

Abstract
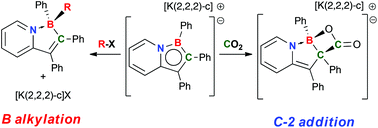
The reactions of 1-bora-7a-azaindenide anions, prepared in moderate to excellent yields by reduction of the appropriate 1-bora-7a-azaindenyl chlorides with KC8 in THF, with alkyl halides and carbon dioxide were studied. With alkyl halides (CH2Cl2, CH3I and BrCH(D)CH(D)tBu), the anions behave as boron anions, alkylating the boron centre via a classic SN2 mechanism. This was established with DFT methods and via experiments utilizing the neo-hexyl stereoprobe BrCH(D)CH(D)tBu. These reactions were in part driven by a re-aromatization of the six membered pyridyl ring upon formation of the product. Conversely, in the reaction of the 1-bora-7a-azaindenide anions with CO2, a novel carboxylation of the C-2 carbon alpha to boron was observed. Computations indicated that while carboxylation of the boron centre was kinetically feasible, the products of B-carboxylation were not thermodynamically favored relative to the observed C-2 carboxylated species, which were formed preferably due to the generation of both C–C and B–O bonds. In these products, the pyridyl ring remains non-aromatic, in part accounting for the observed reversibility of carboxylation.
- This article is part of the themed collection: Philip Power at 65: an icon of organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

