
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studies towards the synthesis of Pd(II)-containing [2] and [3]catenanes in aqueous media†
E. M.
López-Vidal‡
 *a,
A.
Prokofjevs
b,
I. C.
Gibbs-Hall
b,
E. J.
Dale
b,
J. M.
Quintela
a and
C.
Peinador
*a
*a,
A.
Prokofjevs
b,
I. C.
Gibbs-Hall
b,
E. J.
Dale
b,
J. M.
Quintela
a and
C.
Peinador
*a
aDepartamento de Química and Centro de Investigacións Científicas Avanzadas (CICA). Facultade de Ciencias, Universidade da Coruña, 15071, A Coruña, Spain. E-mail: E.M.Lopez.Vidal@bath.ac.uk; carlos.peinador@udc.es
bDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA
First published on 24th January 2018
Abstract
Here is reported the investigation of a synthetic route for the preparation of Pd(II)-containing catenanes in aqueous media. A pseudorotaxane intermediate was prepared, which can potentially be converted into a series of catenanes. From the pseudorotaxane, using a Pd(II)-driven clipping step a dinuclear [3]catenane was obtained in the solid state.
The synthesis of mechanically interlocked molecules (MIMs; i.e. rotaxanes, catenanes, and molecular knots)1,2 has attracted substantial interest among scientists, not only because of their intrinsic beauty, or the substantial synthetic challenge that they signify, but for other more practical reasons like their prospective use in the development of molecular-scale machinery.3,4
Besides the initial statistical and directed approaches used for the synthesis of MIMs,5 template-directed methods have dominated the field since the seminal work by Sauvage et al., who proposed the use of tetrahedral Cu(I) complexes for the preorganization of phenanthroline-based threads and their subsequent conversion into [2]catenanes.2,6 To date, all known weak intermolecular interactions have been used for the template-based strategy (e.g. hydrogen bonding, π–π interactions, etc.).1,2 For instance, donor–acceptor π–π interactions have been extensively used for templating the threading of molecular strands through macrocycles, creating pseudorotaxane architectures that can be subsequently converted into the corresponding MIMs by covalent capture (e.g. olefin metathesis, alkyne homocoupling, Cu(I)-catalyzed azide–alkyne cycloaddition, etc.).7 Even though these template-directed approaches have demonstrated their usefulness, there is still a great need for the development of synthetic strategies capable of producing targeted mechanically interlocked molecules via self-assembly of rationally designed components.
In this context, metal-directed self-assembly8 has proven to be a very useful tool in those cases where transition metal ions work as templating agents by gathering and organizing ligands around them or as active structural units of the MIM,9 serving, for example, as ring closing elements converting pseudorotaxanes into catenanes.10
Making use of the strategies outlined above, π–π interactions and coordination to metal complexes, we have reported in a previous work the preparation of a [2]catenane in aqueous media by following a stepwise metal-directed strategy.11 As shown in Scheme 1a, this approach consists of the threading of the electron-rich dioxoaryl-based molecular axle 12+ through the cavity of a preformed electron-deficient metallacycle, yielding the corresponding pseudorotaxane, followed by a kinetically controlled metal-directed cyclization step of the corresponding pseudorotaxane producing the catenane as the main product.
 | ||
| Scheme 1 Potential topologies arising from a stepwise metal-directed strategy for catenane formation using a Pd(II) square-planar complex with two labile ligands at cis positions (a) or with four labile ligands (b). | ||
Based on this previous work, we present here the results obtained of our attempted synthesis of [2] and [3]catenanes, as well as the double [2]catenane, which could potentially arise from the substitution of the square-planar complex (en)Pd(NO3)2, which has two labile ligands at cis positions,8 with the complex [Pd(CH3CN)4](BF4)2. As shown in Scheme 1b, the use of this tetravalent complex, with four labile ligands, would substantially increase the number of potential topologies obtained after the clipping step upon pseudorotaxane formation. The possibility of the ligand coordinating the metal complex in cis and/or trans would result, potentially, in the formation of 2 isomers of the [2]catenane, 3 isomers of the [3]catenane or a double-[2]catenane. In order to simplify the NMR analysis of the products, we decided to utilise CBPQT4+, a π-deficient receptor,12 instead of the Pt(II) metallacycle previously used, since CBPQT4+ has a higher symmetry order which would result in simpler NMR spectra.
We began our investigations by studying the interactions concerning the different components of the designed system, starting with the first reaction of our intended route, namely, the pseudorotaxane synthesis by self-assembly of CBPQT4+ with the axle 12+, which contains an electron-rich 1,5-dioxonaphthalene subunit. Thus, addition of 1 equiv. of CBPQT·4Cl to an aqueous solution of 1·2Cl (5 mM) resulted in a prominent colour change, from orange to purple, suggesting that a new charge-transfer interaction between the electron-rich 1,5-dioxonaphthalene subunit in 12+ and the electron-poor regions of CBPQT4+ had been established, which is a qualitative indication of the formation of the pseudorotaxane 26+ (Scheme 1b).
The 1H NMR spectrum of the reaction mixture in D2O (Fig. 1) displays signals in good agreement with the expected pseudorotaxane 26+. Consequently, the assembly of 26+ results in an upfield shift of the naphthalene core protons, suggesting that the electron-rich aromatic system is positioned within the cavity of the tetracationic receptor. Consequently, the donor–acceptor π–π interactions also promote the upfield shift of the nuclei Hα and Hβ of the viologen-based box.
 | ||
| Fig. 1 Partial 1H NMR spectra (298 K, 500 MHz, D2O) illustrating salt effects in the formation of 26+ from CBPQT4+ and 12+. (a) CBPQT4+; (b) equimolar solution of CBPQT4+ and 12+ in 0.7 M NaCl; (c) 12+. | ||
Furthermore, both the shielding of Hc (Δδ = −4.73 ppm) and the deshielding (Δδ = 0.22 ppm) of the aromatic protons of the phenylene ring (HPh) can be interpreted as arising from C–H⋯π interactions between both structural elements, providing further evidence in support of the proposed structure of 26+. Moreover, the signals corresponding to the CBPQT4+ subunit within 26+ are duplicated, as compared to those in the free macrocyclic host, which results from the reduced symmetry of the pseudorotaxane. Nevertheless, the 1H NMR spectrum also displays peaks which are attributable to free CBPQT4+ and 12+, suggesting that all three species exist in equilibrium, which is slow compared to the 1H NMR timescale (see Fig. S1†).
Based on the integration of the 1H NMR signals corresponding to 12+, CBPQT4+, and 26+, the equilibrium constant for pseudorotaxane formation in D2O at 298 K is 2.5 ± 0.5 × 103 M−1. As could be reasonably anticipated, dissociation of 26+ into the starting components can be promoted by raising the temperature of the solution (see Fig. S2†). Conversely, increasing the ionic strength of the reaction medium by adding NaCl enhances the hydrophobic effect which shifts the equilibrium towards the formation of 26+ (see Fig. 1b and Fig. S3†). This very same effect was obtained using the non-coordinating neutral salt NaNO3. For instance, the equilibrium constant measured in 0.7 M NaCl solution is 1.6 ± 0.3 × 104 M−1. Considering these results, it should be noted that addition of a neutral salt is necessary in order to avoid the disentangling of the pseudorotaxane in the envisioned clipping step leading to the targeted catenanes.
Our initial attempts to obtain, by different methods, suitable single crystals of 26+ for XRD, from the solution of the pseudorotaxane in 0.7 M NaCl, were completely unsuccessful. In order to modulate the solubility of the species in water, we decided to precipitate the cation as its tetrachlorozincate salt.13 Addition of ZnCl2 (10 equiv.) to the solution of 26+, prepared by mixing CBPQT4+ and 12+ in 0.7 M(aq) NaCl, resulted in precipitation of a purple solid, substantially reducing the amount of 26+ present in solution. To confirm the identity of this purple precipitate, it was collected and subsequently redissolved in D2O. A 1H NMR assay of the resulting solution matched that of the pseudorotaxane 26+, with no changes in the spectrum resulting from interaction of basic pyridine nitrogens with Zn2+. Since addition of an excess of ZnCl2 to the solution of 12+ also did not result in any significant changes to the 1H NMR spectrum, neither 26+ nor 12+ appears to engage in strong N⋯Zn interactions in solution. More significantly, this method allowed us to obtain single crystals suitable for XRD studies. Thus, purple plate-like crystals were produced by storing the liquid fraction of the above-mentioned mixture, prepared by adding 10 equiv. of ZnCl2 to the solution of CBPQT4+ and 12+ in 0.7 M(aq) NaCl, for several days at room temperature. The single crystal structure (Fig. 2) clearly supports the formation of the expected pseudorotaxane, with the electron-rich dioxoaryl moiety of 12+ inserted within the hydrophobic cavity of CBPQT4+ and the geometrical parameters being in good agreement with the establishment of π–π interactions.
 | ||
Fig. 2 Projection of one of the two symmetrically independent pseudorotaxanes [2(ZnCl3)2]4+ within the unit cell of 26+, the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) single crystal of [2(ZnCl3)2][(ZnCl4)2]. single crystal of [2(ZnCl3)2][(ZnCl4)2]. | ||
Unexpectedly, the obtained structure has the formula [2(ZnCl3)2](ZnCl4)2 instead of 2·3(ZnCl4), with the terminal pyridyl N of the axle 12+ capped with anionic ZnCl3− moieties.14 In light of this observation, it is tempting to describe the observed folding of the [2(ZnCl3)2]4+ cation as a consequence of an attractive interaction between the bipyridinium rings within CBPQT4+ and those on the 1(ZnCl3)2 thread (∠planes = 9.4 and 4.6° and dcent = 4.0 and 4.0 Å, respectively, for the two symmetrically independent pseudorotaxanes within the unit cell of the P single crystal). Since this coordination-induced folding is also likely to limit the conformational mobility of the supramolecule, it could also explain the facile crystallization of 2·6Cl upon treatment with ZnCl2.
We then proceeded to explore the products resulting from the self-assembly between 12+ and the Pd(II) complex [Pd(CH3CN)4](BF4)2, which is necessary for the clipping step in our intended synthesis (Scheme 1). Therefore, addition of 1·2Cl (1 equiv.) to an aqueous solution of [Pd(CH3CN)4](BF4)2 (0.5 equiv.) resulted in the self-assembly of the siamese metallacyclophane 3·(BF4)2Cl4 (Scheme 2). The resulting 1H NMR spectrum shows all of the bipyridinium signals in 12+ moving downfield as a result of coordination to the metal center (ΔδHg = 1.01 ppm, ΔδHf = 0.60 ppm, ΔδHe = 0.29 ppm, ΔδHd = 0.28 ppm, see Fig. S9ii†). HR-ESI mass spectrometry further confirmed the identity of 36+ as the self-assembled product (see Fig. S15†).
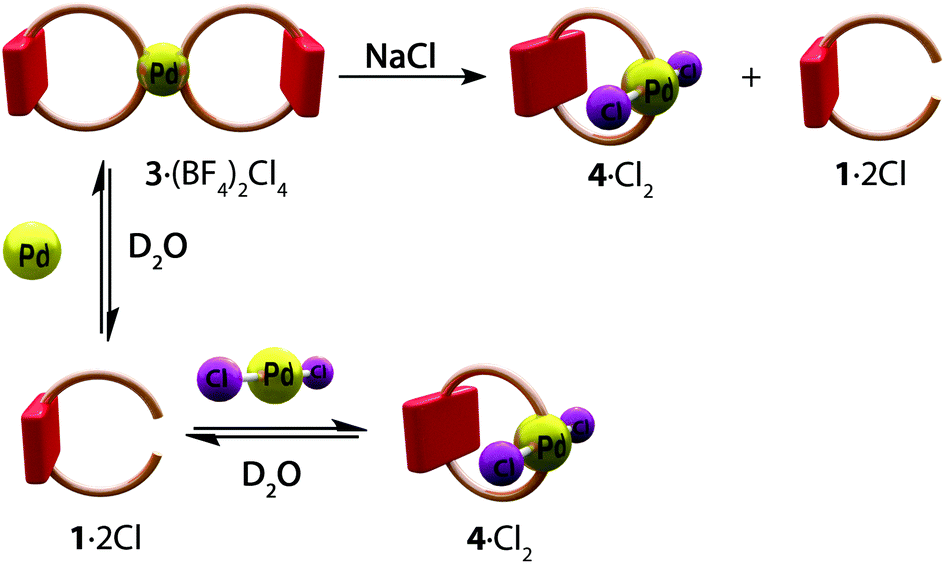 | ||
| Scheme 2 Self-assembly of the metallacyclophanes 36+ and 42+ from 12+ and the Pd(II) complex [Pd(CH3CN)4](BF4)2 in the absence and presence of salts (NaCl or NaNO3). | ||
In order to test the effect of the addition of NaCl and NaNO3 on the self-assembly of the metallacyclophanes, as this would be a prerequisite for further assembly of the targeted catenanes (vide supra), those salts (70 equiv.) were added to solutions of 12+ (1 equiv.) and [Pd(CH3CN)4](BF4)2 (0.5 equiv.) in D2O.
Whilst the addition of NaNO3 did not change the outcome of the self-assembly process (Fig. S9viii†), the NMR spectra resulting from the assay with NaCl show the self-assembly of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 42+, along with the free axle 12+ (Fig. S9v†). This situation was also confirmed by HR-ESI mass spectrometry (see Fig. S21 and S22†). These results are in good agreement with the well-known trans effect of chloride anions, with the added excess of the halide promoting the blocking of two trans positions of the Pd(II) metal center.15
:1 mixture of 42+, along with the free axle 12+ (Fig. S9v†). This situation was also confirmed by HR-ESI mass spectrometry (see Fig. S21 and S22†). These results are in good agreement with the well-known trans effect of chloride anions, with the added excess of the halide promoting the blocking of two trans positions of the Pd(II) metal center.15
Further experiments were carried out with the Pd(II) complex Pd(CH3CN)2Cl2 in place of [Pd(CH3CN)4](BF4)2. The results show that self-assembly does not depend on the complex used, but rather on the salt present in excess in the reaction mixture (see Fig. S9†).
We proceeded with our intended plan for the synthesis of the targeted catenanes by studying the interactions of the pseudorotaxane 26+ with the square planar complex [Pd(CH3CN)4](BF4)2. The 1H NMR spectrum recorded at room temperature after addition of stoichiometric amounts of the metal center to equimolar solutions of 1·2Cl and CBPQT·4Cl (5 mM) in D2O, in either 0.7 M NaNO3 or NaCl, appears to show the formation of very complex reaction mixtures (Fig. S23 and S24i†). Moreover, the identity of the species after addition of the metal center could not be determined by mass spectrometry. Surprisingly, addition of ZnCl2 (10 equiv.) to an equimolar mixture of the axle 1·2Cl, CBPQT·4Cl and [Pd(CH3CN)4](BF4)2 (5 mM) in D2O (0.7 M NaCl) produced changes in the 1H NMR spectrum (Fig. S24ii†). In order to determine the structure of the self-assembled species, several crystallization experiments were carried out using the previous solution. Fortunately, the slow evaporation of this earlier solution allowed us once again to obtain purple single crystals which were appropriate for XRD. The obtained structure revealed the formation of the [3]catenane 5·(ZnCl4)2Cl8 consisting of two pseudorotaxane subunits of 26+ connected by the coordinating bipyridine subunits to two PdCl2 centers (Fig. 3). The formation and crystallization of the obtained trans/trans-[3]catenane can be explained on the basis of two key factors: (i) the in situ blocking of two trans-positions on the complex [Pd(CH3CN)4](BF4)2, and (ii) modulation of the solubility of the resulting catenane by introduction of poorly polarizable [ZnCl4]2− anions in the reaction media (Scheme 3).
 | ||
| Fig. 3 Crystal structure of the [3]catenane 512+. Pd–Cl⋯H–C hydrogen bonds and π–π interactions are represented as green dotted lines. | ||
 | ||
| Scheme 3 Reaction conditions for the preparation of single crystals of the [3]catenane 5 (ZnCl4)2Cl8. | ||
In addition to the expected π–π interactions associated with the CBPQT-dioxoaryl host–guest aggregation, there are also π–π interactions between two viologen-like motifs corresponding to two different CBPQT units. The PdCl2 centers are also involved in stabilizing the structure by means of Pd–Cl⋯H–C hydrogen bonds (Fig. 3).16
Crystals of 5·(ZnCl4)2Cl8 were dissolved in D2O and dilution experiments performed on this solution resulted in 1H NMR spectra containing two sets of signals which confirms the existence of two species in solution as one set of signals becomes more intense at decreasing concentrations. We propose that the nature of this second species corresponds to the [2]catenane 66+ (Scheme S5†) which consists of a lower number of subcomponents compared to 512+, being favored at lower concentrations.
Conclusions
In summary, we have reported herein our studies on the preparation of Pd(II)-containing catenanes in aqueous media using a synthetic route that implies self-assembly of the pseudorotaxane 26+ as a synthetic intermediate followed by a Pd(II)-driven clipping step. The self-assembly of 26+ takes advantage of donor–acceptor π–π interactions between 12+ and CBPQT4+ and the hydrophobic effect, with addition of neutral salts to the reaction mixture increasing pseudorotaxane formation by raising the ionic strength of the medium. Coordination of the terminal N atoms of 12+ to ZnCl3 motifs facilitates pseudorotaxane folding and subsequent crystallization. In order to test the Pd(II)-driven clipping of 12+, this bidentate ligand was reacted with [Pd(CH3CN)4](BF4)2, which produced the metallocycle 36+ or 42+. Remarkably, 36+ can be converted into 42+ by in situ blocking of the two trans positions of [Pd(CH3CN)4](BF4)2 with chloride anions. Although attempts on the self-assembly of the targeted catenanes led to complex mixtures of products in solution, we were able to crystallize a butterfly-like shaped [3]catenane, 5·(ZnCl4)2Cl8, which is one of the few examples of [3]catenanes self-assembled in aqueous media.17Conflicts of interest
There are no conflicts to declare.Author contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Funding sources
This research was supported by the Ministerio de Economía, Industria y Competitividad (Ministerio de Economía y Competitividad FEDER, Grant CTQ2016-75629-P).Acknowledgements
We are enormously grateful to Prof. Sir James Fraser Stoddart (2016 Nobel Laureate in Chemistry) for supervising and hosting part of this research in his laboratory at Northwestern University. We also thank Ms Charlotte Stern (Northwestern University) for the refinement of the crystal structures reported in this work. E. M. L.-V. thanks the Ministerio de Economía, Industria y Competitividad (FPI program).Notes and references
- J.-P. Sauvage and C. Dietrich-Buchecker, Molecular Catenanes, Rotaxanes and Knots, Wiley-VCH, Weinheim, 1999 Search PubMed.
- P. D. Beer, M. R. Sambrook and D. Curiel, Chem. Commun., 2006, 2105–2117 RSC; J. A. Faiz, V. Heitz and J.-P. Sauvage, Chem. Soc. Rev., 2009, 38, 422–442 RSC; J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530–1541 RSC; A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875–882 RSC; K. D. Hanni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC; J. E. Beves, B. A. Blight, D. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260–9327 CrossRef CAS PubMed; R. S. Forgan, J.-P. Sauvage and J. F. Stoddart, Chem. Rev., 2011, 111, 5434–5464 CrossRef PubMed; V. N. Vukotic and S. J. Loeb, Chem. Soc. Rev., 2012, 41, 5896–5906 RSC; G. T. Spence and P. D. Beer, Acc. Chem. Res., 2012, 46, 571–586 CrossRef PubMed.
- B. L. Feringa, Molecular Switches, Wiley-VCH, Weinheim, 2001 Search PubMed; V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines, Wiley-VCH, Weinheim, 2003 Search PubMed.
- C. P. Collier, G. Mattersteig, E. W. Wong, Y. Luo, K. Beverly, J. Sampaio, F. M. Raymo, J. F. Stoddart and J. R. Heath, Science, 2000, 289, 1172–1175 CrossRef CAS PubMed; V. Balzani, A. Credi, F. Raymo and J. F. Stoddart, Angew. Chem., Int. Ed., 2000, 39, 3348–3391 CrossRef PubMed; J.-P. Sauvage, Chem. Commun., 2005, 1507–1510 RSC; E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef PubMed; A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC; C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef PubMed.
- H. L. Frisch and E. Wasserman, J. Am. Chem. Soc., 1961, 83, 3789–3795 CrossRef CAS; G. Schill, Catenanes, Rotaxanes and Knots, Academic Press, New York, 1971 CrossRef; G. Schill and C. Zürcher, Naturwissenschaften, 1971, 58, 40–45 CrossRef.
- C. O. Dietrich-Buchecker and J.-P. Sauvage, Chem. Rev., 1987, 874, 795–810 CrossRef CAS; B. Champin, P. Mobian and J.-P. Sauvage, Chem. Soc. Rev., 2007, 36, 358–366 RSC.
- W. R. Dichtel, O. S. Miljanić, W. Zhang, J. M. Spruell, K. Patel, I. Aprahamian, J. R. Heath and J. F. Stoddart, Acc. Chem. Res., 2008, 41, 1750–1761 CrossRef CAS PubMed; J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC; S. Li, M. Liu, B. Zheng, K. Zhu, F. Wang, N. Li, X.-L. Zhao and F. Huang, Org. Lett., 2009, 11, 3350–3353 CrossRef PubMed.
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef CAS; M. Fujita, Chem. Soc.Rev., 1998, 27, 417–425 RSC; D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef; B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef PubMed.
- M. Fujita, F. Ibukuro, H. Hagihara and K. Ogura, Nature, 1994, 367, 720–723 CrossRef CAS; M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52–55 CrossRef; M. Fujita, Acc. Chem. Res., 1999, 32, 53–61 CrossRef; A. Hori, T. Sawada, K. Yamashita and M. Fujita, Angew. Chem., Int. Ed., 2005, 44, 4896–4899 CrossRef PubMed.
- D. M. Whang, K. M. Park, J. Heo, P. Ashton and K. Kim, J. Am. Chem. Soc., 1998, 120, 4899–4900 CrossRef CAS; K.-M. Park, S.-Y. Kim, J. Heo, D. Whang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2002, 124, 2140–2147 CrossRef PubMed.
- E. M. López-Vidal, M. D. García, C. Peinador and J. M. Quintela, Chem. – Eur. J., 2015, 21, 2259–2267 CrossRef PubMed.
- B. Odell, M. V. Reddington, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1988, 27, 1547–1550 CrossRef.
- C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, P. Turner, M. B. Duriska and R. Willis, Chem. Commun., 2008, 10, 1190–1192 RSC.
- W.-T. Chen, D.-S. Liu, S.-M. Ying, H.-L. Chen and Y.-P. Xu, Inorg. Chem. Commun., 2008, 11, 1212–1214 CrossRef CAS.
- F. R. Hartley, Chem. Soc. Rev., 1973, 2, 163–179 RSC.
- G. Aullón, D. Bellamy, L. Brammer, E. A. Bruton and A. G. Orpen, Chem. Commun., 1998, 653–654 RSC.
- A. Hori, K. Kumazawa, T. Kusukawa, D. K. Chand, M. Fujita, S. Sakamoto and K. Yamaguchi, Chem. – Eur. J., 2001, 7, 4142–4149 CrossRef CAS; F. B. L. Cougnon, N. A. Jenkins, G. D. Pantoş and J. K. M. Sanders, Angew. Chem., Int. Ed., 2012, 51, 1443–1447 CrossRef PubMed; R. S. Forgan, J. J. Gassensmith, D. B. Cordes, M. M. Boyle, K. J. Hartlieb, D. C. Friedman, A. M. Z. Slawin and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17007–17010 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and NMR, HR-ESI MS and X-ray data (PDF). X-ray crystallographic data in CIF format. CCDC 1812494–1812495. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04792d |
| ‡ Current address: Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. |
| This journal is © The Royal Society of Chemistry 2018 |