
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn isolable magnesium diphosphaethynolate complex†‡
Robert J.
Gilliard
Jr.
 ab,
Dominikus
Heift
bc,
Zoltán
Benkő
d,
Jerod M.
Keiser
a,
Arnold L.
Rheingold
e,
Hansjörg
Grützmacher
*b and
John D.
Protasiewicz
*a
ab,
Dominikus
Heift
bc,
Zoltán
Benkő
d,
Jerod M.
Keiser
a,
Arnold L.
Rheingold
e,
Hansjörg
Grützmacher
*b and
John D.
Protasiewicz
*a
aDepartment of Chemistry, Case Western Reserve University, Cleveland, OH 44106, USA. E-mail: Protasiewicz@case.edu
bDepartment of Chemistry and Applied Biosciences, ETH Zurich, CH-8093 Zurich, Switzerland. E-mail: hgruetzmacher@ethz.ch
cDepartment of Chemistry, Durham University, Lower Mountjoy, South Road, Durham, DH1 3LE, UK
dBudapest University of Technology and Economics, H.111 Budapest Szent Gellért tér 4, Hungary
eDepartment of Chemistry, University of California, San Diego, La Jolla, CA 92093, USA
First published on 8th December 2017
Abstract
The reaction of magnesium chloride with two equivalents of sodium phosphaethynolate, Na[OCP]·(dioxane)2.5 (1), yields a magnesium diphosphaethynolate complex, [(THF)4Mg(OCP)2] (3). The formation of compound 3 goes through a monosubstituted chloromagnesium phosphaethynolate Mg(OCP)Cl (2). The structure of 3 was determined via a single crystal X-ray diffraction study. For comparison, we also report the structure of a monomeric sodium phosphaethynolate complex, [Na(OCP)(dibenzo-18-crown-6)] (4).
It has been more than two decades since Becker reported the structure of lithium phosphaethynolate, [(DME)2Li(OCP)] A (Fig. 1).1 The highly reactive nature and limited stability of this compound discouraged detailed studies of the (OCP) anion. Subsequent reports discussed the challenging nature of phosphaethynolate chemistry which was mostly attributed to the instability and low yield of Mx(OCP)y (M = metal) salts.1,2 We recently reported a new method for the large scale preparation of sodium phosphaethynolate, Na(OCP)·(dioxane)x, spawning a resurgence of interest in the synthesis, structure, and reactivity of this 3-atom organophosphorus building block.3 Notably, Na(OCP)·(dioxane)x is a remarkably stable synthon that can be stored under inert atmosphere indefinitely.
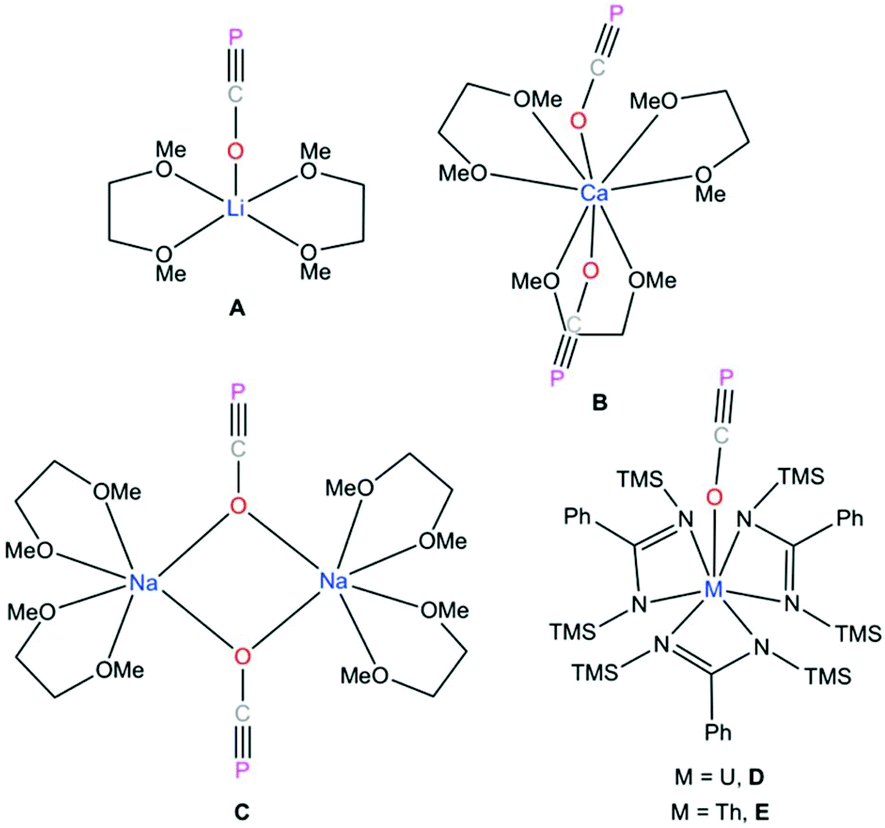 | ||
| Fig. 1 O-Bound metal phosphaethynolate complexes.1,3,17,22 | ||
The phosphaethynolate anion, (OCP)−, may be regarded as the phosphorus analogue of cyanate, (OCN)−.3 Natural Resonance Theory calculations have shown that (OCP)− has three major resonance structures: [O–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P]− (51.7%), [O
P]− (51.7%), [O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CP]− (40.2%) and [OC → P]− (7.1%).4 Recently, reactivity studies involving these salts has led to the formation of novel heterocycles,5–11 main-group small molecules,12–15 transition metal complexes,4,16–19 and phosphorus derivatives of organic molecules.20,21 However, the number of O-bound phosphaethynolate salts are limited as most Mx(OCP)y compounds feature M–P instead of M–O bonds.4,11–16,18,19 Thus, the only structurally characterized examples of complexes with M–O–CP functionalities are of lithium A,1 calcium B,22 sodium C,3 uranium D,17 and thorium E (Fig. 1).17 While the O–CP moiety in compounds A, B, and C are stabilized via dimethoxyethane (DME) coordination, the isolation of compounds D and E relies on the coordination of tris-amidinate ligands.
CP]− (40.2%) and [OC → P]− (7.1%).4 Recently, reactivity studies involving these salts has led to the formation of novel heterocycles,5–11 main-group small molecules,12–15 transition metal complexes,4,16–19 and phosphorus derivatives of organic molecules.20,21 However, the number of O-bound phosphaethynolate salts are limited as most Mx(OCP)y compounds feature M–P instead of M–O bonds.4,11–16,18,19 Thus, the only structurally characterized examples of complexes with M–O–CP functionalities are of lithium A,1 calcium B,22 sodium C,3 uranium D,17 and thorium E (Fig. 1).17 While the O–CP moiety in compounds A, B, and C are stabilized via dimethoxyethane (DME) coordination, the isolation of compounds D and E relies on the coordination of tris-amidinate ligands.
Westerhausen prepared a series of alkaline earth metal phosphaethynolates.22 The synthesis of these compounds involved the reaction of alkali earth metal bis(trimethylsilyl)phosphides M[P(SiMe3)2]2 (M = Mg, Ca, Sr, Ba) with dimethyl carbonate (MeO)2CO in DME to afford M(OCP)x(DME)y and trimethylsilylether MeOSiMe3.22 However, these salts were unstable, even at low temperatures. Storage of B at −30 °C resulted in the formation of single crystals, but attempts to isolate the salt resulted in immediate decomposition. The structure of magnesium phosphaethynolate was formulated to be the cis-diphosphaethynolate [(DME)3Mg(OCP)2], however, samples obtained by this method could not be isolated and characterization was limited to NMR experiments. Indeed, it has proved challenging to prepare diphosphaethynolate complexes where two OCP units bind to the same metal center. During our studies we have found that the formation and stability of Mx(OCP)y salts is highly dependent on the synthetic route and the coordination environment around the metal cation. Herein, we report the synthesis, molecular structure, and computations of a magnesium diphosphaethynolate complex, [(THF)4Mg(OCP)2] (3). Notably, compound 3 is a unique example of a stable metal diphosphaethynolate complex that can be isolated and stored at room temperature. The structure of compound 3 is compared to a novel [Na(OCP)(dibenzo-18-crown-6)] complex (4).
The reaction of magnesium chloride with Na[OCP]·(dioxane)2.5 (1) is extremely sensitive to the reaction conditions. Our initial attempts to synthesize magnesium diphosphaethynolate involved the addition of a DME solution containing two equivalents of 1 to a suspension of anhydrous MgCl2 powder in DME at various temperatures. These reactions failed to yield the desired diphosphaethynolate complex and instead led to the formation of an insoluble gray precipitate. However, upon slow dropwise addition of a THF solution of 1 to a suspension of MgCl2 vigorously stirred in a THF/toluene mixture, a slightly cloudy pink solution containing 3 was obtained (Scheme 1). Immediately after the addition was complete, the solution was filtered and concentrated to give an off-white solid which was collected by filtration. Heating the solid in refluxing THF until completely dissolved followed by slow cooling to room temperature gave colorless microcrystalline 3 in nearly quantitative yield. It is noteworthy that if 1 is added to MgCl2 too rapidly, the yield of 3 is compromised and instead an insoluble dark colored precipitate is formed. Samples of solid 3 analyzed after months of storage under N2 showed no evidence of decomposition by 31P NMR spectroscopy. Interestingly, while Mg(OCP)2 is stable as a THF adduct, if DME is added to the solid, some decomposition is observed (i.e., formation of a small amount of gray precipitate), thereby highlighting the importance of coordination chemistry in the synthesis, stability, and reactivity of these salts.
 | ||
| Scheme 1 Synthesis of magnesium diphosphaethynolate (3). | ||
The X-ray structure of 3 reveals a six-coordinate magnesium atom in an octahedral geometry and the two OCP units are related by a crystallographic inversion center (Fig. 2). This differs from the square antiprismatic geometry in eight-coordinate metal complexes 4 (Fig. 3) and B (Fig. 1).
 | ||
| Fig. 2 Molecular structure of 3 (thermal ellipsoids set at 30% probability; hydrogen atoms omitted for clarity). Selected bond distances [Å] and angles [°]: Mg1–O1: 2.024(16); O1–C1: 1.196(3); C1–P1: 1.572(3). Mg–O1–C1: 142.8(15); O1–C1–P1: 179.6(2); O2–Mg1–O1: 91.4(6). | ||
 | ||
| Fig. 3 Molecular structure of 4 (thermal ellipsoids set at 50% probability; hydrogen atoms omitted for clarity). Selected bond distances [Å] and angles [°]: Na1–O1: 2.290(2); O1–C1: 1.207(4); C1–P1: 1.582(3). Na–O1–C1: 138.1(2); O1–C1–P1: 179.4(3); O8–Na–O1: 169.8(9). | ||
The Ca–OOCP bond distance in B is 2.358(2) Å, which is significantly longer than the comparable Mg1–O1 bond in 3 (2.024(16) Å).22 The M–O–C angle in 3 is 142.8°, which is smaller than in A (170.7°),1D (170.9°),17 and E (176.4°),17 but more comparable to B (154.6°),22C (132.1°),3 and 4 (138.1°). In notable contrast to the monomeric potassium phosphaethynolate reported by Goicoechea5 which contains a K–PPCO bond (3.383 Å), the structure of 4 features an Na–OOCP bond (2.290 Å).
Analysis of the reaction progress by 31P NMR spectroscopy suggest that the formation of compound 3 goes through a monosubstituted chloromagnesium phosphaethynolate 2. Accordingly, MgCl2 was reacted with various equivalents of compound 1 in THF at room temperature and the 31P NMR spectra were recorded. The spectrum of 1 in THF shows a singlet at δ = −392.9 ppm (Fig. 4a). When MgCl2 is reacted with one equivalent of 1, a new peak at δ = −369.5 ppm is formed and compound 1 is completely consumed (Fig. 4b). Although the structure has not been obtained, we assign this peak as the monosubstituted Mg(OCP)Cl compound (2). Significantly, compounds 2 and 3 can be observed upon addition of 1.5 equivalents of 1 to MgCl2 (Fig. 4c). When two equivalents of 1 is added to MgCl2, only the diphosphaethynolate complex 3 can be observed (Fig. 4d). Indeed, these 31P NMR chemical shifts are in the range of metal phosphaethynolate complexes A–E (δ = −334 to −398 ppm).1,3,17,22
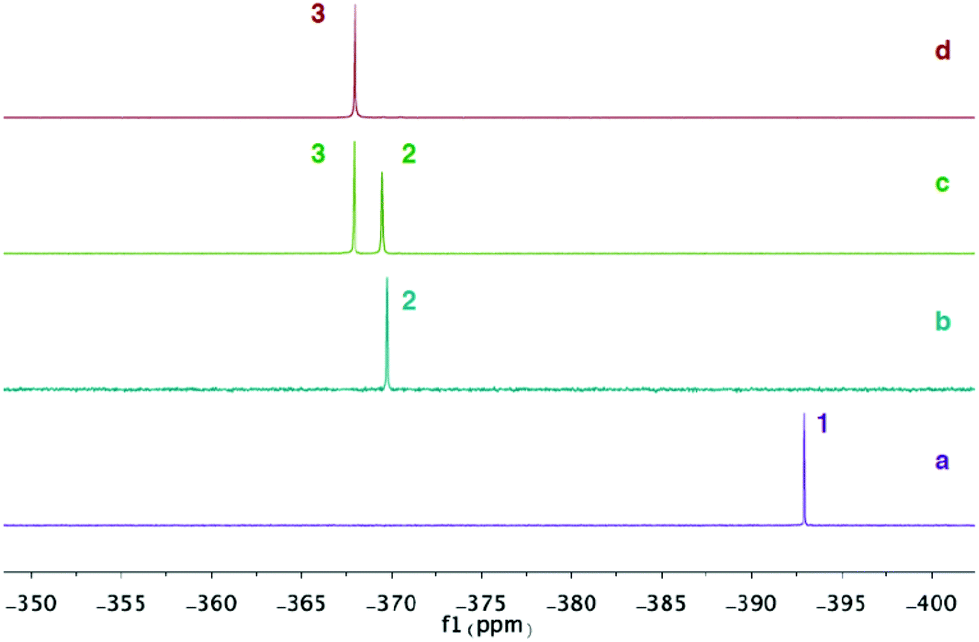 | ||
| Fig. 4 31P{1H} NMR spectra in THF: (a) Na(OCP)·(dioxane)2.5 (1). (b) MgCl2 + 1 equiv. 1. (c) MgCl2 + 1.5 equiv. 1. (d) MgCl2 + 2 equiv. 1. | ||
The differences in the 31P NMR chemical shifts for the sodium and magnesium phosphaethynolate compounds prompted us to perform DFT calculations on the electronic structure of [(THF)4Mg(OCP)2] (3), model species [(THF)5Na(OCP)] (4M) and free OCP− anion in the gas phase (5M) at the M06-2X/6-31+G* level of theory. In 4M and 5M, negative partial charges are located at the O and P atoms [4M: q(O) = −0.79e, q(P) = −0.18e; 5M: q(O) = −0.67e, q(P) = −0.44e]. In notable contrast, in 3, the C and P atoms are essentially neutral [q(C) = −0.04e, q(P) = −0.01e] and the majority of the charge is carried by the O atom [q(O) = −0.88e]. This data is in agreement with the experimental 31P NMR shifts which show that the magnesium salts are more deshielded (less electron density) compared to the sodium salt. While it is known that the OCP− anion can be described as a superposition of the phosphaethynolate and phosphaketenide resonance structures,4 based on the charge distribution of the OCP moiety, 3 represents a rather pure phosphaethynolate-type structure with an O–C single bond and a CP triple bond. This is in stark contrast to 4M where the weighting of the phosphaketenide structure is substantial. The bonding parameters of the computed salts show a similar trend. In 5M, 4M, and 3 the oxygen–carbon bond length increases (5M: 1.199 Å, 4M: 1.219 Å, and 3: 1.238 Å; decreasing double bond character) and the phosphorus–carbon bond length decreases (5M: 1.625 Å, 4M: 1.601 Å, 3: 1.584 Å; increasing triple bond character), which supports the decreasing weight of the allenic resonance structure (see ESI‡ for additional computational data). The low allenic character of the OCP fragment in 3 is also reflected in the experimental IR frequency of 1759 cm−1, which is assigned to the asymmetric stretching vibration of the OCP moiety. This value is clearly smaller than that obtained for the “interaction-free” OCP− anion (1791 cm−1)18 and A (1780 cm−1),3 but in the range of 1 (1755 cm−1)3 and 4 (1765 cm−1).
We have thus described the synthesis, molecular structure, and computations of a magnesium diphosphaethynolate complex (3), which has been obtained via salt metathesis reaction with Na(OCP)·(dioxane)2.5 (1). Although alkaline earth metal diphosphaethynolate compounds have been reported to be extremely reactive species, this route to complex 3 has rendered it stable such that it can be isolated and structurally characterized. Indeed, this parallels the significance of solvent choice in successfully isolating the sodium salt of the OCP anion.3 We also compare the structure of 3 to a monomeric sodium phosphaethynolate (4). Notably, unlike previously reported stable phosphaethynolate compounds, 3 features two OCP units per metal center. Moreover, while 3 is similarly ionic compared to Na(OCP), the P atom is less charged. The synthesis of compound 3 goes through a monosubstituted magnesium complex (2). The clean transformation to 2 by 31P NMR spectroscopy may provide additional opportunities for functionalization at the metal, facilitating interesting new organophosphorus chemistry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation (CHE-1464855), the ETH Zürich and the Swiss National Science Foundation (SNF) for financial support. R. J. G. is grateful to the UNCF-Merck Fellowship program and the Ford Foundation for postdoctoral fellowship awards. D. H. was supported by a European Union COFUND/Durham Junior Research Fellowship under EU grant agreement number 609412. Z. B. appreciates the support of the NKFIH (PD 116329) and the János Balyai Research Fellowship.Notes and references
- G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS.
- G. Becker and K. Hübler, Z. Anorg. Allg. Chem., 1994, 620, 405–417 CrossRef CAS.
- F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H. F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed.
- S. Alidori, D. Heift, G. Santiso-Quinones, Z. Benkő, H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. – Eur. J., 2012, 18, 14805–14811 CrossRef CAS PubMed.
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed.
- X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benko, Z. Li, G. Becker, H. F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 CrossRef CAS PubMed.
- D. Heift, Z. Benko and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 6757–6761 CrossRef CAS PubMed.
- D. Heift, Z. Benkő and H. Grützmacher, Chem. – Eur. J., 2014, 20, 11326–11330 CrossRef CAS PubMed.
- D. Heift, Z. Benkő, H. Grützmacher, A. R. Jupp and J. M. Goicoechea, Chem. Sci., 2015, 6, 4017–4024 RSC.
- T. P. Robinson and J. M. Goicoechea, Chem. – Eur. J., 2015, 21, 5727–5731 CrossRef CAS PubMed.
- M. M. Hansmann, D. A. Ruiz, L. Liu, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 3720–3725 RSC.
- D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 5920–5928 RSC.
- T. P. Robinson, M. J. Cowley, D. Scheschkewitz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 683–686 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, T. Szilv_si, E. Ballestero-Mart_nez, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 4333–4336 CrossRef CAS PubMed.
- (a) L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem., 2016, 1, 147–153 CrossRef CAS; (b) Y. Li, R. K. Siwatch, T. Mondal, Y. Li, R. Ganguly, D. Koley and C.-W. So, Inorg. Chem., 2017, 56, 4112–41120 CrossRef CAS PubMed; (c) R. J. Gilliard Jr., R. Suter, E. Schrader, Z. Benkő, A. L. Rheingold, H. Grützmacher and J. D. Protasiewicz, Chem. Commun., 2017, 53, 12325–12328 RSC.
- L. N. Grant, B. Pinter, B. C. Manor, R. Suter, H. Grützmacher and D. J. Mindiola, Chem. – Eur. J., 2017, 23, 6272–6276 CrossRef CAS PubMed.
- C. Camp, N. Settineri, J. Lefèvre, A. R. Jupp, J. M. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379–6384 RSC.
- L. Liu, D. A. Ruiz, F. Dahcheh, G. Bertrand, R. Suter, A. M. Tondreau and H. Grützmacher, Chem. Sci., 2016, 7, 2335–2341 RSC.
- J. M. Kieser, R. J. Gilliard Jr., A. L. Rheingold, H. Grützmacher and J. D. Protasiewicz, Chem. Commun., 2017, 53, 5110–5112 RSC.
- A. R. Jupp and J. M. Goicoechea, J. Am. Chem. Soc., 2013, 135, 19131–19134 CrossRef CAS PubMed.
- R. Suter, Y. Mei, M. Baker, Z. Benkő, Z. Li and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 1356–1360 CrossRef CAS PubMed.
- M. Westerhausen, S. Schneiderbauer, H. Piotrowski, M. Suter and H. Nöth, J. Organomet. Chem., 2002, 643–644, 189–193 CrossRef CAS.
Footnotes |
| † This manuscript is published in honor of Professor Phil Power's 65th birthday. We are grateful to Phil for his service to the chemistry community. His contributions in synthetic inorganic chemistry continues to inspire us all. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1544892 (3) and 1544893 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt04539e |
| This journal is © The Royal Society of Chemistry 2018 |