
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpirocyclic germanes via transannular insertion reactions of vinyl germylenes into Si–Si bonds†‡
Małgorzata
Walewska
a,
Judith
Baumgartner
 *a,
Christoph
Marschner
*a,
Lena
Albers
*b and
Thomas
Müller
*b
*a,
Christoph
Marschner
*a,
Lena
Albers
*b and
Thomas
Müller
*b
aInstitut für Anorganische Chemie, Technische Universität Graz, Stremayrgasse 9, A-8010 Graz, Austria
bInstitut für Chemie, Carl von Ossietzky Universität Oldenburg, Carl von Ossietzky Straße 9-11, 26129 Oldenburg, European Union
First published on 23rd March 2018
Abstract
The reactions of two cyclic germylene phosphane adducts with monosubstituted acetylenes caused the formation of spirocyclic germanes, which is postulated to occur by double acetylene insertion into germylene attached bonds. Further insertion of the formed cyclic divinylgermylene into transannular Si–Si or Si–Ge bonds provides the spirocyclic germanes. Thermal treatment of two germacyclopropenes, formed by the reaction of the two cyclic germylene phosphane adducts with tolane, also produced spirocyclogermanes. The structures of the latter require, however, a more complicated mechanistic proposal.
Introduction
In addition to the chemistry of carbenes,1,2 that of analogous heavier congeners3–9 has also become increasingly popular in recent years. In addition to constituting an interesting class of new transition metal ligands,10–15 several of these compounds are able to participate in small molecule bond activation reactions.16–19Our own interest in this compound class concentrates on silylated tetrylenes.20–34 In contrast to the well-studied N-heterocyclic heavy carbene analogues, these are not stabilised by π-donation of electron density from the attached nitrogen lone pairs into the empty p-orbitals, which leads to a much higher reactivity. To prevent dimerization or rearrangement reactions the formation of phosphane adducts proved to be a viable concept since the interaction between the tetrylene center and the donor is strong enough to suppress its decomposition but is still weak enough to allow dissociation. This is important, since the introduction of stronger donor molecules such as small N-heterocyclic carbenes (NHC) switches the reactivity pattern of the respective adduct from “free tetrylene” to “tetrylenoid”.6
In a recent communication we reported that the reaction of phenylacetylene with the PMe3 adduct of bis[tris(trimethylsilyl)silyl]germylene (1) occurs by regioselective insertion into a Si–Ge bond, thus yielding a PMe3 adduct of a vinylgermylene (2).33 The reaction of 1 with tolane, however, gives the less unexpected germirene 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 (Scheme 1). The formation of 2 is very promising as it clearly indicates that weak donor adducts of divalent germanium are energetically similar or even more stable than isomeric structures with tetravalent germanium. This is a prerequisite for the potential use of divalent germanium compounds as catalysts in catalytic cycles with the intermediate formation of tetravalent germanes.
33 (Scheme 1). The formation of 2 is very promising as it clearly indicates that weak donor adducts of divalent germanium are energetically similar or even more stable than isomeric structures with tetravalent germanium. This is a prerequisite for the potential use of divalent germanium compounds as catalysts in catalytic cycles with the intermediate formation of tetravalent germanes.
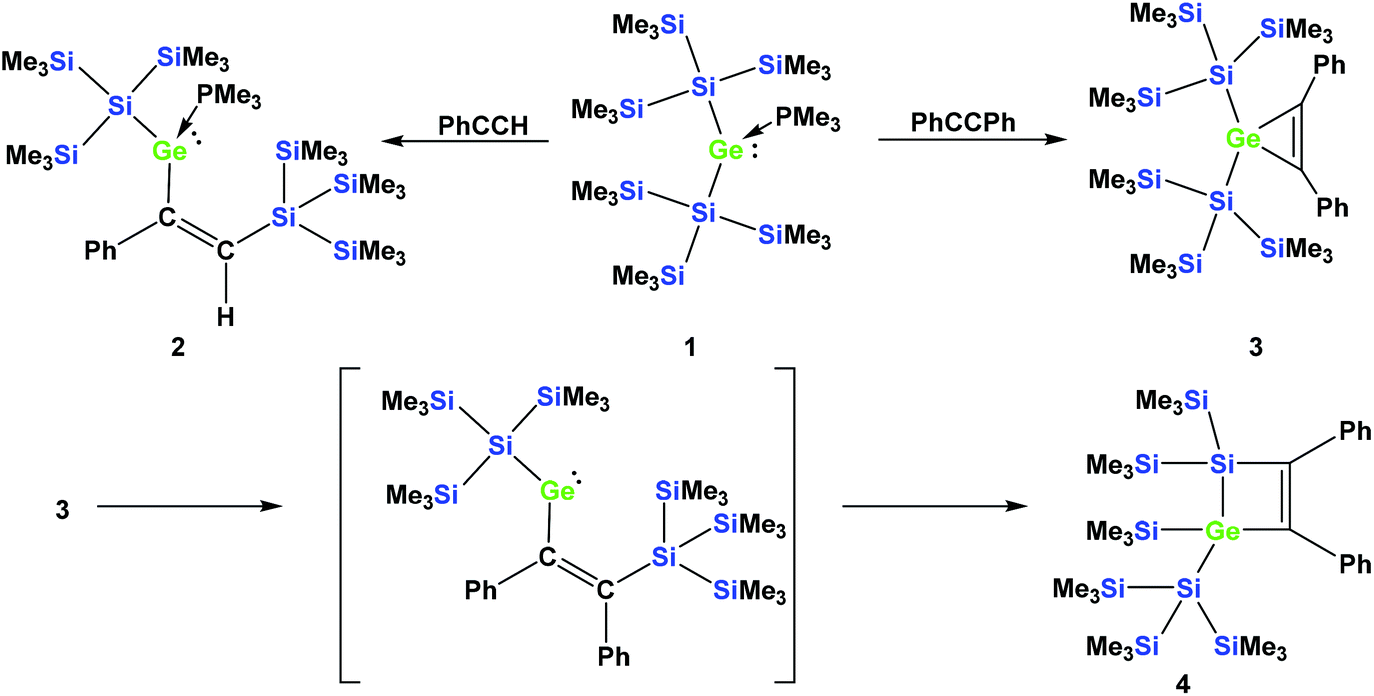 | ||
| Scheme 1 Reactions of germylene adduct 1 with either phenylacetylene (top left) or tolane (top right). Thermally-induced rearrangement of germirene 3 to silagermacyclobutene 4 (bottom part). | ||
In a subsequent study, we could show that the formation of 2 involves the intermediate formation of a germirene similar to 3.34 The reason that 3 itself does not rearrange to the isomeric vinylgermylene is due to the fact that the silyl shift onto the phenyl substituted sp2-hybridised carbon is sterically less favored. Heating compound 3 to 150 °C gives silagermacyclobutene 4, likely occurring via the intermediate formation of a vinylgermylene, which stabilizes by insertion into a Si–SiMe3 bond (Scheme 1).34
A related germirene with two less sterically demanding ethyl substituents, obtained by the reaction of 1 with 3-hexyne, was shown to undergo a rearrangement to a structure analogous to 4 even at ambient temperature.34
A number of related reactions were reported recently for silylenes and stannylenes. Rieger and co-workers reported the insertion of ethylene into the Si–Si(SiMe3)3 bond of a silylated silylene,35 while Kato and Baceiredo described the insertion of an olefin into a Si–H bond of their stabilised silylene also involving the previous formation of a silirene.36 Insertion of tolane into the Sn–B and Ge–B bonds of a stannylene and a germylene, respectively, was reported by Aldridge and co-workers.37 An example where two germylenes react with several different disubstituted alkynes to 1,2-digermacyclobut-3-enes was observed recently by Ketkov, Dostál and co-workers.19 A different approach to vinylgermylenes was communicated by Rivard and co-workers utilizing N-heterocyclic olefins.38 Most recently, Sasamori and co-workers reported the use of a digermyne for the catalytic regioselective cyclotrimerization of terminal alkynes. Their mechanistic explanation for this reaction also involves alkyne insertion into the bonds attached to germylenes.39
Results and discussion
Reactions of cyclic germylene adducts with two alkynes
In order to study whether the unexpected migratory insertion behavior of 1 is a general reactivity feature of silylated germylenes, we further investigated the behavior of cyclic germylene adducts 5a and 5b.28,31 The reaction of the disilylated germylene 5a with two equivalents of phenylacetylene was found to give a mixture of the two spirocyclic compounds 6a and 7a in an approximate ratio of 1:1 (Scheme 2).
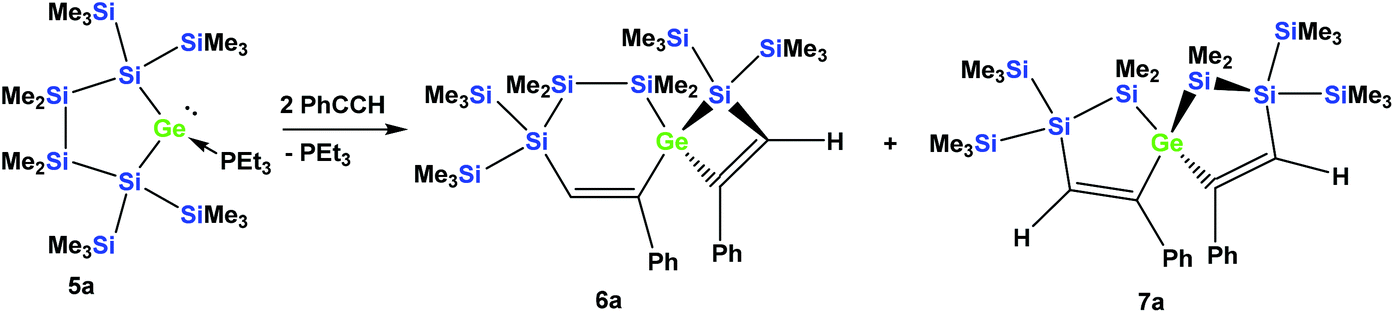 | ||
| Scheme 2 Reaction of cyclic germylene phosphane adduct 5a with phenylacetylene. | ||
However, the reaction of two equivalents of phenylacetylene with the analogous digermylated germylene adduct 5b gave only a single product, namely the asymmetric spirocyclus 6b (Scheme 3).
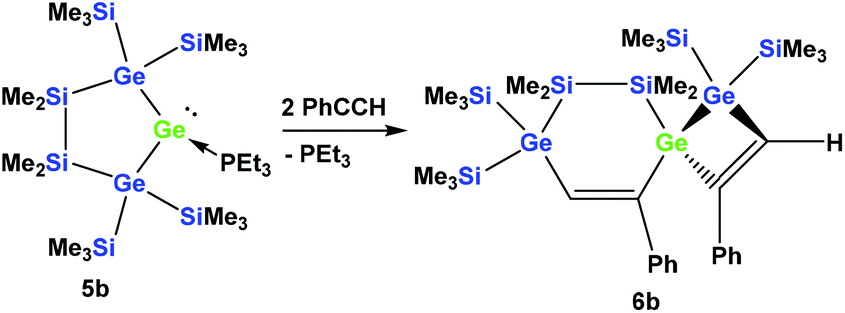 | ||
| Scheme 3 Reaction of digermylated cyclic germylene phosphane adduct 5b with phenylacetylene. | ||
The fact that subtle differences are essential for the reactivity in these reactions was shown by the reaction of 5a with two equivalents of Me3SiCCH, which also gave only a single product, but in this case the symmetric spirocyclic compound 7c was formed (Scheme 4).
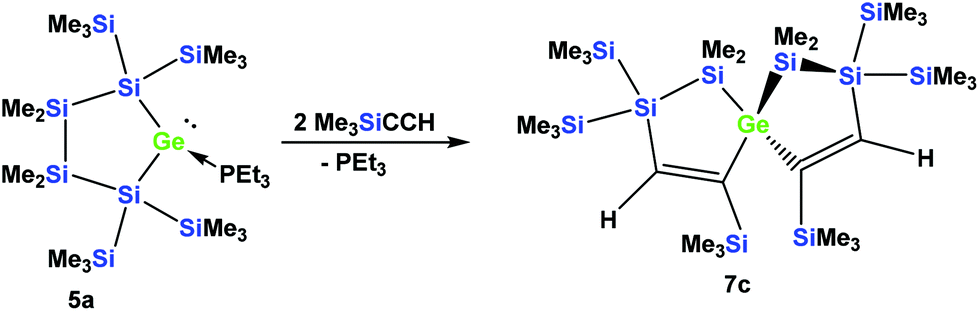 | ||
| Scheme 4 Reaction of cyclic germylene phosphane adduct 5a with trimethylsilylacetylene. | ||
It seems reasonable to assume that all these reactions proceed in a very similar manner. The first steps are likely to be double insertion reactions of the acetylene units into the Si–Ge or Ge–Ge bonds of 5a and 5b to give cyclic divinylated germylenes 9a, 9b, and 9c (Scheme 5). The regiochemistry of these steps is the same as that described above for acyclic germylene adduct 1.32 The resulting nine-membered ring germylenes are likely to be conformationally rather flexible. The subsequent intramolecular insertion of the released free germylenes 9 can occur either into transannular Me2Si–SiMe2 or Me2Si–E(SiMe3)2 (E = Si, Ge) bonds (Scheme 5).
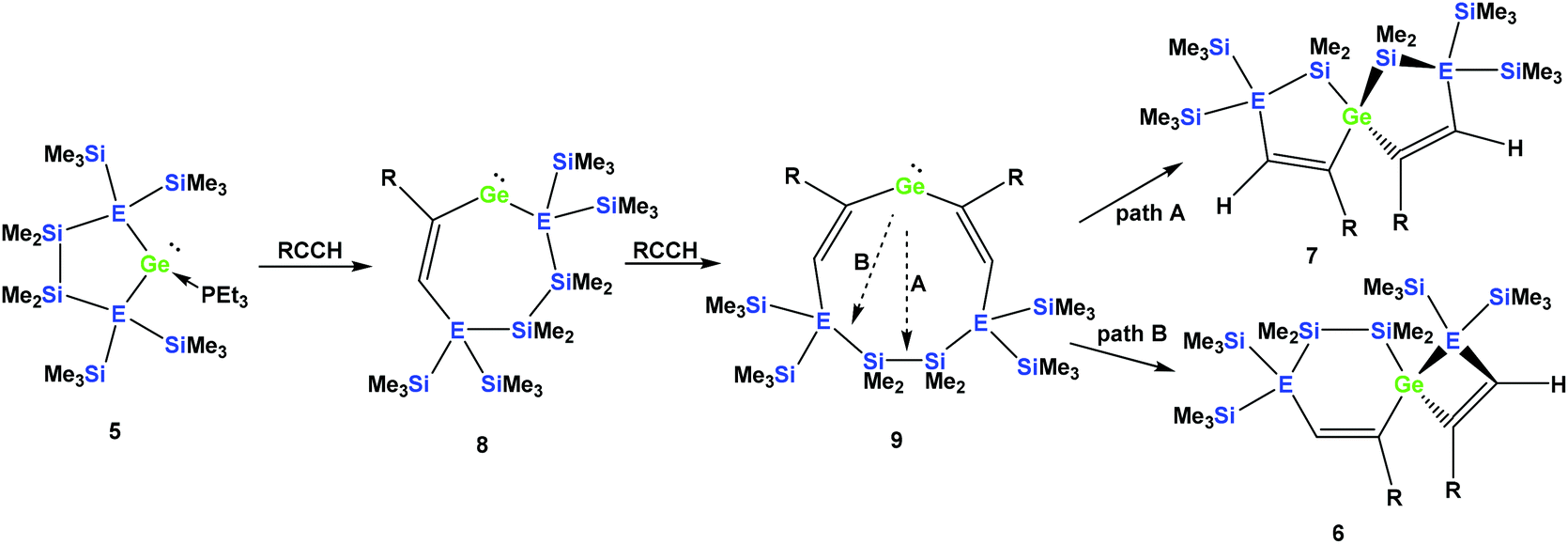 | ||
| Scheme 5 Assumed reaction mechanism of germylene adducts 5a and 5b with monosubstituted acetylenes to spirocyclic compounds (a: E = Si and R = Ph; b: E = Ge and R = Ph). | ||
Attempts to run the reactions of 5a and 5b with only one equivalent of phenylacetylene led to more complicated mixtures. In the case of 5b the 29Si NMR spectrum showed the presence of spirocycle 6b derived from 9b and the starting material 5b. In addition, signals which likely belong to a [3.3]-spirocycle derived from the mono-inserted germylene 8b (Scheme 5) were observed. This suggests that germylenes 5b and 8b are of similar reactivity and compete for phenylacetylene.
Depending on into which bond the germylene insertion occurs, either symmetric [4.4]-spirocyclic compounds like 7a or 7c or [5.3]-spirocyclic ones like 6a or 6b are formed (Schemes 2–5). The initial structural assignment was carried out on the basis of single crystal XRD analyses of 6a, 6b, and 7c. Compounds 6a and 7a were obtained as a mixture and the signals in the 29Si spectrum are not easily assignable. However, the spirocyclic compound 7c, which is very similar to 7a was formed selectively and displays a rather simple 29Si NMR spectrum with five signals (Table 1). Apart from the trimethylsilyl vinyl signal at −6.9 ppm, the two geminal trimethylsilyl groups were observed at −12.1 and −12.7 ppm, and the dimethylsilyl unit was found at −28.2 ppm with the remaining Si(SiMe3)2 signals located at −66.4 ppm. Assuming that the set of very similar signals (−12.1, −12.5, −27.3, and −66.9 ppm) represent compound 7a, which were then subtracted from the spectrum of the mixture of 6a and 7a, the remaining signals can be assigned to be those of 6a (−9.6, −10.9, −11.4, −11.8, −35.1, −46.1, −46.8, and −86.6 ppm).
| Compound | 29Si | 29Si | 29Si | 29Si |
|---|---|---|---|---|
| E(SiMe3) | SiMe2 | Si(SiMe3)2 | Other SiMe3 | |
|
5a31 |
−4.3/−8.5 | −22.3 | −126.0 | |
|
5b31 |
−2.0/−4.1 | −16.9 | n.a. | |
| 6a | −9.6/−10.9/−11.4/−11.8 | −35.1 (GeSiMe2)/−46.1 (SiSiMe2) | −46.8 (GeSiC)/−86.6 (Me2SiSiC) | |
| 7a | −12.1/−12.5 | −27.3 | −66.9 | |
| 6b | −3.3/−3.7/−4.7/−5.0 | −35.5/−37.3 | n.a. | |
| 7c | −12.1/−12.7 | −28.2 | −66.4 | −6.9 |
|
10a31 |
−7.3 | −30.3 | −120.1 | |
| 10b | −1.5 | −23.4 | n.a. | |
| 12a | −2.9/−7.6/−9.3/−12.5 | −22.6/−35.3 | −60.1 (CSi(SiMe3)2)/−119.1 (SiSi(SiMe3)2) | |
| 13a | −8.6/−8.7/−9.5/−9.6 | 9.9, 3.8 | −94.9 (GeSi(SiMe3)2)/−108.2 (SiSi(SiMe3)2) | |
| 13b | −2.5/−2.9/−3.2/−4.5 | 3.5 (GeSiC)/18.4 (GeSiGe) | n.a. |
The four signals between −9.6 and −11.8 ppm are associated with the trimethylsilyl groups and the signal at −86.6 ppm belongs to the Si(SiMe3)2 in the six-membered ring. The other Si(SiMe3)2 unit is assigned to the signal at −46.8 ppm, which shows the rather typical strong down-field shift in cyclotetrasilane rings.40,41 The two remaining signals at −35.1 and −46.1 ppm are those of the SiMe2 groups in the six-membered ring, with the one at −35.1 being attached to the central germanium atom, which usually leads to a down-field shifted signal compared to a related unit attached to a silicon atom. This assignment of the signals of 6a is fully consistent with what we observe for compound 6b, which is isostructural to 6a.
Compared to 6a, for 6b the four trimethylsilyl signals are shifted some 6 ppm down-field (Table 1), while the SiMe2 signals appear at −35.5 and −37.3 ppm, reflecting the fact that in contrast to the situation of 6a, both groups are attached to a germanium atom.
The striking dependence of the product formation on subtle differences in the reactants that are shown in Schemes 2–4 prompted us to investigate these reactions in detail using DFT calculations at the B3LYP-D3 level.42 Based on our previous investigations,33 we supposed that cyclic bis-vinylgermylenes 9 (Scheme 5 and Fig. 1) are key compounds for the formation of the two different germanium spiro-cycles 7 (via path A, Fig. 1) and 6 (via path B). In both cases, the intramolecular insertion reaction of the dicoordinated germanium atom into the remote Si–Si or Si–E (E = Si, Ge) in compounds 9 was calculated to be a strongly exergonic process (see Fig. 1).
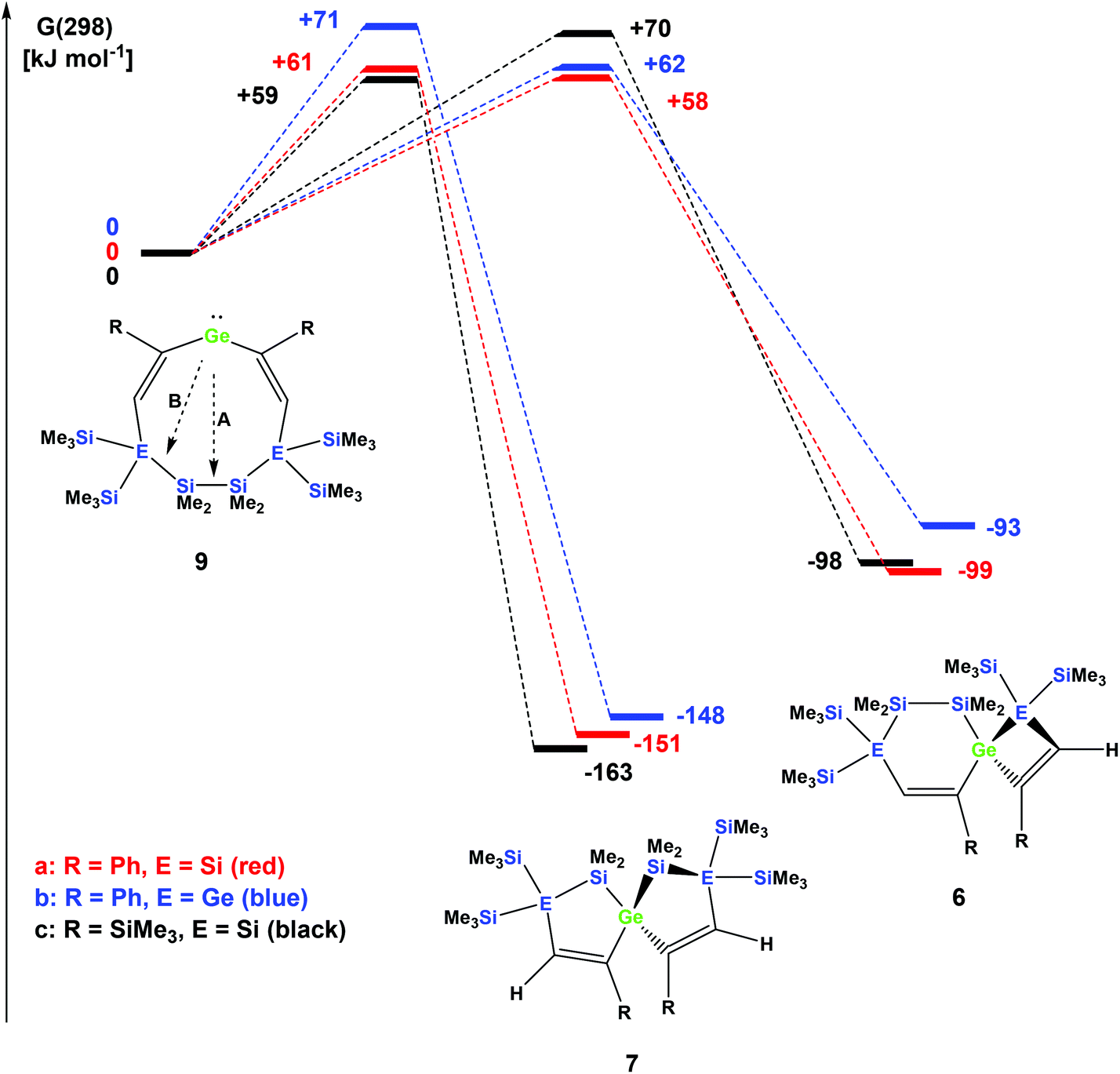 | ||
| Fig. 1 Calculated reaction coordinates for the intramolecular bond insertion reaction of germylenes 9; A: insertion into the Si–Si bond to give [4.4] germanium spirocycles 7; B: insertion into the Si–E bond to give [5.3] germanium spirocycles 6 (free energy differences at 298 K G(298), at B3LYP-D3/6-311G(d,p)(Si,C,H); def2tzvp(Ge)). | ||
As expected, the formation of the less strained [4.4]-spirocyclic compounds 7 is in each case thermodynamically favored over the [5.3]-spirocyclic isomers 6 (ΔΔG (7a/6a) = 52 kJ mol−1), (ΔΔG (7b/6b) = 55 kJ mol−1), and (ΔΔG (7c/6c) = 65 kJ mol−1) (Fig. 1). Therefore, the computational results indicated that the generation of [5.3]-spirocycles 6 is only possible under kinetic control. We found that both types of insertion reactions proceeded in one concerted step via the transition states TS(9/7) and TS(9/6) (Fig. 1 and Table 2).
| Compound | TS germylene 9/spiro [5.5] 7 | Spiro [5.5] 7 | TS germylene 9/spiro [6.4] 6 | Spiro [6.4] 6 |
|---|---|---|---|---|
| R = Ph, E = Si | +54 (+61) | −152 (−151) | +61 (+58) | −101 (−99) |
| R = Ph, E = Ge | +59 (+71) | −150 (−148) | +57 (+62) | −103 (−93) |
| R = SiMe3, E = Si | +52 (+59) | −165 (−163) | +64 (+70) | −104 (−98) |
The calculated barriers ΔGǂ for both processes are between 58 and 71 kJ mol−1, in qualitative agreement with the reactions that proceed under ambient conditions (see Fig. 1). In view of the high barriers for the back reaction (155–168 kJ mol−1 for 6 → 9, and 211–222 kJ mol−1 for 7 → 9, Fig. 1), the establishment of a thermodynamic equilibrium between the three isomers 9–6 is unlikely. For the phenyl-substituted germylene 9a the energy difference ΔΔGǂ between both competing reaction paths is merely 3 kJ mol−1 in favor of the formation of the [5.3]-spirocycle 6a. Based on a basic Boltzmann treatment this would suggest the formation of a product mixture of the two isomers 6a and 7a of approximately 23:77. This is in qualitative agreement with the experimental results that the reaction of germylene 5a with phenylacetylene provides a mixture of the isomers 6a and 7a (Scheme 2). For the reaction of germylene 5b with phenylacetylene the bis-vinylgermylene 9b is the most likely intermediate. The intramolecular insertion of the dicoordinated germanium atom into the weaker Ge–Si (path B, Scheme 5) is favored over the insertion into the Si–Si bond (path A) by ΔΔGǂ = 9 kJ mol−1. Under the conditions of kinetic control at room temperature, this energy difference suggests a nearly exclusive formation of the less stable [5.3] germanium spirocycle 6b (ratio 7b:6b = 3:97), in perfect agreement with the experimental results. The situation is different for the reaction of germylene 5a with trimethylsilylacetylene (Scheme 4). In the case of the corresponding cyclic bis-vinylgermylene 9c, we found for the generation of the more stable [4.4]-spirocycle 7c a barrier which is by ΔΔGǂ = 11 kJ mol−1 lower than that predicted for the competing reaction channel to give the [5.3] isomer 6c (Fig. 1). Therefore, our computations suggest the almost exclusive formation of the more stable isomer 7c (ratio 7c:6c = 99:1) in accordance with the experiment.
Rearrangement reactions of spirocyclic germacyclopropenes
Cyclic germylene adducts 5a and 5b can be reacted with tolane and the expected spirocyclic germacyclopropenes 10a34 and 10b are obtained. Further attempts to react 5a and 5b with bis(trimethylsilyl)acetylene, however, did not give the expected germacyclopropenes.
As was described above for the temperature-induced rearrangement of the germacyclopropene 3,34 compound 10a was also subjected to 100 °C to facilitate the insertion step of the olefin group into the Ge–Si bond. We expected that upon the formation of the seven-membered cyclic germylene 11a, the insertion into a transannular Si–Si bond would give the [3.3]-spirocyclic compound 12a with the two bis(trimethylsilyl)silylene units in different rings (Scheme 6).
 | ||
| Scheme 6 Expected thermally-induced reaction pathway of 10a to give [3.3]-spirocyclic vinylgermane 12a. | ||
According to the 29Si NMR spectroscopic analysis of the reaction solution, two products were formed with two signal sets of eight symmetry non-equivalent 29Si signals each (Fig. 2). Although it was not possible to separate the mixture of crystalline products, we were nevertheless able to obtain a crystal of one of the main products which was subjected to a single crystal XRD study. According to this analysis a spirocyclic product was obtained again.
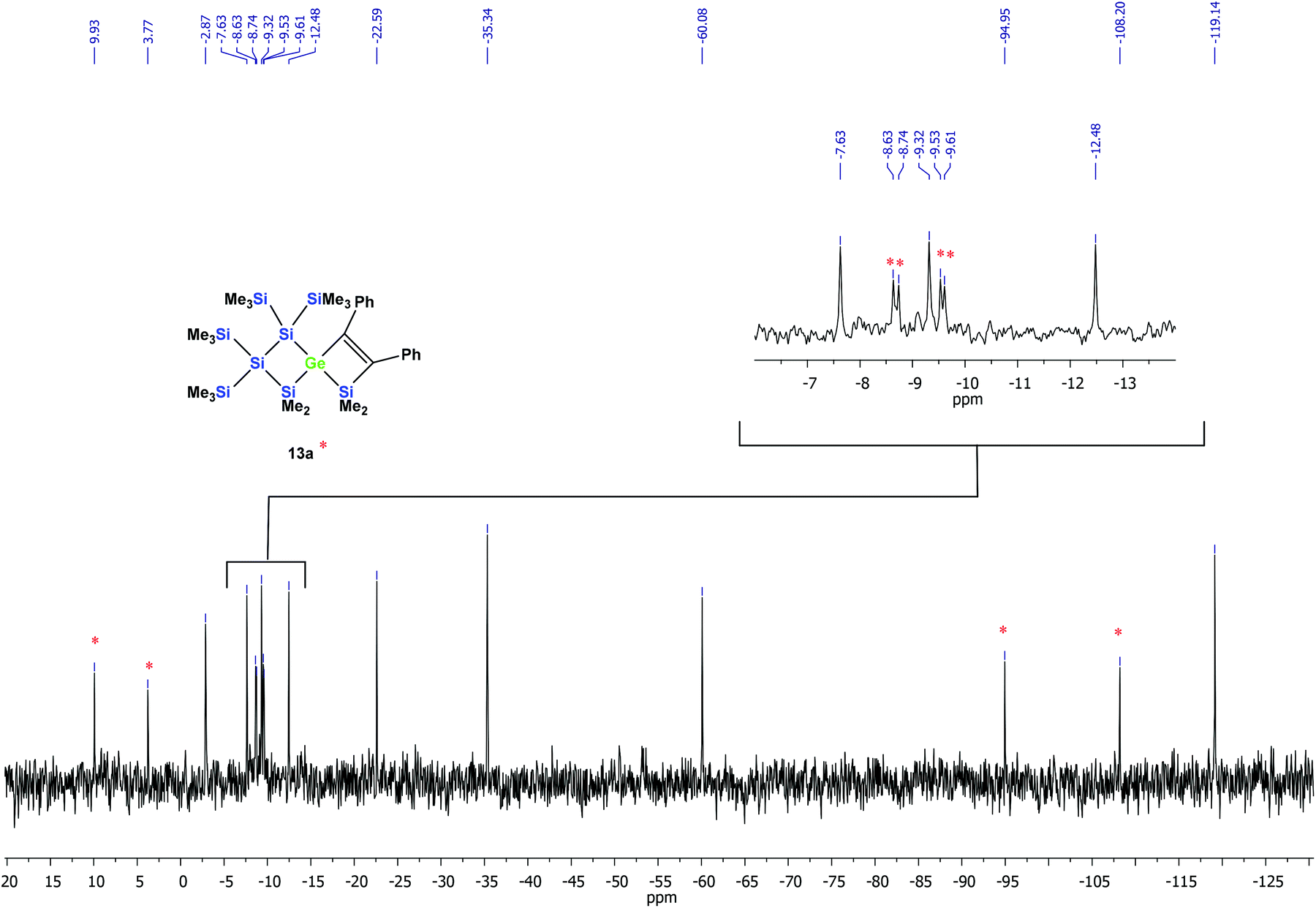 | ||
| Fig. 2 29Si{1H} INEPT NMR spectrum of the thermally promoted reaction of compound 10a. The peaks assigned to compound 13a are marked with an asterisk. | ||
However, the product was not the expected 12a following the pathway as outlined above (Scheme 6). Despite the fact that the crystal structure solution showed indeed the formation of a [3.3]-spirocyclic vinylgermane (13a), it is not the expected but an isomeric one. Compound 13a features both bis(trimethylsilyl)silylene and one dimethylsilylene units in one ring and another dimethylsilylene and 1,2-diphenylethylene in the other one (Scheme 7). However, analysis of the 29Si{1H} NMR spectrum of the mixture suggests that in addition to 13a another product is present in which the SiMe2 units are part of a five-membered ring. Dimethylsilylene units are quite sensitive to the ring-size and their 29Si chemical shift is typically around −10 ppm when incorporated into a four-membered ring and can be found further up-field in larger rings.
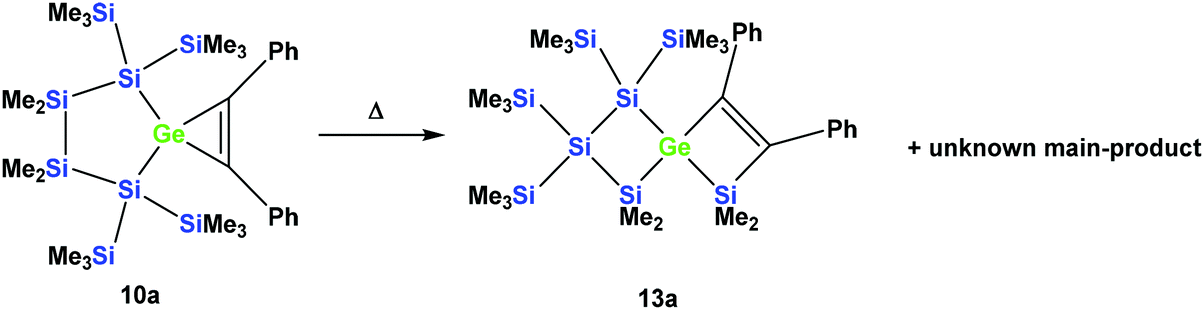 | ||
| Scheme 7 Thermal rearrangement of the [4.2]-spirocyclic germacyclopropene 10a to the [3.3]-spirocyclic vinylgermane 13a and an unknown product. | ||
Also subjecting digermylgermacyclopropene 10b, easily available by the reaction of 5b with tolane, to a thermally-induced rearrangement gave only a single product (13b), which is isosteric to 13a and features the two additional germanium atoms bearing two trimethylsilyl groups each (Scheme 8). Scheme 9 (top part) shows a mechanistic proposal for the formation of 13a and 13b. We assume that indeed the reactions first follow the steps outlined above: (1) insertion of the 1,2-diphenylacetylene unit into the Ge–E bond to give 11a and 11b and (2) germylene insertion into the transannular Si–E bond to give 12a for the silicon case and 12b for E = Ge (Scheme 9). At the stage of 12a and 12b we assume that a cycloreversion reaction leads to the formation of the isomeric seven-membered cyclic vinylgermylenes 14a and 14b (Scheme 9). Also, if now a reversal of the germacyclopropene-vinylgermylene rearrangement occurs, the resulting germacyclopropenes (15a and 15b) have the possibility to shift the acetylene unit to the Ge–SiMe2 bond, this way forming germylenes 16a and 16b (Scheme 9). If the latter molecules (16a/16b) now insert into the transannular Me2Si–SiMe2 bonds the observed spirocyclic germanes 13a and 13b are formed (Scheme 9, top part). If the insertion of the germylene (11a or 14a) occurs instead of the endocyclic Si–Si bond into an exocyclic Si–SiMe3 bond either 17a or 18a would be formed (Scheme 9, bottom part). As both compounds feature five-membered rings consistent with the 29Si NMR resonances in Fig. 2 they are likely candidates for the unknown compound as shown in Scheme 7. This assignment is further supported by 29Si NMR chemical shift calculations. For a test set that consists of the isolated and well-characterised compounds 6a, 7a, 10a, 13a and 13b, we found that the GIAO/M06L/6-311G(2d,p)42 method allows a reliable prediction of the 29Si NMR chemical shifts for this family of oligosilanyl compounds (see ESI, Fig. S3‡). The characteristic 29Si NMR chemical shifts that were predicted for the [3.3]-spirocyclic compound 12a deviate significantly from the experimental data of the unknown compound. The predicted 29Si NMR chemical shifts for the annulated bicycles 17a and 18a, however, agree well with the experimental data (see Fig. S3‡ in the ESI).
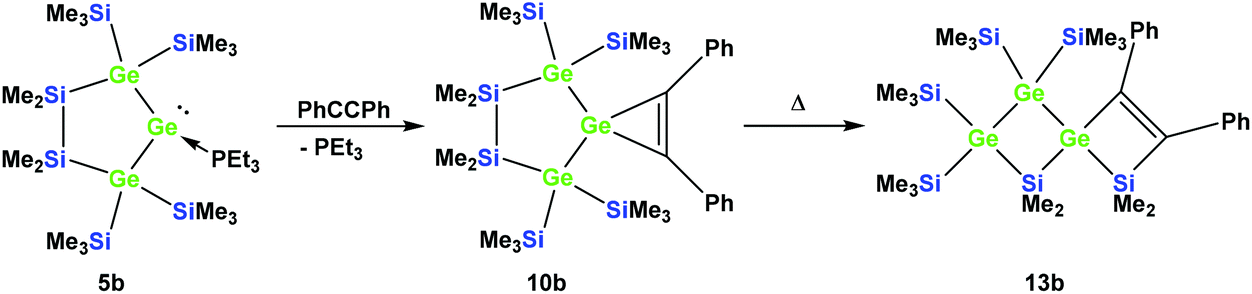 | ||
| Scheme 8 Reaction of 5b with tolane gave the expected [4.2]-spirocyclic germacyclopropene 10b, which upon heating rearranged into the [3.3]-spirocyclic vinylgermane 13b. | ||
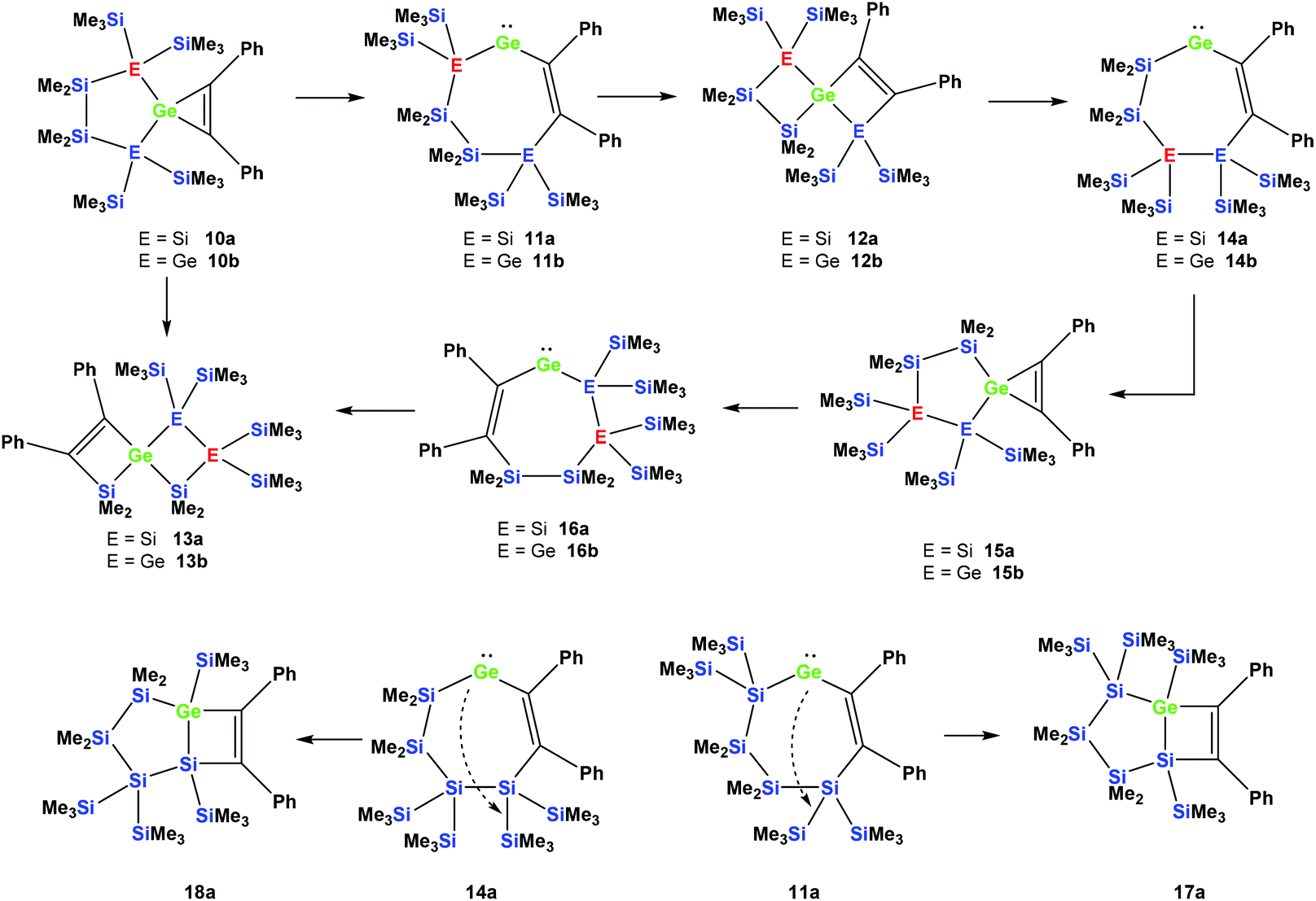 | ||
| Scheme 9 Mechanistic rationale for the formation of 13a and 13b from 10a and 10b, respectively (top part), as well as for the formation of compounds with five-membered rings such as compounds 17a and 18a (bottom part). | ||
Recently, we have reported a structure very similar to 17a, which was formed in the reaction of compound 5a with 1,4-bis(trimethylsilyl)butadiyne.31 We interpreted the formation of this product as a cycloaddition reaction of an alkyne across a cyclic silagermene.31 Given the evidence of the chemistry described in the current paper, we might be inclined to revise this initial mechanistic explanation.
Quantum mechanical calculations at the B3LYP-D342 level of theory support the mechanistic proposal for the formation of 17 or 18. For both cases, E = Si and E = Ge, the energy diagrams are very similar (see Fig. S1 and S2 in the ESI‡): All spirocycles (10a, 10b, 12a, 12b, 15a, 15b, 13a, and 13b) and the ring-annulated compounds 17a and 18a are lower in energy and well separated from the seven-membered ring germylenes (11a, 11b, 14a, 14b, 16a, and 16b). The reason for this energetic preference is clearly connected to the formation of two additional bonds in the bicyclic compounds involving tetra-coordinated germanium. It is, however, noteworthy that for both investigated potential energy surfaces, the energy difference between Ge(II) and Ge(IV) compounds is small enough to allow an easy thermal switch between both coordination modes.
The ratio and the different constitutions of the obtained products are probably determined by the subtle interplay between the kinetic and thermodynamic factors. Although a thorough investigation of all barriers involved in the rearrangements suggested in Scheme 9 is beyond the scope of the present investigation, it seems reasonable to assume that thermodynamic factors such as ring strain and bond strengths are important for the product formation. The [2.4]-spirocyclic compounds 10 and 15 that involve unsaturated three-membered rings are clearly destabilized compared to the four-membered [3.3]-spirocycles 12 and 13. In addition, for the pair 10a/13a an estimate based on the calculated strengths of the Si–Si, Si–Ge, Si–C and Ge–C bonds43 favors compounds 13 by 11 kJ mol−1.
Interestingly, when dispersion interactions are neglected by using only the B3LYP functional, the seven-membered rings (11a, 11b, 14a, 14b, 16a, and 16b) lose notable relative stability (ΔΔG = 20–50 kJ mol−1), whereas the relative energies of the spirocyclic compounds (10a, 10b, 12a, 12b, 15a, 15b, 13a, and 13b) only show little changes of ΔΔG = 1–8 kJ mol−1 (see Fig. S1 and S2 in the ESI‡). This fact indicates the importance of stabilizing dispersion contributions during this rearrangement, without which the barriers would be too high to be overcome under the experimental conditions.
If the formation of spirocyclic compounds 13a and 13b really follows the mechanism outlined in Scheme 9, three different vinylgermylenes are involved. All attempts to capture one of these germylenes as a stable adduct by the addition of several different NHCs failed. The presence of NHCs did not change the outcome of the reaction at all.
X-Ray crystallography
Compounds 6a (Fig. 3), 6b (Fig. 4), 7c (Fig. 5), 13a (Fig. 6), and 13b (Fig. 7) all obtained by the conversion of the cyclic germylenes 5a or 5b were characterized by single crystal X-ray diffraction analysis. The structure of 10a was determined previously.31 Although we obtained a fair number of structures of different germanium spirocyclic compounds (6a, 6b, 7c, 10a, 13a, and 13b), they show no big differences regarding their structural features and do not appear to be very strained. The longest Ge–Si distances were found as expected in the four-membered rings with 2.4276(15) Å in 6a and 2.4217(12) Å in 13a not differing much from the Ge–Ge distances of 2.4644(11) and 2.4758(11) Å in 13b.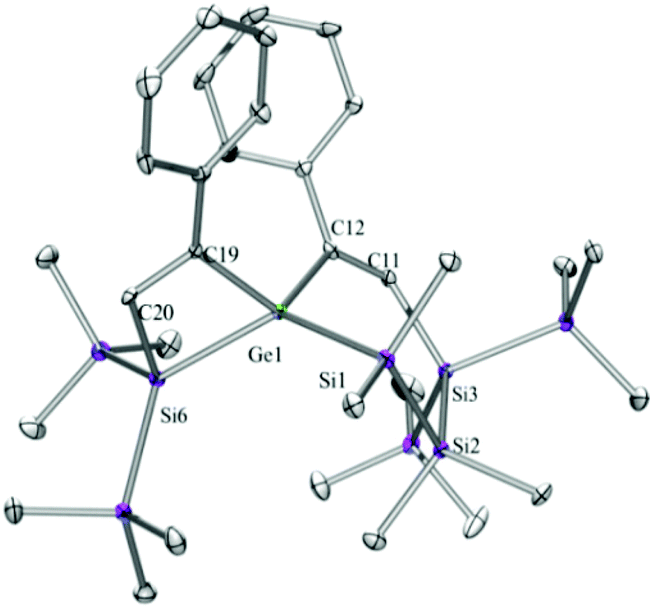 | ||
| Fig. 3 Molecular structure of 6a (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å and angles in degrees). Ge(1)–C(12) 1.987(5), Ge(1)–C(19) 2.001(5), Ge(1)–Si(1) 2.3899(15), Ge(1)–Si(6) 2.4276(15), Si(1)–C(1) 1.882(5), Si(1)–Si(2) 2.3271(19), Si(3)–C(11) 1.889(5), Si(3)–Si(4) 2.3441(19), C(11)–C(12) 1.342(7), C(19)–C(20) 1.346(7), C(12)–Ge(1)–C(19) 116.7(2), C(12)–Ge(1)–Si(1) 103.51(14), C(19)–Ge(1)–Si(1) 111.31(14), C(12)–Ge(1)–Si(6) 120.67(14), C(19)–Ge(1)–Si(6) 72.70(14), Si(1)–Ge(1)–Si(6) 128.48(5), C(12)–C(11)–Si(3) 134.7(4), C(11)–C(12)–Ge(1) 123.5(4), C(20)–C(19)–Ge(1) 103.6(3), C(19)–C(20)–Si(6) 108.6(4). | ||
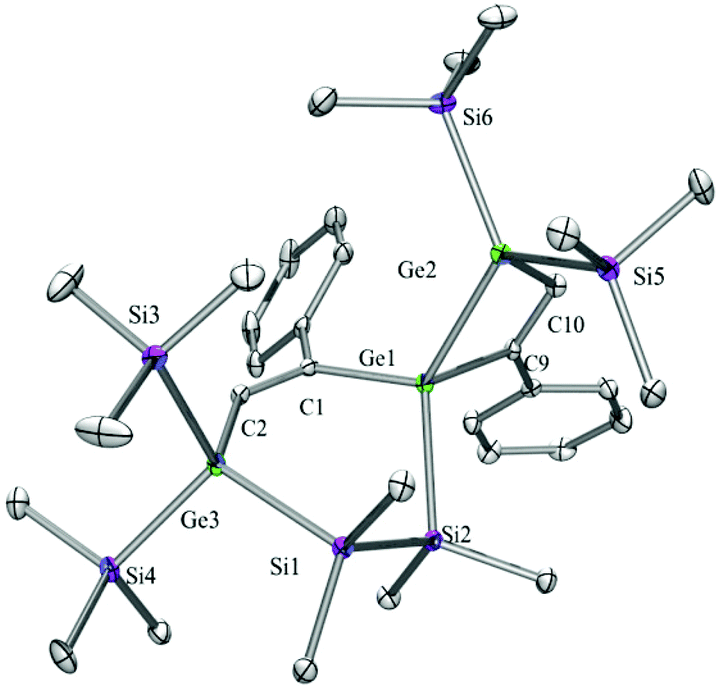 | ||
| Fig. 4 Molecular structure of 6b (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å and angles in degrees). Ge(1)–C(1) 1.989(3), Ge(1)–C(9) 1.995(3), Ge(1)–Si(2) 2.3937(9), Ge(1)–Ge(2) 2.4741(6), Ge(2)–C(10) 1.976(3), Ge(2)–Si(6) 2.4025(9), Ge(3)–C(2) 1.973(3), Ge(3)–Si(3) 2.3809(9), Si(2)–C(20) 1.869(3), Si(2)–Si(1) 2.3275(12), C(1)–Ge(1)–C(9) 116.33(11), C(1)–Ge(1)–Si(2) 104.54(8), C(9)–Ge(1)–Si(2) 111.73(8), C(1)–Ge(1)–Ge(2) 119.71(7), C(9)–Ge(1)–Ge(2) 74.07(8), Si(2)–Ge(1)–Ge(2) 127.29(3), C(10)–Ge(2)–Si(5) 109.22(8), C(10)–Ge(2)–Si(6) 109.10(8). | ||
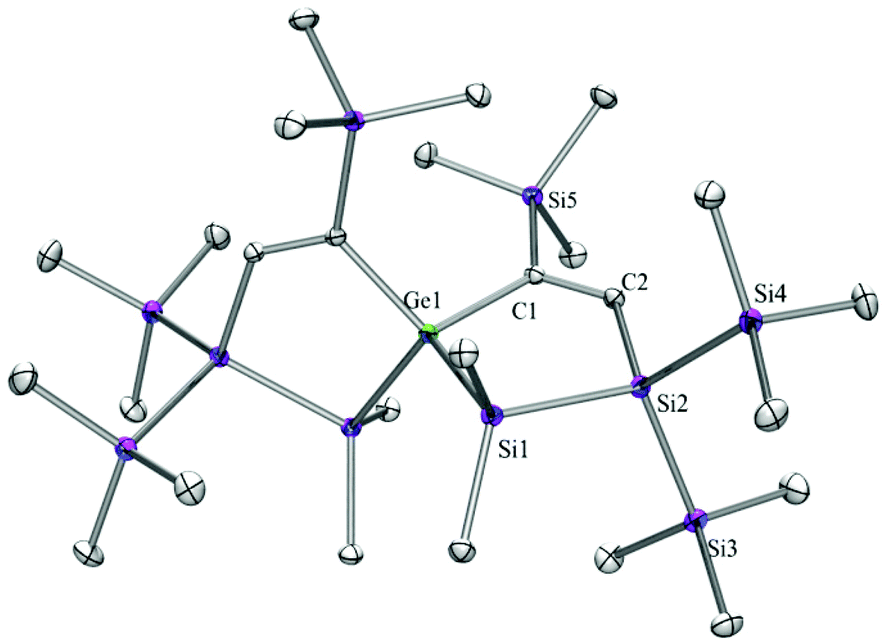 | ||
| Fig. 5 Molecular structure of 7c (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å and angles in degrees). Ge(1)–C(1) 1.9870(11), Ge(1)–Si(1) 2.4017(5), Si(1)–C(3) 1.8805(13), Si(1)–Si(2) 2.3387(6), Si(2)–C(2) 1.8953(12), C(1)–C(2) 1.3518(17), C(1)–Ge(1)–Si(1) 99.81(4), C(1)–C(2)–Si(2) 126.64(9). | ||
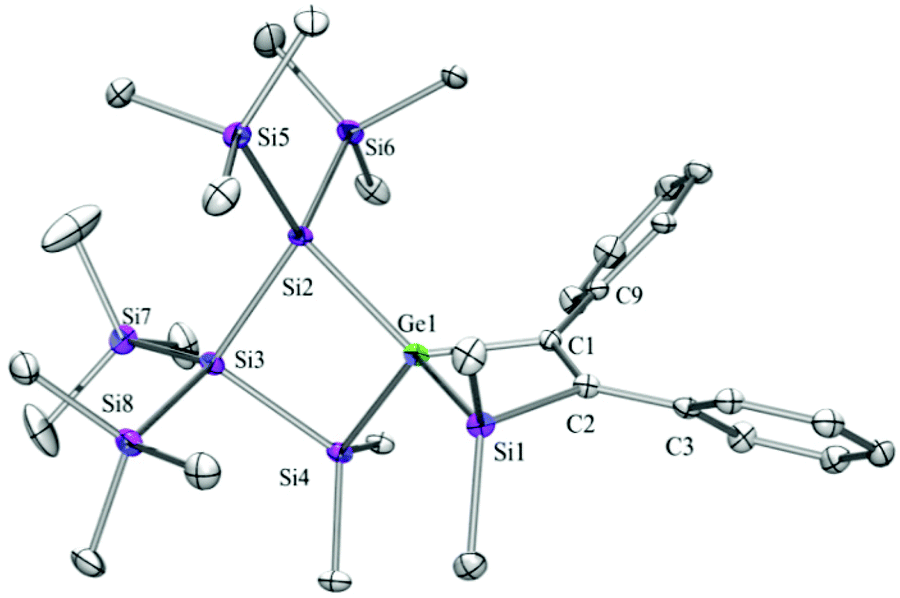 | ||
| Fig. 6 Molecular structure of 13a (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å and angles in degrees). Ge(1)–C(1) 2.002(4), Ge(1)–Si(1) 2.3758(12), Ge(1)–Si(4) 2.3984(12), Ge(1)–Si(2) 2.4217(12), Si(1)–C(2) 1.892(4), Si(2)–Si(5) 2.3605(16), Si(2)–Si(3) 2.3818(15), Si(3)–Si(4) 2.3557(16), C(1)–C(2) 1.361(5), C(1)–C(9) 1.476(5), C(2)–C(3) 1.475(5), C(1)–Ge(1)–Si(1) 72.15(11), C(1)–Ge(1)–Si(4) 121.30(10), Si(1)–Ge(1)–Si(4) 119.68(4), C(1)–Ge(1)–Si(2) 129.02(11), Si(1)–Ge(1)–Si(2) 131.33(4), Si(4)–Ge(1)–Si(2) 88.17(4), C(2)–Si(1)–Ge(1) 77.59(11), Si(3)–Si(2)–Ge(1) 86.42(4), Si(4)–Si(3)–Si(2) 90.12(5), C(2)–C(1)–Ge(1) 105.2(3), C(1)–C(2)–Si(1) 104.7(3). | ||
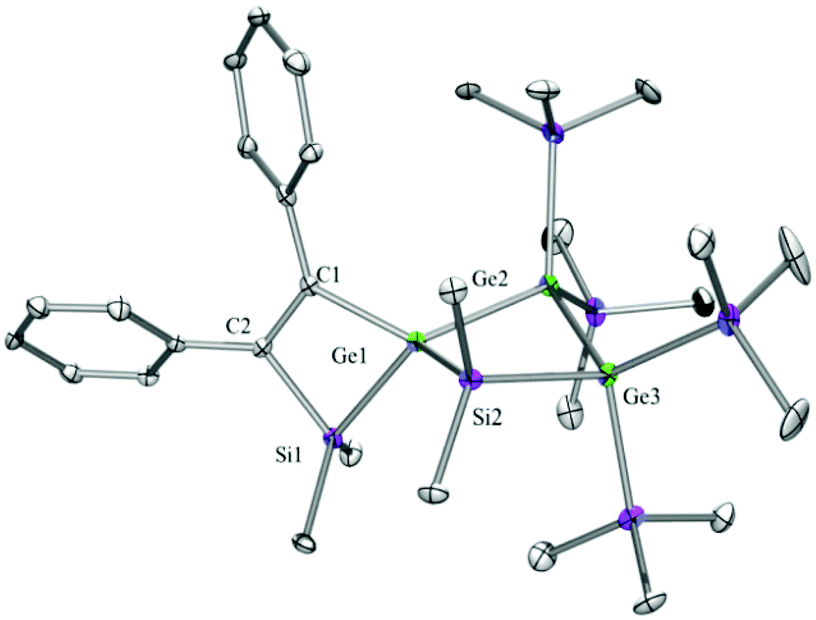 | ||
| Fig. 7 Molecular structure of 13b (thermal ellipsoid plot drawn at the 30% probability level). All hydrogen atoms are omitted for clarity (bond lengths in Å and angles in degrees). Ge(1)–C(1) 1.988(7), Ge(1)–Si(1) 2.376(2), Ge(1)–Ge(2) 2.4644(11), Ge(2)–Ge(3) 2.4758(11), Si(1)–C(2) 1.895(7), Si(2)–C(17) 1.873(7), C(1)–C(2) 1.380(10), C(1)–C(9) 1.471(10), C(1)–Ge(1)–Si(1) 72.7(2), C(1)–Ge(1)–Ge(2) 128.1(2), Si(1)–Ge(1)–Ge(2) 130.56(6), Ge(1)–Ge(2)–Ge(3) 85.47(4), C(2)–Si(1)–Ge(1) 77.5(2), C(2)–C(1)–Ge(1) 105.0(5), C(1)–C(2)–Si(1) 104.4(5). | ||
The Ge–C and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths are within a small range for all these compounds: Ge–C between 2.002(4) Å in 13a and 1.958(2) Å in 10a and the CC between 1.380(10) Å in 13b and 1.340(4) Å in 6b. The two ring planes with the germanium atom at their intersection engage an angle of about 84° in spiro[4.2] 10a (84.9°) and in spiro[5.3] 6a (84.1°) and 6b (84.7°) and about 88° in spiro[3.3] 13a (88.1°) and 13b (88.2°) and in spiro[4.4] 7c (88.1°). The four-membered ring systems containing a double bond (6a, 6b, and 13a) are largely planar, whereas the second four-membered ring in 13a is folded by some 30°. The five-membered rings in 7c and 10a engage the expected envelope and the six-membered rings in 6a and 6b a twisted boat conformation.
C bond lengths are within a small range for all these compounds: Ge–C between 2.002(4) Å in 13a and 1.958(2) Å in 10a and the CC between 1.380(10) Å in 13b and 1.340(4) Å in 6b. The two ring planes with the germanium atom at their intersection engage an angle of about 84° in spiro[4.2] 10a (84.9°) and in spiro[5.3] 6a (84.1°) and 6b (84.7°) and about 88° in spiro[3.3] 13a (88.1°) and 13b (88.2°) and in spiro[4.4] 7c (88.1°). The four-membered ring systems containing a double bond (6a, 6b, and 13a) are largely planar, whereas the second four-membered ring in 13a is folded by some 30°. The five-membered rings in 7c and 10a engage the expected envelope and the six-membered rings in 6a and 6b a twisted boat conformation.
Conclusions
The reactions of the two cyclic germylene phosphane adducts 5a and 5b with phenylacetylene and trimethylsilylacetylene at ambient temperatures led to the formation of spirocyclic compounds 6a–7c. In contrast to the acyclic case described earlier,33,34 no intermediate formation of the phosphane adducts of cyclic mono- or divinylgermylenes was observed. Instead, germylene insertion into the transannular bonds caused the formation of spirocyclic germanes. The result of DFT calculations indicated that the formation of the different spirocyclic compounds is controlled by the reaction kinetics of the intramolecular insertion of the germylene into the remote Si–Si bonds.There is no clear energetic differentiation between the insertion into the two different endocyclic Si–Si bonds in germylene 9a (Fig. 1), leading to the formation of the [4.4]-spirocyclic germasilane 7a along with the [3.5]-spirocycle 6a. For germylene 9b, the insertion reaction into the weaker Ge–Si bond is clearly favored over insertion into the central Si–Si bond, and thus leads to the exclusive formation of the less stable [3.5]-spirocyclic compound 6b. In contrast, α-trimethylsilyl substitution as in the intermediate germylene 9c favors insertion into the central Si–Si bond leading to the more stable [4.4]-spirogermasilane 7c.
Thermal treatment of germacyclopropenes 10a and 10b, which were obtained by the reaction of phosphane adducts 5a and 5b, produced spirocyclogermanes (13a and 13b) and annelated bicyclic germanes (17a or 18a). Therefore, the thermolysis of germacyclopropenes provides straightforward access to several classes of new bicyclic silagerma-heterocycles. It is, however, noteworthy that these reactions that involve a cascade of ring-opening and ring-closure reactions via germylene (Ge(II)) and germane (Ge(IV)) intermediates proceed in solution at temperatures below 150 °C. The results of the DFT calculations confirm that the bicyclic Ge(IV) products are more stable than the suggested cyclic germylene intermediates, but the calculated energy differences between both families of compounds are relatively small (less than 120 kJ mol−1). These thermal rearrangements provide examples that the introduction of destabilizing factors to Ge(IV) compounds such as ring strain allows reaching an energetic regime that favors reversible reductive elimination to Ge(II) compounds. In consequence, this would lead to model systems which allow a reversible Ge(II)/Ge(IV) interconversion based on vinylgermylenes and spiro-germacyclopropenes and -germacyclobutenes. To this point, we would like to emphasize that Sasamori and co-workers recently communicated a catalytic cyclotrimerization reaction of alkynes based on a vinylgermylene catalyst.39
Experimental section
General remarks
All reactions involving air-sensitive compounds were carried out under an atmosphere of dry nitrogen or argon using either Schlenk techniques or a glove box. All solvents were dried using a column-based solvent purification system.44 The chemicals were obtained from different suppliers and used without further purification. Compounds 5a28 and 5b28 were prepared following the reported procedures.
The 1H (300 MHz), 13C (75.4 MHz), and 29Si (59.3 MHz) NMR spectra were recorded on a Varian INOVA 300 spectrometer in C6D6 and are referenced to tetramethylsilane (TMS) for 1H, 13C and 29Si. To compensate for the low isotopic abundance of 29Si the INEPT pulse sequence45,46 was used for the amplification of the signal. Elemental analysis was carried out using a Heraeus Vario Elementar instrument.
X-Ray structure determination
For X-ray structure analyses the crystals were mounted onto the tip of glass fibers, and data collection was performed with a BRUKER-AXS SMART APEX CCD diffractometer using graphite-monochromated Mo Kα radiation (0.71073 Å). The data were reduced to F2o and corrected for absorption effects with SAINT47 and SADABS,48,49 respectively. The structures were solved by direct methods and refined by the full-matrix least-squares method (SHELXL97).50 If not noted otherwise all non-hydrogen atoms were refined with anisotropic displacement parameters and all hydrogen atoms were located in calculated positions to correspond to the standard bond lengths and angles. All diagrams were drawn with 30% probability thermal ellipsoids and all hydrogen atoms were omitted for clarity.Crystallographic data (excluding structural factors) for the structures of compounds 6a, 6b, 7c, 10a, 13a, and 13b reported in this paper have been deposited with the Cambridge Crystallographic Data Center as supplementary publication nos. CCDC 1450265 (6a), 1450272 (6b), 1450270 (7c), 1450269 (10a), 1450273 (13a), and 1450271 (13b).‡ Figures of the solid-state molecular structures were generated using Ortep-3 as implemented in WINGX51 and rendered using POV-Ray 3.6.52
5,5,6,6-Tetramethyl-3,9-diphenyl-1,1,7,7-tetrakis(trimethylsilyl)-1,5,6,7-tetrasila-4-germaspiro[3.5]nona-2,8-diene (6a) and 1,1,6,6-tetramethyl-4,9-diphenyl-2,2,7,7-tetrakis(trimethylsilyl)-1,2,6,7-tetrasila-5-germaspiro[4.4]nona-3,8-diene (7a)
A solution (2 mL THF) of 5a (130 mg, 0.20 mmol) and phenylacetylene (41 mg, 0.40 mmol) was stirred for 18 h at r.t. The solvent was removed under reduced pressure and the products were extracted with pentane (3 × 5 mL). The solvent was removed and an inseparable colorless mixture of 6a and 7a (131 mg) in an approximate ratio of 1:1 was obtained. NMR: 6aδ in ppm: −9.6 (SiMe3), −10.9 (SiMe3), −11.4 (SiMe3), −11.8 (SiMe3), −27.3 (SiMe2), −35.1 (GeSiMe2), −46.1 (SiSiMe2), −46.8 (GeSiC), −86.6 (Me2SiSiC) and 7aδ in ppm: −12.1 (SiMe3), −12.5 (SiMe3), −27.3 (SiMe2) and −66.9 (CSiSiMe2). Although it was not possible to separate 6a and 7a, the quantitative inspection of the crystal batch under the microscope revealed good crystals of 6a, which were subjected to single crystal XRD analysis.
5,5,6,6-Tetramethyl-3,9-diphenyl-1,1,7,7-tetrakis(trimethylsilyl)-5,6-disila-1,4,7-trigermaspiro[3.5]nona-2,8-diene (6b)
A solution (2 mL THF) of 5b (37 mg, 0.05 mmol) and phenylacetylene (10 mg, 0.10 mmol) was stirred for 2 h at r.t. The solvent was removed under reduced pressure and the product was extracted with pentane (3 × 5 mL). The solvent was removed and crystallization from pentane at −35 °C gave colorless crystalline 6b (38 mg, 90%). Mp = 101–102 °C. NMR: δ in ppm: 1H: 7.78 (s, 1H, C–H), 7.58 (d, 2H, o-H), 7.41 (s, 1H, C–H), 7.07–7.00 (m, 4H, Ph), 6.95–6.85 (m, 2H, Ph), 0.54 (s, 3H, SiMe2), 0.52 (s, 3H, SiMe2), 0.43 (s, 9H, SiMe3), 0.41 (s, 3H, SiMe2), 0.40 (s, 9H, SiMe3), 0.38 (s, 3H, SiMe2), 0.32 (s, 9H, SiMe3), 0.31 (s, 9H, SiMe3). 13C: 169.7, 162.5, 149.4, 146.6, 142.6, 140.1, 126.5, 126.1, 125.9, 2.6 (SiMe3), 2.2 (SiMe3), 1.8 (SiMe3), 1.5 (SiMe3), −2.0 (SiMe2), −3.1 (SiMe2), −3.8 (SiMe2), −5.1 (SiMe2). 29Si: −3.3 (SiMe3), −3.7 (SiMe3), −4.7 (SiMe3), −5.0 (SiMe3), −35.5 (SiMe2), −37.3 (SiMe2). Anal. calcd for C32H60Ge3Si6 (831.25): C 46.24, H 7.28. Found: C 46.01, H 7.34.1,1,6,6-Tetramethyl-2,2,4,7,7,9-hexakis(trimethylsilyl)-1,2,6,7-tetrasila-5-germaspiro[4.4]nona-3,8-diene (7c)
A mixture of 5a (65 mg, 0.10 mmol) and trimethylsilylacetylene (20 mg, 0.20 mmol) was stirred in THF (2 mL) for 18 hours at r.t. The solvent was removed under reduced pressure and the residue was extracted with pentane (3 × 5 mL).The solvent was evaporated to give 7c as a white solid (63 mg, 86%). Crystallization from pentane at −35 °C gave colorless crystals suitable for X-ray diffraction analysis. Mp = 113–116 °C. NMR: δ in ppm: 1H: 7.80 (s, 2H, CH), 0.55 (s, 12H, SiMe2), 0.28 (s, 18H, SiMe3), 0.21 (s, 18H, SiMe3), 0.20 (s, 18H, SiMe3). 13C: = 176.6, 158.2, 1.2 (SiMe3), 1.1 (SiMe3), 0.3 (SiMe3), −0.3 (SiMe2), −1.4 (SiMe2). 29Si: −6.9 (SiMe3), −12.1 (SiMe3), −12.7 (SiMe3), −28.2 (SiMe2), −66.4 (Si(SiMe3)3). Anal. calcd for C26H68GeSi10 (734.31): C 42.53, H 9.33 Found: C 42.24, H 9.03.
5,5,6,6-Tetramethyl-1,2-diphenyl-4,4,7,7-tetrakis(trimethylsilyl)-5,6-disila-3,4,7-trigermaspiro[2.4]hept-1-ene (10b)
The same procedure as that for the preparation of 10a using 5b (74 mg, 0.10 mmol) and diphenylacetylene (18 mg, 0.10 mmol) was performed. Crystallization from pentane at −35 °C gave yellow crystalline 10b (52 mg, 64%). Mp = 168–169 °C. NMR: δ in ppm: 1H 7.82 (m, 4H): 7.19 (m, 4H,), 7.00 (m, 2H, p-H), 0.46 (s, 12 H, SiMe2), 0.28 (s, 36 H, SiMe3). 13C: 147.7, 135.4, 131.6, 128.8, 128.2, 127.1, 3.0 (SiMe3), −2.2 (SiMe2). 29Si: −1.5 (SiMe3), −23.4 (SiMe2). Anal. calcd for C30H58Ge3Si6 (805.21): C 44.98, H 6.59. Found: C 44.98, H 6.58.1,1,5,5-Tetramethyl-6,7-diphenyl-2,2,3,3-tetrakis(trimethylsilyl)-1,2,3,5-tetrasila-4-germaspiro[3.3]hept-6-ene (13a) and an unknown by-product
A solution of 10a (50 mg, 0.07 mmol) in toluene (1 mL) was heated up to 100 °C for 18 h. The solvent was removed under reduced pressure and the residue was extracted with pentane (3 × 5 mL). After the removal of the solvent, an inseparable mixture of colorless crystalline 13a (minor component, identified by single crystal XRD) and an unknown compound was obtained in a ratio of about 1:2. Based on the signal intensities and calculated 29Si NMR shifts we assign the following 29Si NMR shifts to 13a: δ in ppm: 29Si: 9.9 (SiMe2), 3.8 (SiMe2), −8.6 (SiMe3), −8.7 (SiMe3), −9.5 (SiMe3), −9.6 (SiMe3), −94.9 (GeSi(SiMe3)2), −108.2 (SiSi(SiMe3)2). The remaining signals in the 29Si spectrum: −2.9 (GeSiMe3), −7.6 (SiMe3), −9.3 (SiMe3), −12.5 (SiMe3), −22.6 (SiMe2), −35.3 (SiMe2), −60.1 (CSi(SiMe3)2), −119.1 (SiSi(SiMe3)2) suggest a compound with the SiMe2 groups being part of a five-membered ring.
Although we were not able to separate the crystals, it was nevertheless possible to use a single crystal of 13a for crystal structure analysis.
1,1,5,5-Tetramethyl-6,7-diphenyl-2,2,3,3-tetrakis(trimethylsilyl)-1,5-disila-2,3,4-trigermaspiro[3.3]hept-6-ene (13b)
A solution (C6D6 1 mL) of 10b (35 mg, 0.04 mmol) in a closed tube was heated up to 150 °C for 18 h. The solvent was removed under reduced pressure and the residue was extracted with pentane (3 × 5 mL). The extract was concentrated to 0.5 mL and stored at −35 °C. Colorless crystals (20 mg, 58%) of 13b were obtained. Mp = 168–170 °C. NMR: δ in ppm: 1H:, 7.28 (m, 2H, o-H), 7.21–7.09 (m, 4H), 7.04–6.91 (m, 4 H), 0.94 (s, 3H, SiMe2), 0.91 (s, 3H, SiMe2), 0.64 (s, 3H, SiMe2), 0.60 (s, 3H, SiMe2), 0.44 (s, 18 H, 2 × SiMe3), 0.34 (s, 9 H, SiMe3), 0.21 (s, 9H, SiMe3). 13C: 170.6, 156.9, 144.0, 141.0, three signals overlapping with C6D6, 126.6, 126.1, 126.0, 5.4 (SiMe2), 3.7 (SiMe3), 3.7 (SiMe3), 3.6 (SiMe3), 3.5 (SiMe3), 1.9 (SiMe2), 1.6 (SiMe2), 0.2 (SiMe2). 29Si: 18.4 (SiMe2), 3.5 (SiMe2), −2.5 (SiMe3), −2.9 (SiMe3), −3.2 (SiMe3), −4.5 (SiMe3). Anal. calcd for C30H58Ge3Si6 (805.21): C 44.75, H 7.26. Found: C 44.88, H 7.17.Conflicts of interest
The authors state that there are no conflicts to declare.Acknowledgements
Support for this study was provided by the Austrian Fonds zur Förderung der wissenschaftlichen Forschung (FWF) via the projects P-22678 (C. M.) and P-30955 (J. B.). The simulations were performed at the HPC Clusters HERO and CARL, located at the University of Oldenburg (Germany, European Union) and funded by the DFG through its Major Research Instrumentation Program (INST 184/108-1 FUGG/INST 184/157-1 FUGG) and the Ministry of Science and Culture (MWK) of the Lower Saxony State.The authors are indebted to a reviewer pointing out inconsistencies in the interpretation of the 29Si NMR spectrum of the thermal treatment of compound 10a.
Notes and references
- N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer, Berlin, Heidelberg, 2007 Search PubMed.
- P.-C. Chiang and J. W. Bode, in N-Heterocyclic Carbenes, ed. S. Díez-González, RSC, Cambridge, 2011, pp. 399–435 Search PubMed.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- C. Marschner, Eur. J. Inorg. Chem., 2015, 3805–3820 CrossRef CAS.
- A. V. Zabula and F. E. Hahn, Eur. J. Inorg. Chem., 2008, 5165–5179 CrossRef CAS.
- W.-P. Leung, K.-W. Kan and K.-H. Chong, Coord. Chem. Rev., 2007, 251, 2253–2265 CrossRef CAS.
- E. Rivard, Chem. Soc. Rev., 2016, 45, 989–1003 RSC.
- L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef.
- B. Blom, M. Stoelzel and M. Driess, Chem. – Eur. J., 2013, 19, 40–62 CrossRef CAS PubMed.
- J. Baumgartner and C. Marschner, Rev. Inorg. Chem., 2014, 34, 119–152 CrossRef CAS.
- B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134–148 RSC.
- R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed.
- M. Okazaki, H. Tobita and H. Ogino, Dalton Trans., 2002, 493–506 Search PubMed.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CrossRef CAS PubMed.
- Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed.
- J. Böserle, G. Zhigulin, P. Štěpnička, F. Horký, M. Erben, R. Jambor, A. Růžička, S. Ketkov and L. Dostál, Dalton Trans., 2017, 46, 12339–12353 RSC.
- K. W. Klinkhammer and W. Schwarz, Angew. Chem., Int. Ed. Engl., 1995, 34, 1334–1336 CrossRef CAS.
- K. Klinkhammer, Polyhedron, 2002, 21, 587–598 CrossRef CAS.
- N. Katir, D. Matioszek, S. Ladeira, J. Escudié and A. Castel, Angew. Chem., Int. Ed., 2011, 50, 5352–5355 CrossRef CAS PubMed.
- H. Arp, J. Baumgartner, C. Marschner and T. Müller, J. Am. Chem. Soc., 2011, 133, 5632–5635 CrossRef CAS PubMed.
- H. Arp, J. Baumgartner, C. Marschner, P. Zark and T. Müller, J. Am. Chem. Soc., 2012, 134, 10864–10875 CrossRef CAS PubMed.
- H. Arp, J. Baumgartner, C. Marschner, P. Zark and T. Müller, J. Am. Chem. Soc., 2012, 134, 6409–6415 CrossRef CAS PubMed.
- H. Arp, C. Marschner, J. Baumgartner, P. Zark and T. Müller, J. Am. Chem. Soc., 2013, 135, 7949–7959 CrossRef CAS PubMed.
- J. Hlina, H. Arp, M. Walewska, U. Flörke, K. Zangger, C. Marschner and J. Baumgartner, Organometallics, 2014, 33, 7069–7077 CrossRef CAS PubMed.
- J. Hlina, J. Baumgartner, C. Marschner, L. Albers and T. Müller, Organometallics, 2013, 32, 3404–3410 CrossRef CAS PubMed.
- J. Hlina, J. Baumgartner, C. Marschner, P. Zark and T. Müller, Organometallics, 2013, 32, 3300–3308 CrossRef CAS PubMed.
- J. Hlina, J. Baumgartner, C. Marschner, L. Albers, T. Müller and V. Jouikov, Chem. – Eur. J., 2014, 20, 9357–9366 CrossRef CAS PubMed.
- M. Walewska, J. Hlina, J. Baumgartner, T. Müller and C. Marschner, Organometallics, 2016, 35, 2728–2737 CrossRef CAS PubMed.
- M. Walewska, J. Hlina, W. Gaderbauer, H. Wagner, J. Baumgartner and C. Marschner, Z. Anorg. Allg. Chem., 2016, 642, 1304–1313 CrossRef CAS.
- M. Walewska, J. Baumgartner and C. Marschner, Chem. Commun., 2015, 51, 276–278 RSC.
- M. Walewska, J. Baumgartner, C. Marschner, L. Albers and T. Müller, Chem. – Eur. J., 2016, 22, 18512–18521 CrossRef CAS PubMed.
- D. Wendel, W. Eisenreich, C. Jandl, A. Pöthig and B. Rieger, Organometallics, 2016, 35, 1–4 CrossRef CAS.
- R. Rodriguez, D. Gau, Y. Contie, T. Kato, N. Saffon-Merceron and A. Baceiredo, Angew. Chem., Int. Ed., 2011, 50, 11492–11495 CrossRef CAS PubMed.
- A. V. Protchenko, M. P. Blake, A. D. Schwarz, C. Jones, P. Mountford and S. Aldridge, Organometallics, 2015, 34, 2126–2129 CrossRef CAS.
- C. Hering-Junghans, P. Andreiuk, M. J. Ferguson, R. McDonald and E. Rivard, Angew. Chem., Int. Ed., 2017, 56, 6272–6275 CrossRef CAS PubMed.
- T. Sugahara, J.-D. Guo, T. Sasamori, S. Nagase and N. Tokitoh, Angew. Chem., Int. Ed., 2018, 57, 3499–3503 CrossRef CAS PubMed.
- Y. S. Chen and P. P. Gaspar, Organometallics, 1982, 1, 1410–1412 CrossRef CAS.
- H. Wagner, A. Wallner, J. Fischer, M. Flock, J. Baumgartner and C. Marschner, Organometallics, 2007, 26, 6704–6717 CrossRef CAS.
- M. Frisch, et al., The Gaussian 09 program was used. Gaussian 09, Revision B.01/D.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed. For detailed description, see the ESI.‡.
- H. Wagner, J. Baumgartner, T. Müller and C. Marschner, J. Am. Chem. Soc., 2009, 131, 5022–5023 CrossRef CAS PubMed.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- G. A. Morris and R. Freeman, J. Am. Chem. Soc., 1979, 101, 760–762 CrossRef CAS.
- B. J. Helmer and R. West, Organometallics, 1982, 1, 877–879 CrossRef CAS.
- SAINTPLUS: Software Reference Manual, Version 6.45, Bruker-AXS, Madison, WI, 1997–2003 Search PubMed.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. M. Sheldrick, SADABS. Version 2.10, Bruker AXS Inc., Madison, USA, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- POVRAY 3.6, Persistence of Vision Pty. Ltd, Williamstown, Victoria, Australia, 2004. Available online: http://www.povray.org/download/(accessed on 09.07.2008) Search PubMed.
Footnotes |
| † Dedicated to Phil Power, a main-group chemist extraordinaire and a great person. |
| ‡ Electronic supplementary information (ESI) available: Tables containing the crystallographic information of compounds 6a, 6b, 7c, 10a, 13a, and 13b and the absolute and relative energies of compounds 6–9 and 10–16; figures with relative energy diagrams and the correlation of calculated 29Si NMR shifts; xyz-files of the calculated structures. CCDC 1450265, 1450269–1450273. For the ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt00315g |
| This journal is © The Royal Society of Chemistry 2018 |