
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDestruction of chemical warfare agent simulants by air and moisture stable metal NHC complexes†
Catherine
Weetman
a,
Stuart
Notman
b and
Polly L.
Arnold
 *a
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, King's Buildings, Edinburgh, EH9 3FJ, UK. E-mail: Polly.Arnold@ed.ac.uk
bDefence Science Technology Laboratory (DSTL), Porton Down, Salisbury, SP4 0JQ Wilts, UK
First published on 31st January 2018
Abstract
The cooperative effect of both NHC and metal centre has been found to destroy chemical warfare agent (CWA) simulants. Choice of both the metal and NHC is key to these transformations as simple, monodentate N-heterocyclic carbenes in combination with silver or vanadium can promote stoichiometric destruction, whilst bidentate, aryloxide-tethered NHC complexes of silver and alkali metals promote breakdown under mild heating. Iron–NHC complexes generated in situ are competent catalysts for the destruction of each of the three targetted CWA simulants.
Introduction
Since the first deployment of chemical warfare agents (CWAs) during the First World War, research into efficient decontamination routes has become necessary for immediate applications, and also for the destruction of CWA stockpiles as set out in the 1992 Chemical Weapons Convention Treaty (demiliterisation).1–5 Whilst a variety of fast and effective decontamination routes have been identified, these generally contain a mixture of flammable solvents and corrosive reagents and can be impractical for transportation or decontamination of expensive equipment. For example, DS2 is a combination of nucleophilic diethylenetriamine/ethylene glycol/monomethyl ether/sodium hydroxide mixtures and whilst it is a broadband decontaminant it is highly corrosive towards many materials.6 Alternate decontamination solutions containing basic hypochlorite (bleach)-based microemulsions are very effective for sulfur mustard decontamination7,8 however these are also deemed too corrosive and environmentally unacceptable for widespread use.9 Therefore new reagents that have a low environmental impact and can be used in different situations on different classes of warfare agents are desirable.10Sulfur mustard (HD), bis-2-chloroethyl sulfide, is a potent vesicant with a long environmental persistence that makes decontamination challenging.6,11–13 Generally destruction of HD can occur via three different pathways (Scheme 1). Currently known dehydrohalogenation procedures can be problematic due to the reversible addition of HCl14,15 and considered too slow in comparison to the other decontamination pathways16 whilst at ambient temperatures hydrolysis is limited by the low aqueous solubility of HD.17 Oxidation is currently one of the most effective pathways of decontamination; however care must be taken to form the mono-oxidation product, as the sulfone is a potent vesicant in its own right.
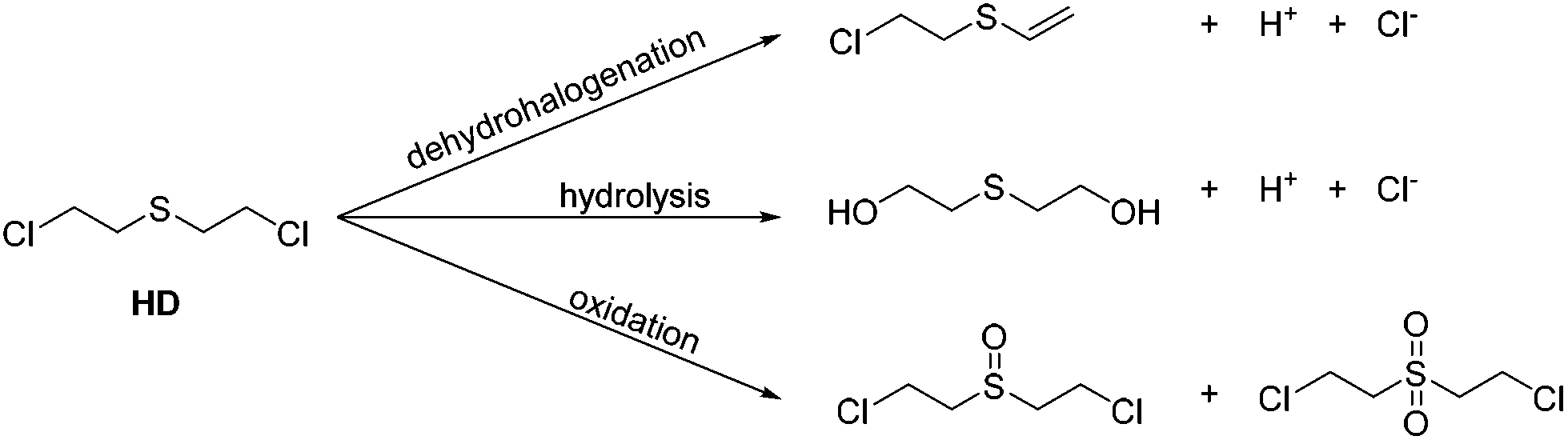 | ||
| Scheme 1 Three pathways for sulfur-mustard destruction. | ||
The other major class, nerve agents, are organophosphonate compounds that were originally developed as pesticides until their highly toxic nature was recognised. Nerve agents can be divided into two main classes: non-persistent G- (e.g. Sarin) and persistent V- (e.g. VX) series, where the persistence relates to the relative ease of degradation or propensity to remain in the environment.
Traditional routes for decontamination of VX have centred arounds its susceptibility towards nucleophilic attack, but again the control of product formation is imperative. In the simplest case addition of H2O yields several products,18,19 but the formation of EA-2192 is of concern as it is nearly as toxic as VX and is less reactive towards further nucleophilic attack (Scheme 2).10
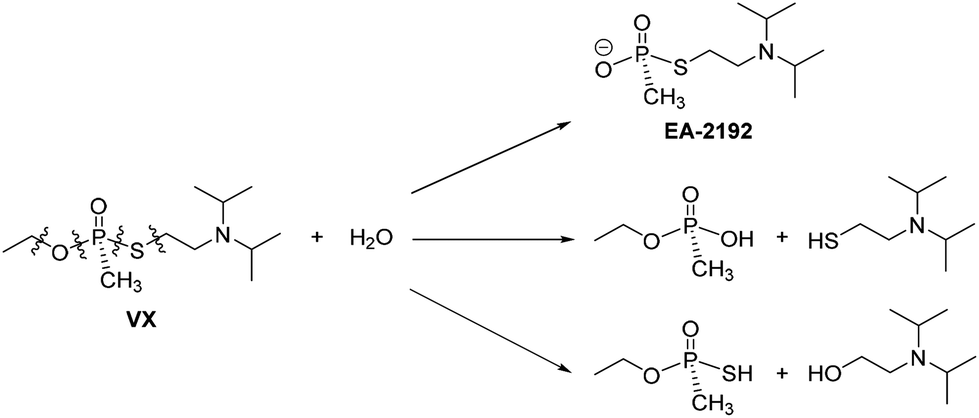 | ||
| Scheme 2 Pathways for VX destruction by hydrolysis. | ||
N-Heterocyclic carbenes (NHCs) have transformed modern organometallic chemistry.20 The strongly Lewis basic σ-donor ligands have been shown to bind to many metals leading to widespread applications, notably their use in homogeneous catalysis with late transition metals for alkene metathesis and C–C coupling reactions.21 In relation to decontamination and demiliterisation of CW agents, the range of reactivity available to NHCs and their highly tuneable nature makes them attractive candidates for these processes due to their ability to act as strong nucleophiles in organocatalysis, as well as organic bases.22 Oxidative CWA destruction pathways can also be considered in combination with right choice of metal centre. Many coinage metal NHC complexes made for medicinal applications display very high water stability,23 while homogeneous catalysts based on Pd and V show both a high stability towards oxidation24–28 and great efficacy as oxidation catalysts,29–31 in particular vanadium doped silica has been previously shown to be effective for the oxidation of CEES. The redox couple of VO2+/VO2+ (vanadyl) complexes is +1.00 V,32 and it has been demonstrated that the complex VOCl3(IMes) is air-stable.33
We have been studying the cooperative reactivity of metal–NHC complexes in which the metal–C bond is relatively labile, and can give rise to reactions of both the metal and NHC. We have previously shown a range of carbon–element bond formation and cleavage reactions.34–36
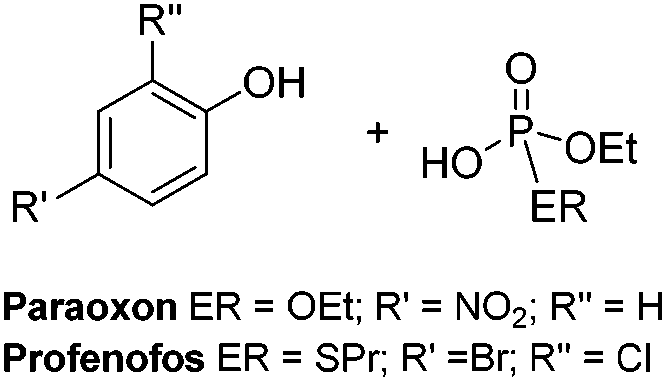
Herein we report the effectiveness of a range of air and moisture stable metal NHC complexes for the stoichiometric and catalytic destruction of both H- (mustards) and V- (nerve) type chemical warfare agent (CWA) simulants shown in Fig. 1. The use of simulants is required due to strictly controlled production, acquisition, retention or use of warfare agents under Schedule 1 of the Chemical Weapons Convention (CWC). Previous research has shown 2-chloroethyl ethyl sulphide (CEES, referred to as half mustard) is an effective simulant for HD,37 whilst paraoxon-ethyl and profenofos are used to simulate VX as these contain the desirable P–O and P–S bond cleavages, respectively. Whilst these simulants are less toxic than the actual CWAs, they are still considered to be very toxic (paraoxon-ethyl: LD50 in Rat = 0.716 mg kg−1) and must be handled with great care. Reactions are carried out on a small scale with no more than 5 μL of the chosen CWA simulant, to manage the risks associated with these compounds.
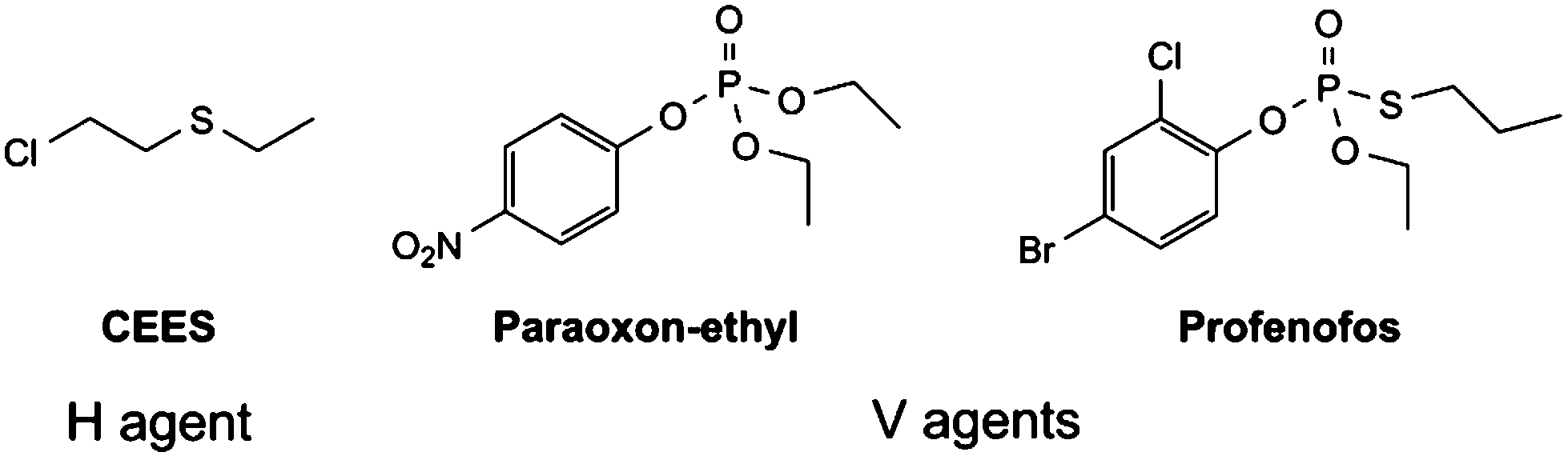 | ||
| Fig. 1 CWA simulants targeted for decontamination. | ||
Results and discussion
Candidate metal–NHC complexes
Copper and silver NHC complexes were initially targeted due to their high water-stability and ability to undergo redox reactions, thus providing a good baseline for how metal–NHC complexes may react with CWA simulants. This focused on the use of previously reported, commercially available unsaturated NHCs (denoted IR, and derived from the imidazolium chloride salts IPr·HCl, IMes·HCl and ICy·HCl) and saturated NHCs (denoted SIR, and derived from the imidazolinium salts SIPr·HCl, and SIMes·HCl) and a bidentate aryloxide-functionalised imidazolium salt (HOArIPr·HCl), Fig. 2. The silver NHC complexes AgCl(IR) (R = Dipp 1-Ag, Mes 2-Ag, Cy 3-Ag), AgCl(SIR) (R = Dipp 4-Ag, Mes 5-Ag)38 and corresponding copper NHC complexes CuCl(IR) (R = Dipp 1-Cu, Mes 2-Cu, Cy 3-Cu), CuCl(SIR) (R = Dipp 4-Cu, Mes 5-Cu)39 are readily obtained from reactions with the corresponding metal oxide. Due to the potential for oxidation destruction pathways the use of the highly oxidising but remarkably air-stable vanadyl NHC complexes VOCl3(IR) (R = Dipp 1-V, Mes 2-V, Cy 3-V), Fig. 2, (1–3)-V were isolated from the stoichiometric addition of VOCl3 to the free carbene.33,40 It is of note that whilst the saturated analogues of the vanadium NHC complexes were also isolated they were found to be unstable towards moisture, and therefore were not used in this study (see ESI†).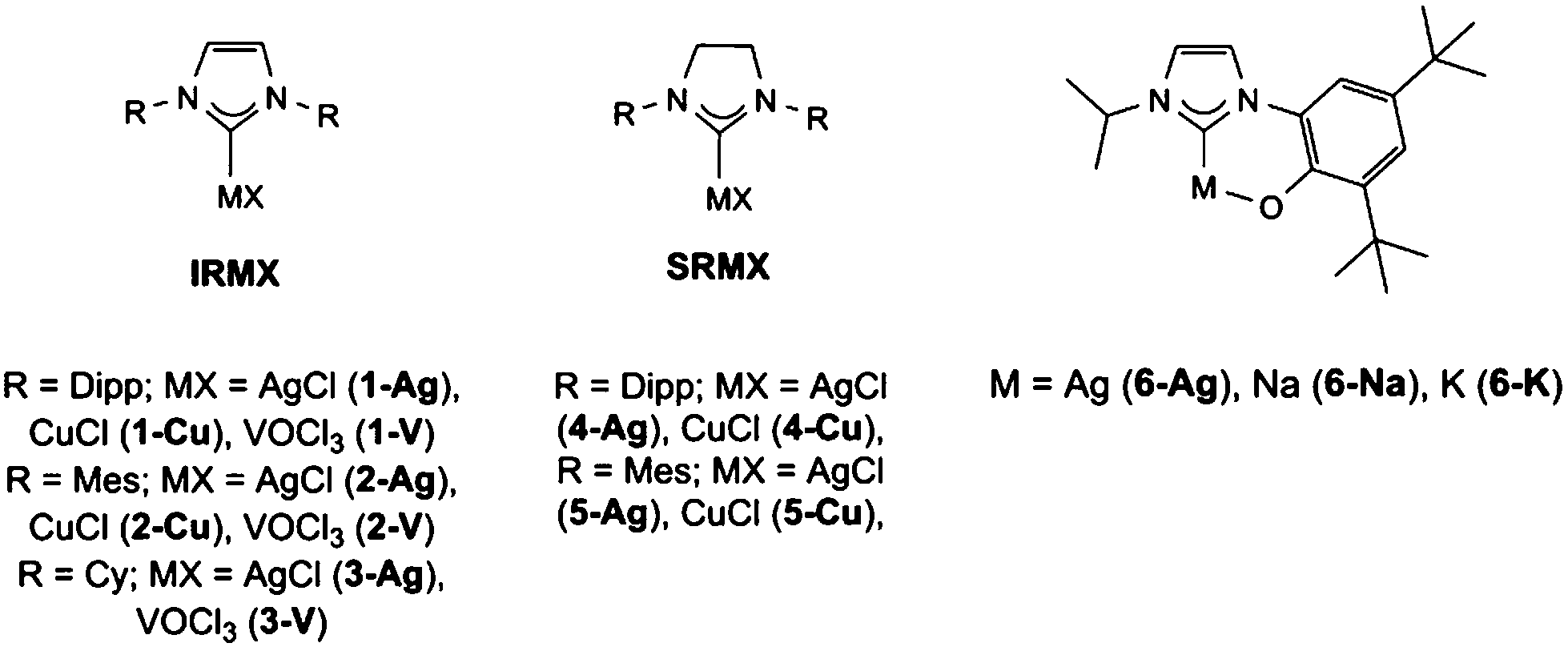 | ||
| Fig. 2 M–NHC complexes used in this study. | ||
In addition, metal complexes of the bidentate aryloxide NHC were also made; 6-Ag was made from silver(I)oxide,41 and the sodium (6-Na) and potassium (6-K) complexes were made from the corresponding MOtBu (M = Na or K) salt in situ prior to use.
Reactions of each compound shown in Fig. 2 with each of the three CWA simulants were monitored by 1H NMR spectroscopy. Reactions used stoichiometric, excess, or catalytic amounts of the complexes and were accelerated by either heat or photolysis.
Destruction by group 11 metal–NHC complexes
Stoichiometric simulant degradation reactions were followed in 0.5 mL of CDCl3. Reactions were monitored at intervals by a combination of NMR spectroscopy (1H and 31P where relevant), UV-Vis spectroscopy and GC mass spectrometry. As shown in Scheme 3, complexes (1–5)-Ag showed no reactivity at room temperature or after prolonged heating at 80 °C with any of the simulants. However, upon exposure to low-intensity UV light (254 nm, 6 W) the reactions with CEES showed the conversion of CEES into new compounds after 1 h, according to 1H NMR spectroscopic analysis. No reaction of the V agent simulants was observed even after prolonged exposure to either 254 nm or 364 nm UV radiation.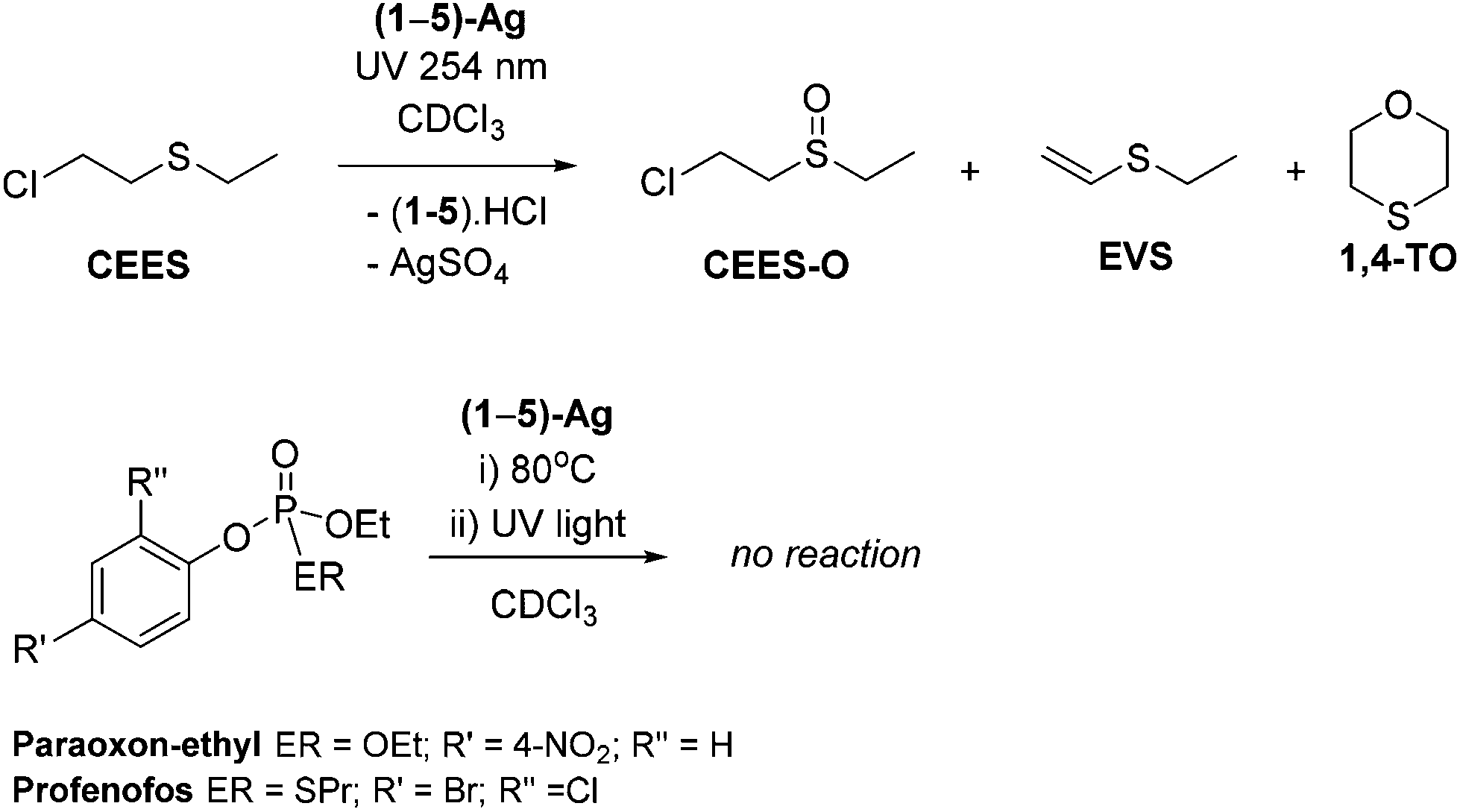 | ||
| Scheme 3 Reactions of (1–5)-Ag with CWA simulants showing the major decontamination products. | ||
Full destruction of CEES was observed after 5 h when using an excess of 1-Ag (5 eq.). The 1H NMR spectra show several new species in solution. Through a combination of HSQC, HMBC, COSY and DOSY 2D NMR spectroscopy experiments the major product was identified as the mono-oxidation product, CEES-O (Scheme 3). The two other major products are attributed to the dehydrohalogenation of CEES and include ethyl(vinyl)sulfide (EVS) and 1,4-thioxane (1,4-TO) which results from the dehydrohalogenation of partially hydrolysed CEES.10 An approximate product distribution was 80% CEES-O: 8% EVS: 10% 1,4-TO, with several minor products accounting for the remaining 2%.
The fate of complex 1-Ag was also determined by NMR spectroscopy and ICP-OES spectrometry as the imidazolium chloride salt IPr·HCl, and a silver- and sulfur- containing material which was observed to precipitate during the reaction. The scale of these reactions (demanded for safety reasons) make unambiguous analysis difficult, but it is most likely to be silver sulfate.
Kinetic analyses of the reactions of (1–5)-Ag with CEES were carried out to examine the influence of the sterics and electronics of the NHC on the reaction. Reactions between (1–5)-Ag and CEES in CDCl3 in a quartz tube were monitored by 1H NMR spectroscopy with mesitylene as an internal standard. NMR spectra were recorded for each sample after 30 minutes periods of UV (254 nm) radiation. The data fitted to a first-order dependence on CEES (Fig. 3), show that 1-Ag is the fastest of all the silver NHC complexes (Fig. S6–10†).
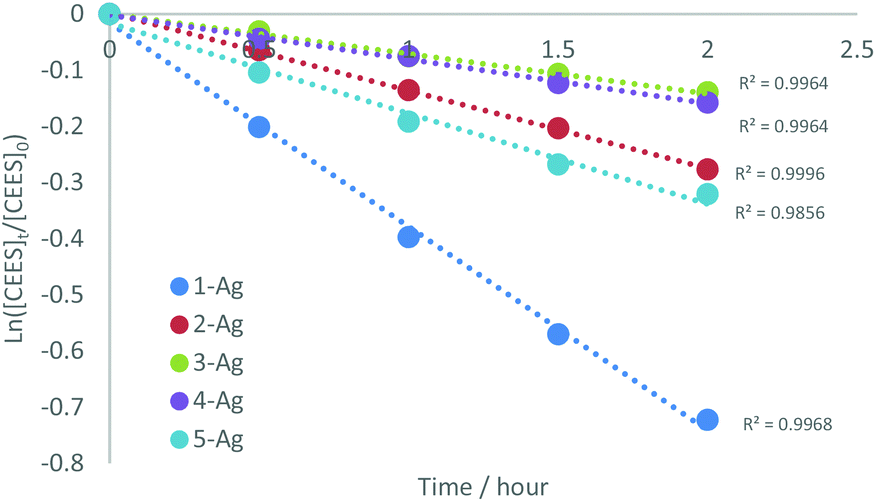 | ||
| Fig. 3 Kinetic rate data for the decomposition of CEES by (1–5)-Ag showing first order dependence on CEES and that 1-Ag is by far the fastest reagent for its destruction. | ||
It is proposed that the photolysis cleaves the silver–NHC bond, thus releasing the silver ion, a potent oxidant for CEES, and the free carbene which is active for the dehydrohalogenation of CEES, giving two routes for the destruction of the CWA simulant, Scheme 4a. Reactions of equimolar amounts of Ag2O and AgCl and CEES were also carried out as control reactions, however they were insoluble in the reaction medium and no decomposition was observed at 80 °C or under photolysis. It is suggested that the NHC plays an important role by both helping solubilise the metal cation whilst also acting as a reagent in its own right.
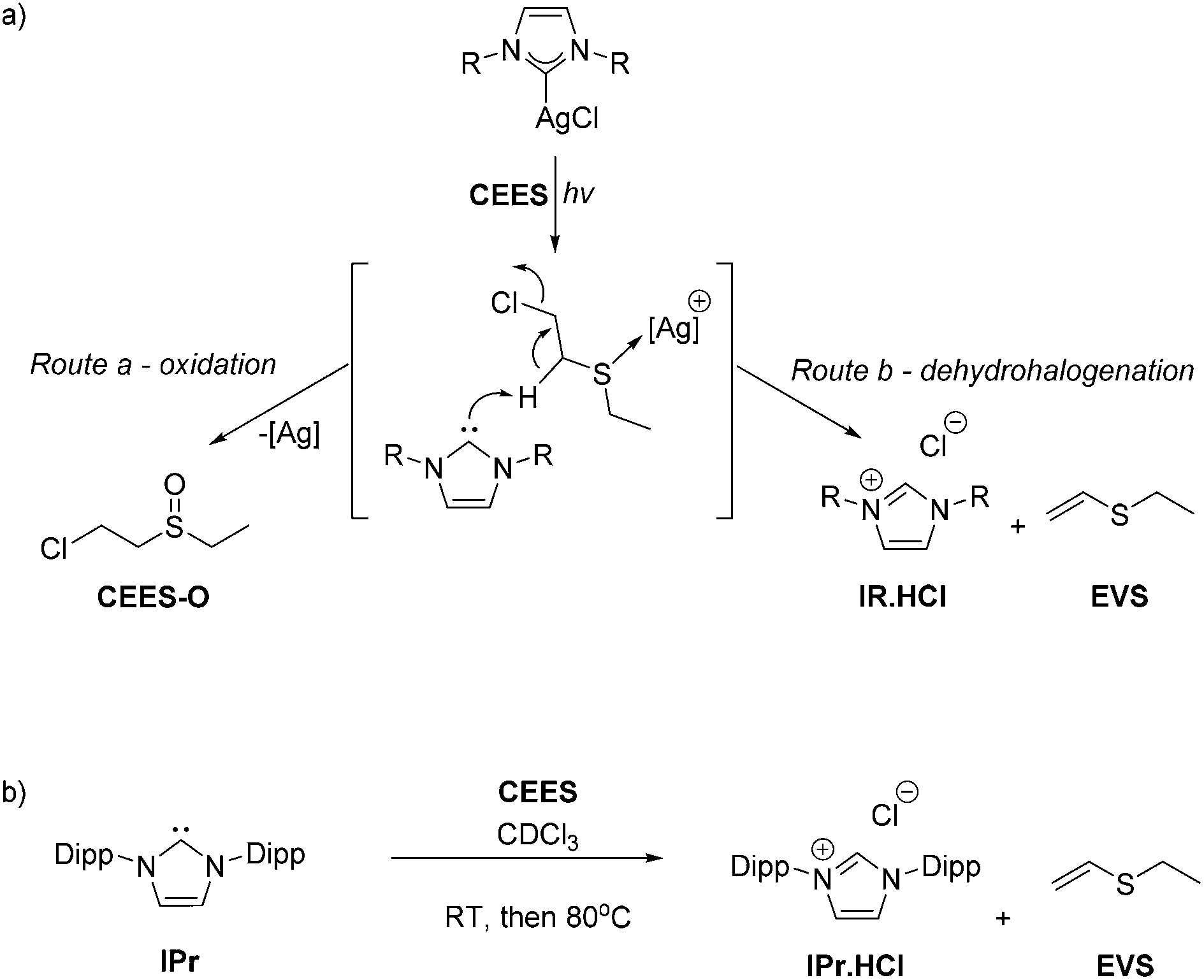 | ||
| Scheme 4 Destruction of CEES mediated by the silver cation (oxidation, route a) or by the released NHC IPr (dehydrohalogenation, route b). | ||
This hypothesis is backed up by the products formed from the reaction of the free base NHC (Scheme 4b). The equimolar reaction between IPr and CEES at room temperature shows 10% conversion to EVS, the dehydrohalogenation product, within 5 minutes. However, storage for a further hour at room temperature or at 80 °C for 24 h showed no further destruction of CEES, presumably because the remaining NHC decomposed during prolonged contact with air and moisture. A trial reaction using artificial sunlight from a commercially available SAD lamp provided ca. 40% decomposition of CEES within the same 5 h time frame.
The analysis of observed rates gives a reactivity in decreasing order for the NHC of IPr > ICy > IMes > SIPr > SIMes. Saturated NHCs (SIR) are better σ-donors than the unsaturated (IR) analogues.42 Thus although the free (SIR) carbene could be considered more reactive for the dehydrohalogenation reaction, the efficacy of complexes (1–5)-Ag appears to be dominated by the reaction that cleaves the Ag–NHC bond.
To further confirm this cooperative effect reactions between the metal and the NHC reactions were carried out using the analogous copper NHC complexes. Following the same protocol for the silver complexes, the Cu(I) analogues (1–5)-Cu were combined with 1 equivalent of CEES in the presence of UV light (254 nm). After 1 h the 1H NMR spectra confirm that the Cu–NHC bonds have been broken, showing broad, paramagnetically-shifted resonances which are attributed to the formation of Cu(II) ions arising from aerobic oxidation after release into solution. GC-MS data indicate that no degradation of CEES has occurred in any of the reactions studied. Prolonged exposure of the reactions to UV light show only background reactivity, ca. 10% decomposition of CEES. Since the Cu+1/Cu0 redox couple is significantly less oxidising than the Ag+1/Ag0 couple (+0.52 V vs. +0.80 V),32 these observations suggest that both the oxidising metal and the basic NHC are responsible for the CEES destruction. We note that the degradation products arise from both oxidation and dehydrohalogenation, suggesting that the metal cation also plays a role in facilitating dehydrohalogenation. Presumably coordination of the sulfur to the silver cation renders the CEES active towards attack by the liberated NHC.
Destruction by vanadium NHC complexes
Complexes (1–3)-V produce efficient stoichiometric destruction of CEES in 5 hours under UV light (254 nm), Scheme 5a. The reactions appear to involve reduction by the vanadium centre as the 1H NMR spectra of the reaction mixtures show contact-shifted resonances due to the paramagnetic vanadium centre, and must be separated from vanadium-containing materials for identification. Reactions monitored by GC-MS also show the conversion of CEES to new products (Scheme 5a). The major decomposition product is CEES-O (60%), with 2-chloroethylethyldisulfide (CEESS) a minor product. Two features are notable: there is no evidence of dehydrohalogenation, and hexachloroethane (PCA) is also formed during the reaction.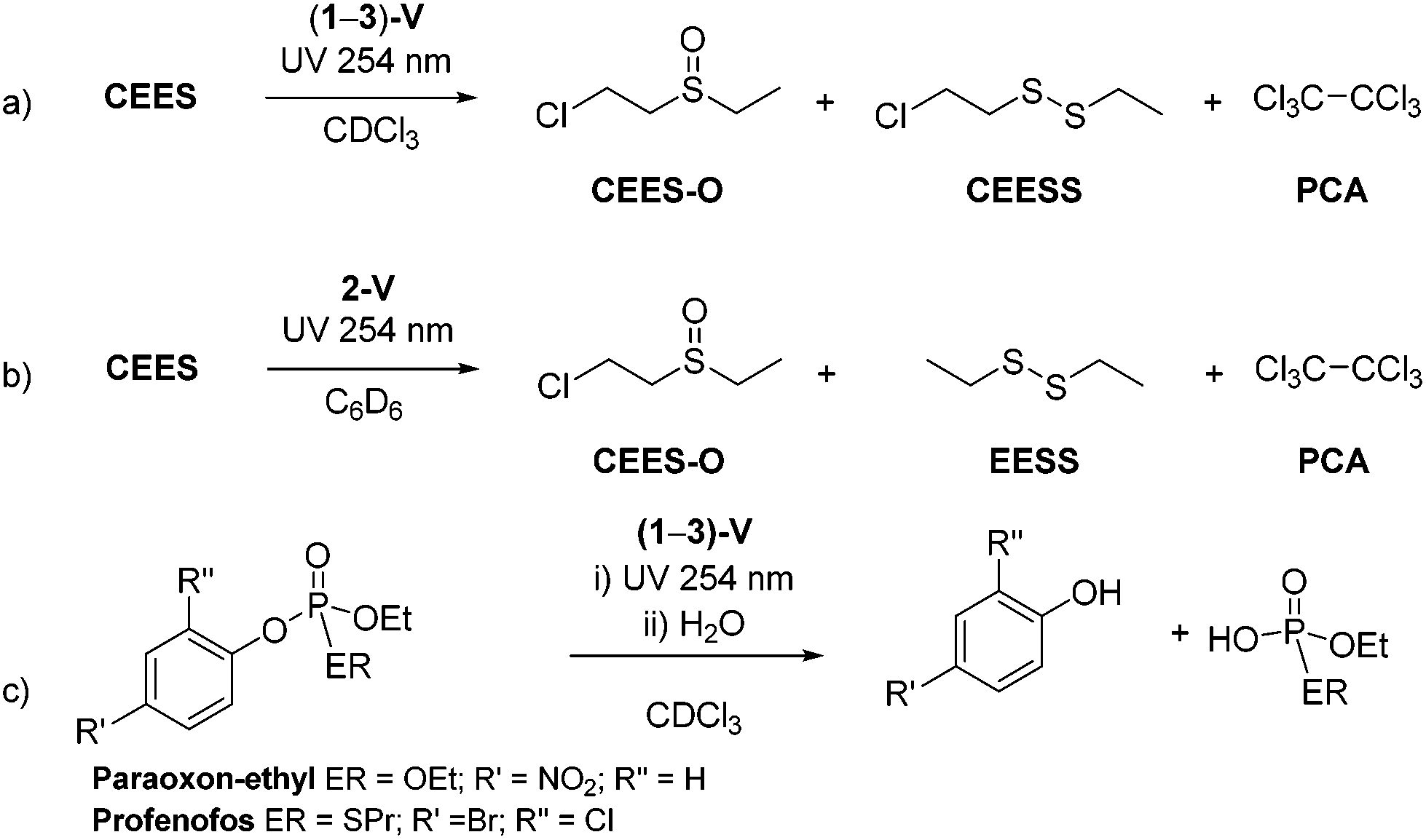 | ||
| Scheme 5 Reactions of (1–3)-V with CWA simulants showing the major decontamination products. | ||
The observation of PCA production is notable since it is usually observed to form in samples of CEES that have been stored for long periods of time (tens of years to the best of our knowledge),17 although there are only a few literature reports of the chlorination of CEES. In these cases the di, tri, and tetrachlorinated sulfides are reported to form, along with hexachloroethane which results from the reaction with excess chlorine radicals.43 Here however, repetition of the reactions between 2-V and CEES in C6D6 confirm the PCA derives from solvent decomposition, as after 3 h, the new decomposition product diethyldisulfide (EESS) is formed (Scheme 5b). VOCl3 has been recently shown to effect the oxychlorination of cyclohexane,44 but if anything, is only a minor contributor here (see ESI† for proposed mechanism).
The complexes (1–3)-V also react with the two V-agent simulants shown in Fig. 1. Scheme 5c shows the reaction of (1–3)-V with both paraoxon-ethyl and profenofos yield substituted phenol products arising from P–OAr bond cleavage and subsequent hydrolysis through trace moisture in the reaction mixture. These were identified by GC-MS trace after 3 h of exposure to UV light (254 nm) (Scheme 5c), with complete decomposition of both V agent simulants observed after 24 h.
Destruction by complexes of tethered-NHCs
To test the hypothesis that a combination of metal and NHC afford enhanced dehydrohalogenation of the H-agent, three different metal complexes of a hemilabile, bidentate aryloxy-NHC, 6-Ag, 6-Na and 6-K shown in Fig. 2, were made and tested (see ESI† for details).No reaction between stoichiometric quantities of 6-Ag and CEES was observed at room temperature by 1H NMR spectroscopy, but after 1 h at 80 °C, 25% conversion to the product of dehydrohalogenation, EVS, was observed. This was characterised by the characteristic alkene hydrogens of the product (Scheme 6a). The complete conversion of CEES to EVS can be achieved using a catalytic amount of 6-Ag if additional Ag2O is added within 5 h, thus rendering the system catalytic in NHC. The proposed mechanism for this reaction is outlined in Scheme 7, showing how the combination of the silver centre and the NHC can be effective for the destruction of CEES.
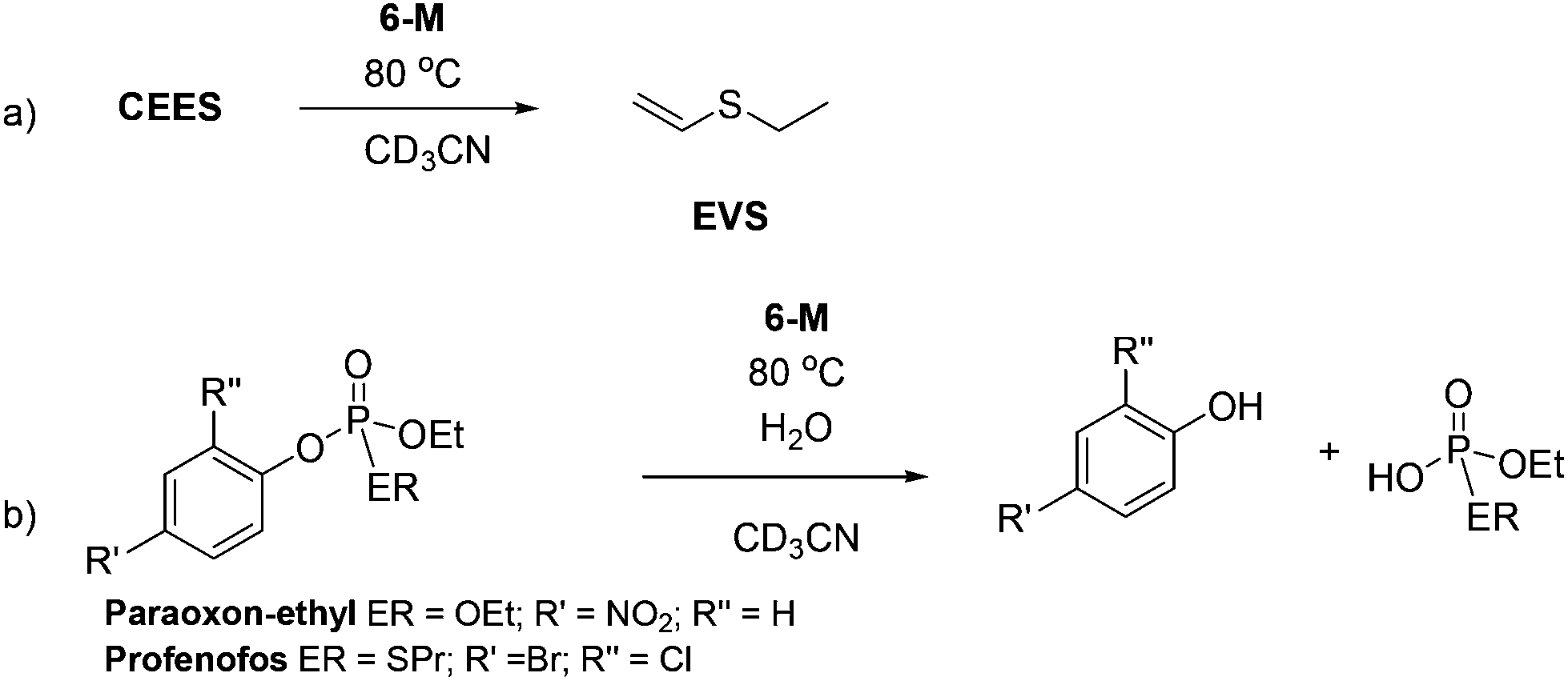 | ||
| Scheme 6 Reactions of 6-M with CWA simulants showing the major decontamination products. | ||
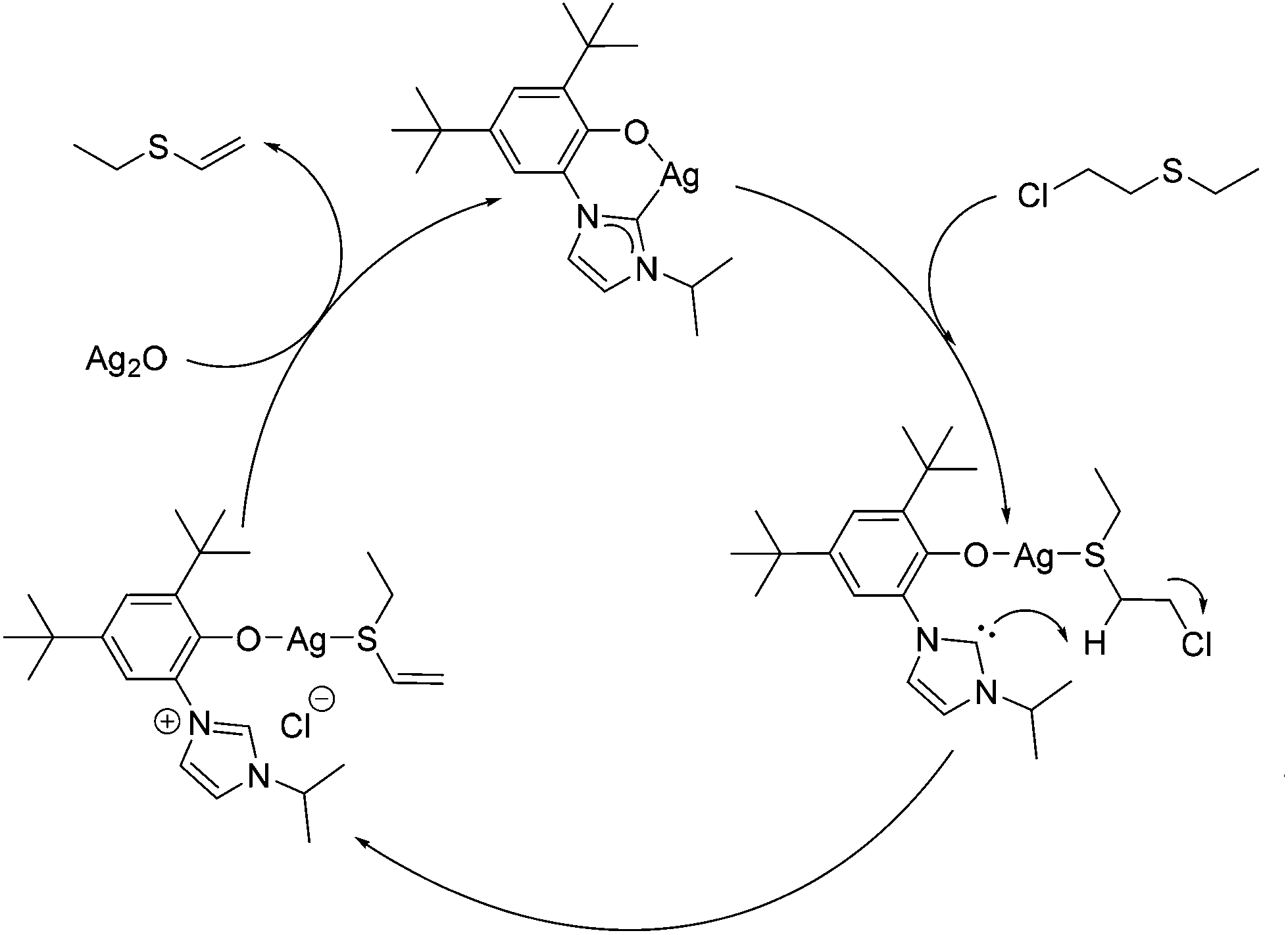 | ||
| Scheme 7 Proposed mechanism for the destruction of CEES catalysed by 6-Ag. | ||
Furthermore, similar reactivity can be obtained with the alkali metal NHC complexes 6-Na and 6-K, which give 30% and 15% destruction of CEES in 1 h, respectively (at 80 °C). Again, the selective formation of EVS was identified by 1H NMR spectroscopy. The kinetic data (Fig. S27†) suggest a small induction period (possibly due to complex formation) and first order dependence on base. It is possible to double the rate of reaction by doubling the amount of alkali metal base. Once formed, these Group 1 salts show moderate moisture instability; decomposition of 6-K occurs over one hour after exposure to air so whilst cheaper, they are less desirable as decontamination reagents than the silver complexes.
The reactivity of complexes 6-M towards both paraoxon-ethyl and profenofos nerve simulants under analogous reaction conditions also proved successful; cleavage of P–OAr bond was identified by 31P NMR spectroscopy after 20 h at 80 °C (Scheme 6b). A proposed mechanism is outlined in Scheme 8, in which the nucleophilic carbene attacks the phosphorus centre, leading to M–OAr bond formation then P–OAr bond cleavage.
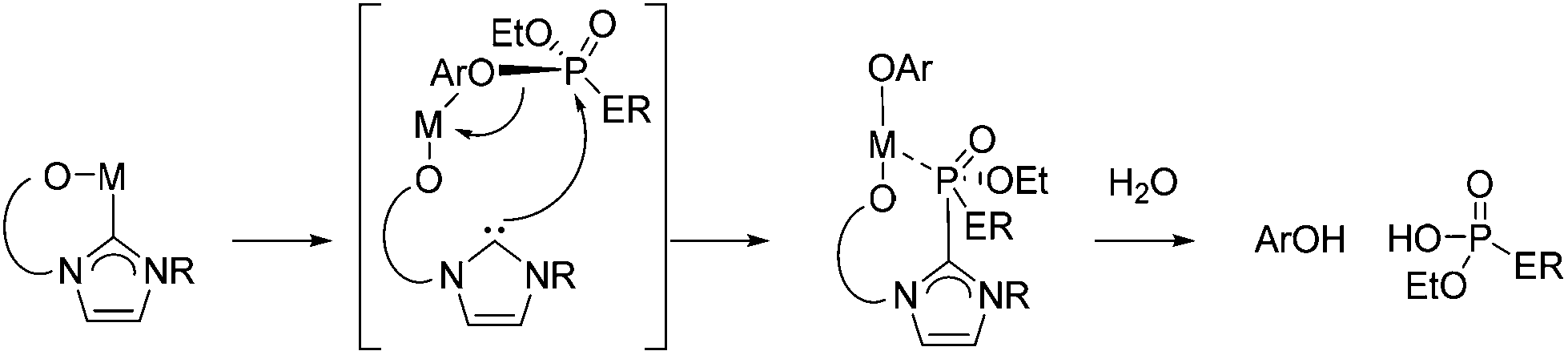 | ||
| Scheme 8 Proposed mechanism for the destruction of nerve agents with 6-M complexes. | ||
Catalytic CWA destruction
NHC complexes of earth-abundant iron and nickel have shown widespread uses in homogeneous catalysis. Their combination with the simple bases KOtBu was studied as a potential destruction method. Thus, a series of catalytic reactions in which the active catalyst is generated in situ, from the use of air stable components (NiCl2, FeBr2 and FeCl3) with imidazolium salts (IPr·HCl, IMes·HCl and ICy·HCl) was carried out. Use of equimolar amounts of MCln and IR·HCl with a fourfold excess of KOtBu leads to the formation of NHC–MXn (where X = Cl or OtBu) in situ, to which each of the chosen CWA simulants were then added 20-fold excess (i.e. 5 mol% catalyst loading w.r.t. simulant) and monitored over a period of 24 h at 80 °C using a combination of UV/vis, GC-MS and NMR spectroscopy.In all cases catalytic destruction of the chosen simulants was observed after mild heating at 80 °C for 24 hours. In terms of CEES destruction, use of the Fe(III) complex results in oxidation to CEES-O as the major product (Table 1, entries 3, 6 and 9) whilst EESS and PCA were identified as the major products for Ni(II) and Fe(II) reactions (Table 1, entries 1, 2, 4, 5, and 7), except for FeBr2 reactions in which oxybis(ethylsulfane) (BES) was identified (Table 1, entry 8; Scheme 9). In the case of the nerve simulants P–OAr cleavage was identified as the major product through GC-MS analysis.
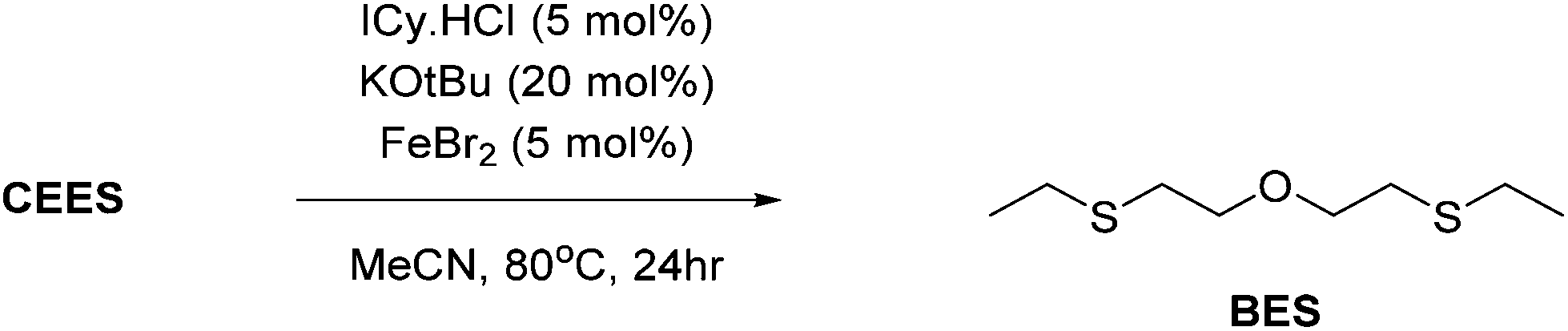 | ||
| Scheme 9 Catalytic breakdown of CEES using an in situ generated NHC–Fe(II) catalyst. | ||
| Entry | IR·HCl | MXn | CWA simulant | ||
|---|---|---|---|---|---|
| CEES | Paraoxon-ethyl | Profenofos | |||
| Reaction conditions used 1 μmol of NHC·HCl and metal salt, with 4 μmol of KOtBu and 20 μmol of CWA simulant in 1 mL of MeCN at 80 °C. | |||||
| 1 | IPr·HCl | NiCl2 | EESS |
|
|
| 2 | IPr·HCl | FeBr2 | EESS | ||
| 3 | IPr·HCl | FeCl3 | CEES-O | ||
| 4 | IMes·HCl | NiCl2 | EESS | ||
| 5 | IMes·HCl | FeBr2 | EESS | ||
| 6 | IMes·HCl | FeCl3 | CEES-O | ||
| 7 | ICy·HCl | NiCl2 | EESS | ||
| 8 | ICy·HCl | FeBr2 | BES | ||
| 9 | ICy·HCl | FeCl3 | CEES-O | ||
Attempts to render the (1–3)-V systems catalytic proved to be unsuccessful, but the reactions underline the capability of the oxidising metal in these CWA destructions.
Conclusions
Air- and moisture stable NHC complexes have been shown to be effective for the destruction of CWA simulants. The cooperative effect of both the NHC and metal centre is vital to the success of these decontamination reactions. Comparison of the simple commercially available imidazoline/imidazolinium silver NHC complexes, (1–5)-Ag, these were found to provide efficient destruction under UV conditions in 5 h, primarily to the oxidation product, CEES-O, and also to the dehydrohalogenation product. The choice of metal centre is key for these transformations as reactions with complexes (1–3)-V provide complete destruction of CEES within 5 h and allowed for destruction of nerve simulants through cleavage of P–OAr bond to form substituted phenols. Preliminary studies using artificial sunlight (from a commercially available SAD lamp) also show reasonable levels of CEES decomposition within the same 5 h timeframe, presumably a result which could be improved with suitable ligand tuning.Examination of the cooperative effect of metal centre and NHC through use of a hemi-labile bidentate aryloxide tethered NHC complex (6-M) shows clean destruction to EVS at 80 °C within a few hours. The rate of destruction is increased, and the reaction rendered catalytic in NHC via addition of further equivalents of alkali metal base or silver(I) oxide. Catalytic destruction of all three CWA simulants can also be achieved using in situ generated iron or nickel NHC complexes. This new class of CWA simulant degradants show promise, and further work is in progress to improve reactivity while retaining the air-stability of the metal–NHC complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DSTL for financial support, and the EPSRC for funding through EP/J018139/1 and the UK Catalysis Hub EP/K014714/1. This project has also received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 740311). We thank Karlotta van Rees and Peter Saghy for assistance with the synthesis of the silver and copper complexes, Euan Doidge for ICP-OES analysis, University of Edinburgh NMR spectroscopy and Mass spectrometry departments for support.Notes and references
-
J. B. Tucker, War of Nerves: Chemical Warfare from World War I to Al-Qaeda, Pantheon Books, New York, 2006 Search PubMed
.
-
K. Coleman, A History of Chemical Warfare Pelgrave, Macmillan, Basingstoke, 2005 Search PubMed
-
E. Croddy, Chemical and Biological Warfare, Springer-Verlag, New York, 2001 Search PubMed
-
J. Paxman and R. Harris, A Higher Form of Killing: The Secret History of Chemical and Biological Warfare, Arrow, London, 2001 Search PubMed
-
J. L. McWilliams and R. J. Steel, Gas! The Battle for Ypres, 1915, Vanwell Publishing Limited, Canada, 1985 Search PubMed
- Y. C. Yang, J. A. Baker and J. R. Ward, Chem. Rev., 1992, 92, 1729–1743 CrossRef CAS
- F. M. Menger and A. R. Elrington, J. Am. Chem. Soc., 1991, 113, 9621–9624 CrossRef CAS
- F. M. Menger and A. R. Elrington, J. Am. Chem. Soc., 1990, 112, 8201–8203 CrossRef CAS
- I. A. Fallis, P. C. Griffiths, T. Cosgrove, C. A. Dreiss, N. Govan, R. K. Heenan, I. Holden, R. L. Jenkins, S. J. Mitchell, S. Notman, J. A. Platts, J. Riches and T. Tatchell, J. Am. Chem. Soc., 2009, 131, 9746–9755 CrossRef CAS PubMed
- B. M. Smith, Chem. Soc. Rev., 2008, 37, 470–478 RSC
- E. Marshall, Science, 1984, 224, 130–132 CAS
- J. P. Fitch, E. Raber and D. R. Imbro, Science, 2003, 302, 1350–1354 CrossRef CAS PubMed
- L. M. Eubanks, T. J. Dickerson and K. D. Janda, Chem. Soc. Rev., 2007, 36, 458–470 RSC
- G. W. Wagner, P. W. Bartram, O. Koper and K. J. Klabunde, J. Phys. Chem. B, 1999, 103, 3225–3228 CrossRef CAS
- G. W. Wagner, O. B. Koper, E. Lucas, S. Decker and K. J. Klabunde, J. Phys. Chem. B, 2000, 104, 5118–5123 CrossRef CAS
- Y. Y. Liu, C. T. Buru, A. J. Howarth, J. J. Mahle, J. H. Buchanan, J. B. DeCoste, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 13809–13813 CAS
- N. B. Munro, S. S. Talmage, G. D. Griffin, L. C. Waters, A. P. Watson, J. F. King and V. Hauschild, Environ. Health Perspect., 1999, 107, 933–974 CrossRef CAS PubMed
- Y. J. Jang, K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2015, 115, PR1–PR76 CrossRef PubMed
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS
- D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS PubMed
- J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed
- S. P. Nolan, Acc. Chem. Res., 2011, 44, 91–100 CrossRef CAS PubMed
- S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778–787 CrossRef CAS PubMed
- I. J. B. Lin and C. S. Vasam, Coord. Chem. Rev., 2007, 251, 642–670 CrossRef CAS
- P. L. Arnold, M. S. Sanford and S. M. Pearson, J. Am. Chem. Soc., 2009, 131, 13912–13913 CrossRef CAS PubMed
- D. A. Bulushev, L. Kiwi-Minsker, V. I. Zaikovskii and A. Renken, J. Catal., 2000, 193, 145–153 CrossRef CAS
- Y. Maeda, N. Kakiuchi, S. Matsumura, T. Nishimura, T. Kawamura and S. Uemura, J. Org. Chem., 2002, 67, 6718–6724 CrossRef CAS PubMed
- A. Lattanzi, A. Senatore, A. Massa and A. Scettri, J. Org. Chem., 2003, 68, 3691–3694 CrossRef CAS PubMed
-
W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, USA, 93rd edn, 2012 Search PubMed
- C. D. Abernethy, G. M. Codd, M. D. Spicer and M. K. Taylor, J. Am. Chem. Soc., 2003, 125, 1128–1129 CrossRef CAS PubMed
- Z. R. Turner, R. Bellabarba, R. P. Tooze and P. L. Arnold, J. Am. Chem. Soc., 2010, 132, 4050–4051 CrossRef CAS PubMed
- P. L. Arnold, Z. R. Turner, R. Bellabarba and R. P. Tooze, J. Am. Chem. Soc., 2011, 133, 11744–11756 CrossRef CAS PubMed
- P. L. Arnold, T. Cadenbach, I. H. Marr, A. A. Fyfe, N. L. Bell, R. Bellabarba, R. P. Tooze and J. B. Love, Dalton Trans., 2014, 43, 14346–14358 RSC
- V. Kumar and E. V. Anslyn, Chem. Sci., 2013, 4, 4292–4297 RSC
- P. de Fremont, N. M. Scott, E. D. Stevens, T. Ramnial, O. C. Lightbody, C. L. B. Macdonald, J. A. C. Clyburne, C. D. Abernethy and S. P. Nolan, Organometallics, 2005, 24, 6301–6309 CrossRef CAS
- F. Lazreg, D. B. Cordes, A. M. Z. Slawin and C. S. J. Cazin, Organometallics., 2015, 34, 419–425 CrossRef CAS
- S. Zhang, W. C. Zhang, D. D. Shang, Z. Q. Zhang and Y. X. Wu, Dalton Trans., 2015, 44, 15264–15270 RSC
- D. D. Yang, Y. G. Tang, H. B. Song and B. Q. Wang, Organometallics, 2015, 34, 2012–2017 CrossRef CAS
- V. H. L. Wong, A. J. P. White, T. S. A. Hor and K. K. Hii, Chem. Commun., 2015, 51, 17752–17755 RSC
- F. G. Mann and W. J. Pope, J. Chem. Soc., 1922, 121, 594–603 RSC
- W. F. Wu, Z. H. Fu, X. Wen, Y. J. Wang, S. Zou, Y. Meng, Y. C. Liu, S. R. Kirk and D. L. Yin, Appl. Catal., A, 2014, 469, 483–489 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for the compounds; kinetic data for the catalysis. Crystal structure data for the vanadium–NHC decomposition product. CCDC 1580495. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04805j |
| This journal is © The Royal Society of Chemistry 2018 |