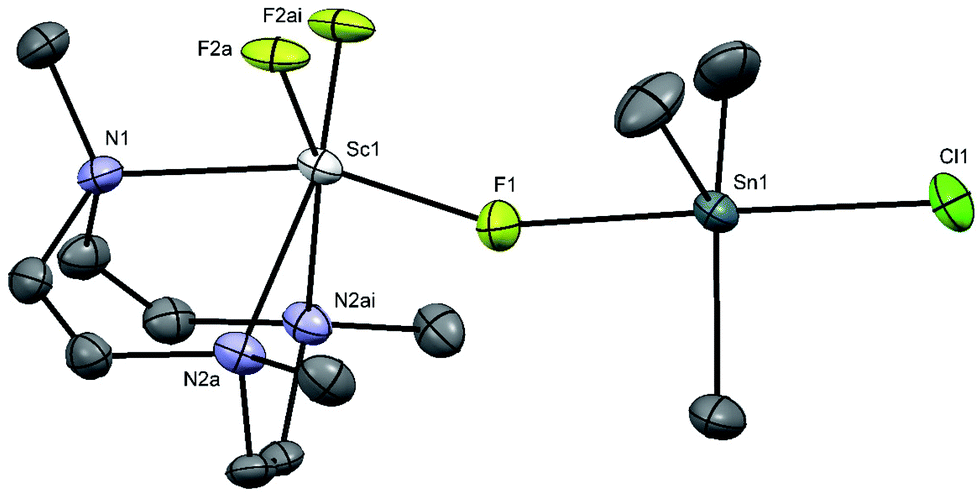 | ||
| Fig. 10 Crystal structure of [Sc(Me3-tacn)F2(μ-F)SnMe3Cl] with atom numbering scheme. Ellipsoids are shown at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Sc1–N1 = 2.334(6), Sc1–N2 = 2.351(4), Sc1–F1 = 1.980(6), Sc1–F2a = 1.92(2), Sn1–F1 = 2.307(6), Sn1–Cl1 = 2.502(2), Sn1–C1 = 2.124(7), Sn1 = C2a = 2.153(3), N2–Sc1–N2 = 75.36(18), N1–Sc1–N2 = 74.92(14), F2a–Sc1–N1 = 94.6(10), F1–Sc1–N1 159.7(3), F1–Sn–Cl1 = 178.7(2). | ||
The only similar complex to have been reported is [ScL(μ-F)2(SnMe3Br)2] (L− = N,N′′-(1,3-dimethyl-1,3-propanediylidine)bis(N′,N′-diethyl-1,2-ethanediamine)), made from [ScLBr2] and Me3SnF.13
The 45Sc and 19F{1H} NMR spectra of [Sc(Me3-tacn)F2(μ-F)SnMe3Cl] and [ScF3(Me3-tacn)] in CH3CN are identical, as are the 1H resonances of the Me3-tacn moieties, whilst the 1H NMR resonance of the Me3SnCl in the former complex is consistent with the free organotin, indicating the adduct is dissociated in solution. Me3SnCl is a weak Lewis acid and complexes such as [Me3SnCl(Ph3PO)] also appear largely dissociated in solution.45 However, attempts to crystallise [ScF3(Me3-tacn)] from solutions of [Sc(Me3-tacn)F2(μ-F)SnMe3Cl] failed with the bimetallic species reformed in the isolated solid. This contrasts with [ScL(μ-F)2(Me3SnBr)2] which decomposes when the mother liquor is removed.13 Gently heating a finely ground sample of [Sc(Me3-tacn)F2(μ-F)SnMe3Cl] (40 °C) under vacuum leads to partial removal of the Me3SnCl, but complete removal of the tin species could not be achieved without decomposition of the scandium moiety.
The reaction of [ScCl3(terpy)] with three equivalents of Me3SnF gave a white solid identified by microanalysis and multinuclear NMR as [Sc(terpy)F(μ-F)2(SnMe3Cl)2]. The 19F{1H} NMR (CD3CN) δ = −37.7 (br s, [2F]), −53.3 (br s, [F]) and 45Sc NMR δ = 64 (s, br) are consistent with the formulation. However, in contrast to [Sc(Me3-tacn)F2(μ-F)SnMe3Cl], [Sc(terpy)F(μ-F)2(SnMe3Cl)2] is unstable in solution and slowly deposits a white insoluble solid.
The reaction of [YCl3(Me3-tacn)] with Me3SnF in CH3CN resulted in decomposition, and no YF3 complex was identified.
Unlike the trichloro analogues, which are very hydrolytically sensitive, solutions of [ScF3(Me3-tacn)] and [ScF3(BnMe2-tacn)] in water at neutral pH are stable for many weeks, monitored by 19F and 45Sc NMR spectroscopy, and to heating aqueous solutions at 80 °C for several hours. The complexes are decomposed by excess F−, and by Na3PO4 and Na2CO3 (which have a pH ∼ 9), but are stable to a 10-fold excess of Cl− or MeCO2−.
Conclusions
The [ScF3(Me3-tacn)] and [ScF3(BnMe2-tacn)], which represent the first reported examples of scandium fluoride complexes with neutral co-ligands, have been prepared by Cl/F exchange reactions from the corresponding chlorides, using [NMe4]F. Attempts to isolate similar complexes of Y or La, or [ScF3(terpy)] have been unsuccessful. In these cases fluorination is accompanied by dissociation of the neutral ligands and formation of insoluble MF3. Highly unusual [Sc(terpy)F(μ-F)2(SnMe3Cl)2] and [Sc(Me3-tacn)F2(μ-F)SnMe3Cl] have been isolated using Me3SnF as the fluoride source. The high stability of [ScF3(R3-tacn)] in water over a range of temperatures and pHs, as well as towards common ions such as Cl− or MeCO2−, indicates that are worth further examination to determine their potential as possible carriers for 18F in PET imaging radio-tracers.The decomposition of analogous Y and La trichloride complexes upon attempted fluorination, indicates that the R3-tacn or terpy ligands are insufficiently strongly bound to these metal ions to prevent their dissociation and formation of MF3 polymer. Since Y and La are larger than Sc, tetra-azamacrocycles (cyclen or cyclam) may be better suited. Alternatively, anionic pendant arm ligands such as the H2-R-NOTA type, which will bind more strongly, may be better for these oxophilic metal centres. These possibilities will be investigated in future work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC and GE Healthcare for funding through a CASE studentship to F. M. M., and we thank EPSRC for support through EP/L505651/1.References
- Scandium, ed. C. T. Horovitz, Academic Press, London, 1976 Search PubMed.
- F. A. Hart, Comprehensive Coordination Chemistry I, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, vol. 3, p. 1060 Search PubMed.
- S. A. Cotton, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 3, p. 93 Search PubMed; K. J. Fisher, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004, vol. 4, p. 1 Search PubMed.
- J. M. Sears and T. J. Boyle, Coord. Chem. Rev., 2017, 340, 154 CrossRef CAS.
- P. M. Zeimentz, S. Arndt, B. R. Elvidge and J. Okuda, Chem. Rev., 2006, 106, 2404 CrossRef CAS PubMed.
- J. Chu, X. Han, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2014, 136, 10894 CrossRef CAS PubMed.
- S. A. Bartlett, G. Cibin, A. J. Dent, J. Evans, M. J. Hanton, G. Reid, R. P. Tooze and M. Tromp, Dalton Trans., 2013, 42, 2213 RSC and refs therein.
- Z. Xie, K. Chui, Q. Yang, T. C. W. Mak and J. Sun, Organometallics, 1998, 17, 3937 CrossRef CAS.
- W. Huang and P. L. Diaconescu, Organometallics, 2017, 36, 89 CrossRef CAS.
- J. Chen, Q. Gao, A. Sanson, X. Jiang, Q. Huang, A. Carnera, C. G. Rodriguez, L. Olivi, L. Wang, L. Hu, K. Lin, Y. Ren, Z. Lin, C. Wang, L. Gu, J. Deng, J. P. Attfield and X. Xing, Nat. Commun., 2017, 8, 14441 CrossRef CAS PubMed; L. Hu, J. Chen, A. Sanson, H. Wu, C. G. Rodriguez, L. Olivi, Y. Ren, L. Fan, J. Deng and X. Xing, J. Am. Chem. Soc., 2016, 138, 8320 CrossRef PubMed.
- L. Han, Y. Wang, L. Guo, L. Zhao and Y. Tao, Nanoscale, 2014, 6, 5907 RSC; J. Xie, X. Xie, C. Mi, Z. Gao, Y. Pan, Q. Fan, H. Su, D. Jin, L. Huang and W. Huang, Chem. Mater., 2017, 29, 9758 CrossRef CAS.
- D. Brown, Halides of the lanthanides and actinides, Wiley, NY, 1968 CrossRef CAS PubMed; M. Leblanc, V. Maisonneuve and A. Tressaud, Chem. Rev., 2015, 115, 1191 CrossRef CAS PubMed.
- A. M. Neculai, D. Neculai, G. B. Nikiforov, H. W. Roesky, C. Schlicker, R. Herbst-Irmer, J. Magull and M. Noltemeyer, Eur. J. Inorg. Chem., 2003, 3120 CrossRef CAS.
- F. Bottomley, D. E. Paez and P. S. White, J. Organomet. Chem., 1985, 291, 35 CrossRef CAS.
- S. L. Benjamin, W. Levason and G. Reid, Chem. Soc. Rev., 2013, 42, 1460 RSC.
- N. J. Hill, W. Levason, M. C. Popham, G. Reid and M. Webster, Polyhedron, 2002, 21, 1579 CrossRef CAS; N. J. Hill, W. Levason, M. C. Popham, G. Reid and M. Webster, Polyhedron, 2002, 21, 445 CrossRef.
- M. Carravetta, M. Concistre, W. Levason, G. Reid and W. Zhang, Inorg. Chem., 2016, 55, 12890 CrossRef CAS PubMed.
- K. Chansaenpak, B. Vabre and F. P. Gabbai, Chem. Soc. Rev., 2016, 45, 954 RSC.
- R. Bhalla, C. Darby, W. Levason, S. K. Luthra, G. McRobbie, G. Reid, G. Sanderson and W. Zhang, Chem. Sci., 2014, 5, 381 RSC; W. Levason, S. K. Luthra, G. McRobbie, F. M. Monzittu and G. Reid, Dalton Trans., 2017, 46, 14519 RSC.
- R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, G. Reid and G. Sanderson, Chem. – Eur. J., 2015, 21, 4688 CrossRef CAS PubMed.
- P. Laverman, W. McBride, R. Sharkey, A. Eek, L. Joosten, W. Oyen, D. Goldberg and O. Boerman, J. Nucl. Med., 2010, 51, 454 CrossRef CAS PubMed; W. McBride, C. D'Souza, R. Sharkey, H. Karacay, E. Rossi, C. Chang and D. Goldberg, Bioconjugate Chem., 2010, 21, 1331 CrossRef PubMed; W. McBride, R. Sharkey, H. Karacay, C. D'Souza, E. Rosso, P. Laverman, C. Chang, O. Boerman and D. Goldberg, J. Nucl. Med., 2009, 50, 991 CrossRef PubMed; S. Lutje, G. M. Franssen, R. M. Sharkey, P. Laverman, E. A. Rossi, D. M. Goldberg, W. J. G. Oyen, O. C. Boerman and W. J. McBride, Bioconjugate Chem., 2014, 25, 335 CrossRef PubMed; W. J. McBride, D. M. Goldberg and R. M. Sharkey, J. Nucl. Med., 2014, 55, 1043 CrossRef PubMed.
- L. E. Manzer, Inorg. Synth., 1982, 21, 1579 Search PubMed.
- P. Sobota, J. Utko and S. Szafert, Inorg. Chem., 1994, 33, 5203 CrossRef CAS.
- K. Wieghardt, P. Chaudhuri, B. Nuber and J. Weiss, Inorg. Chem., 1982, 21, 3086 CrossRef CAS; M. J. Belousoff, M. B. Duriska, B. Graham, S. R. Batten, B. Moubaraki, K. S. Murray and L. Spiccia, Inorg. Chem., 2006, 45, 3746 CrossRef PubMed.
- K. O. Christe, W. W. Wilson, R. D. Wilson, R. Bau and J. A. Feng, J. Am. Chem. Soc., 1990, 112, 7619 CrossRef CAS.
- S. Hajela, W. P. Schaefer and J. E. Bercaw, J. Organomet. Chem., 1997, 532, 45 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. C. Larson and R. B. Von Dreele, Generalized Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR, 2004, p. 86 CrossRef CAS; B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef CAS.
- R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, F. M. Monzittu, J. Palmer, G. Reid, G. Sanderson and W. Zhang, Dalton Trans., 2015, 44, 9569 RSC.
- R. Bhalla, J. Burt, A. L. Hector, W. Levason, S. K. Luthra, G. McRobbie, F. M. Monzittu and G. Reid, Polyhedron, 2016, 106, 65 CrossRef CAS.
- S. L. Benjamin, W. Levason, D. Pugh, G. Reid and W. Zhang, Dalton Trans., 2012, 41, 12548 RSC.
- M. J. D. Champion, W. Levason and G. Reid, J. Fluorine Chem., 2014, 57, 19 CrossRef.
- W. Levason, M. L. Matthews, B. Patel, G. Reid and M. Webster, Dalton Trans., 2004, 3305 RSC.
- M. J. D. Champion, P. Farina, W. Levason and G. Reid, Dalton Trans., 2013, 42, 13179 RSC.
- S. Bambirra, A. Meetsma, B. Hessen and A. P. Bruins, Organometallics, 2006, 25, 3486 CrossRef CAS.
- C. J. Kepert, L. Weimin, B. W. Skelton and A. H. White, Aust. J. Chem., 1994, 47, 365 CrossRef CAS.
- 45Sc is 100% abundant and has I = 7/2, Ξ = 29.24 MHz, Dc = 1710, Q = −0.22 × 10−28 m2; J. Emsley, The Elements, Oxford, 1989 Search PubMed.
- Multinuclear NMR, ed. J. Mason, Plenum, NY, 1987 Search PubMed; Transition metal nuclear magnetic resonance, ed. P. S. Pregosin, Elsevier, Amsterdam, 1991 Search PubMed.
- N. J. Hill, W. Levason, M. C. Popham, G. Reid and M. Webster, Polyhedron, 2002, 21, 1579 CrossRef CAS; L. Deakin, W. Levason, M. C. Popham, G. Reid and M. Webster, J. Chem. Soc., Dalton Trans., 2002, 2439 Search PubMed.
- E. F. Murphy, R. Murugavel and H. W. Roesky, Chem. Rev., 1997, 97, 3425 CrossRef CAS PubMed.
- P. Chaudhary, M. Bieringer, P. Hazendonk and M. Gerken, Dalton Trans., 2015, 44, 19651 RSC.
- J. L. Lefferts, K. C. Molloy, M. R. Hossain, D. Van der Helm and J. J. Zuckerman, J. Organomet. Chem., 1982, 240, 349 CrossRef CAS.
- R. Bhalla, W. Levason, S. K. Luthra, G. McRobbie, G. Reid, G. Sanderson and W. Zhang, Chem. Commun., 2014, 50, 12675 RSC.
- K. S. Pedersen, M. A. Sørensen and J. Bendix, Coord. Chem. Rev., 2015, 299, 1 CrossRef CAS.
- M. F. Davis, W. Levason, R. Ratnani, G. Reid, T. Rose and M. Webster, Eur. J. Inorg. Chem., 2007, 306 CrossRef CAS and refs therein.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic parameters are given in Table S1. The ESI also contains IR and NMR spectra for the complexes and figures showing the π-stacking and H-bonding in the terpy complexes. CCDC 1590538–1590545. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt00480c |
| This journal is © The Royal Society of Chemistry 2018 |