
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bi and trinuclear complexes in palladium carboxylate-assisted C–H activation reactions
Jiří
Váňa
 *,
Jiří
Hanusek
and
Miloš
Sedlák
*,
Jiří
Hanusek
and
Miloš
Sedlák
Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 53210 Pardubice, Czech Republic. E-mail: jiri.vana@upce.cz
First published on 22nd December 2017
Abstract
The role of polynuclear species in C–H activations assisted by palladium carboxylates has not been clear so far. The summary of the key findings covering this issue shows its important role under certain conditions. However, much more effort is necessary for a deeper understanding of the whole issue.
The detailed understanding of reactions involving transition metals requires exact knowledge of the structure of reactants, catalytic species and reaction intermediates. Unfortunately, the ability of metals to form polynuclear complexes does not make it straightforward.1 Thus, most of the reaction schemes use simplified reactions that reduce the mechanism to mononuclear species. Such a simplified view is useful for a basic understanding of the processes occurring during the reactions. However, for further development in the area of transition metal chemistry the precise nature of the key reaction species should be known. We have chosen the C–H activation reactions catalyzed by palladium(II) carboxylates as the illustration of this issue.
Most of the reaction schemes as well as mechanistic studies are based on monopalladium species. Nowadays, the AMLA/CMD (ambiphilic metal–ligand assisted/concerted metalation deprotonation) mechanisms are used for the explanation of the C–H activation step in reactions involving palladium carboxylates (Scheme 1).2 In principle, a molecule of substrate coordinates to the palladium carboxylate in the first reaction step. Next, in the concerted step the reaction center is attacked by a palladium atom while one of the coordinating carboxylates starts to make a new bond with the releasing proton. This leads to the formation of monopalladacycles that in most cases dimerize to form stable C–H activated binuclear complexes.3 However, the ability of palladium carboxylates to form polynuclear complexes opens the way for other reaction mechanisms based on bi or trimetallic species.
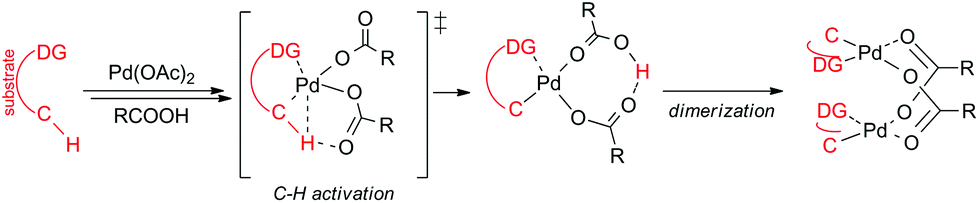 | ||
| Scheme 1 Simplified reaction mechanism of remote group directed C–H activation reaction. | ||
It can be expected that the nuclearity of the species involved in the reaction pathway is influenced by the nuclearity of starting materials. Thus, the first question to be answered is: what is the structure of the catalyst or precatalyst entering into the reaction with the substrate?
Structure of palladium(II) carboxylates
Palladium acetate alone or its combination with carboxylic acids occupies one of the leading positions among transition metal catalysts or catalytic systems. In 1965, Wilkinson4 proposed and in 1970, Skapski5a determined its structure to be trimeric cyclic Pd3(OAc)61 in the solid state (Fig. 1). Subsequent research showed that the trimeric cyclic structure remains preserved in the solution. However, there is possible solvent-dependent equilibration with its monomeric or dimeric form.6 Since then, most of the reaction mechanism proposals involve the formation of monopalladium species that enter into further molecular transformations.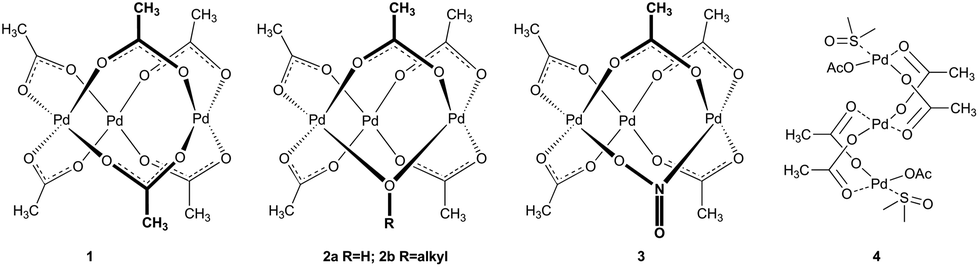 | ||
| Fig. 1 Products of the reaction of palladium acetate with nucleophiles. | ||
However, in the course of time and increasing popularity of palladium acetate chemists observed different reactivities of catalysts dependent on how the palladium acetate was prepared. This nontrivial behavior7 led to a series of studies in which a polymeric form and possible impurities formed by substitution of one acetate ligand for OH−2a, OR−2b and NO2−3 were isolated (Fig. 1).8 These species showed different reactivities in comparison with pure trimeric cyclic palladium acetate. Further questions concerning the structure and nuclearity of precatalysts arose by the introduction of reaction protocols using the combination of palladium acetate with other carboxylic acids. The most favored is the protocol popularized by Fujiwara using the addition of trifluoroacetic acid (TFA) to the reaction system.9 Batsanov showed Pd3(OTFA)6 to be a cyclic trimer in the solid phase5b while Wilkinson observed palladium trifluoroacetate to be monomeric in ethylacetate.4 Furthermore, the monopalladium [Pd(OTFA)2(HOAc)2], [Pd(OTFA)]+ and [(OTFA)2Pd(TFA)3]+ complexes were proposed to be active species in different reactions.10 On the other hand, sequential ligand exchange leading to the formation of cyclic trinuclear Pd3(OTFA)6 in dichloromethane was recently described in detail.11 Next, coordinating solvents like DMSO or acetone cause the opening of the cyclic trinuclear species giving the linear ones 4 (Fig. 1) or cause fragmentation to mono and dipalladium species.4,12 These observations suggest that the composition and nuclearity of the precatalyst is highly influenced by solvent and the purity of starting materials.
Binuclear species
The existence of palladacycles in binuclear dimeric forms motivated research to study the possibility of C–H activation proceeding via binuclear species. Musaev et al. in their computational study thoroughly examined the structures of the dimeric palladium acetate catalyst as well as their reactivity in the C–H activation of various substrates.13 They found that the C–H activation reaction pathway involving binuclear complexes is a little bit disfavored when compared to the mononuclear one. Next, they explained a preferred formation of dimeric palladacycles from monomeric ones due to ligand interactions (π–π stacking) and bridging acetate interactions.Different results were shown in the work of Vicente, Jones et al.14 that were further extended by Granell, Martínez et al.15 The authors experimentally studied the C–H activation of primary benzylamines in the solid state and solution (Scheme 2). They showed that the reaction of benzylamine (BnNH2) with palladium acetate leads to the formation of the monometallic nonactivated precomplex Pd(OAc)2(BnNH2) 5 that upon treating with palladium acetate dimerizes to the precomplex [Pd(OAc)(μ-OAc)(BnNH2)]26. The following C–H activation step proceeds solely on these dimers in a cooperative manner where the metalation by the first palladium atom is the rate limiting step. The C–H activated product is initially formed in its “open book” form 7 and then transforms to the stable complex 8.
 | ||
| Scheme 2 C–H activation of benzylamines. | ||
Recently, Musaev, Lewis et al. thoroughly examined the effects of mono-N-protected amino acids (MPAA) in the C–H activation of dimethylbenzylamines (DMBA-s) (Scheme 3).16 The binuclear complexes 9 analogous to 8 containing bridging MPAA instead of acetates were synthesized. Furthermore, it was shown that these dimeric complexes in the presence of iodobenzene dichloride undergo subsequent C–H functionalization to give other chlorinated dimeric palladacycles 10. The fact that a bi-palladium reactant is transformed to a bi-palladium product led authors to computationally examine and compare the potential mononuclear and binuclear reaction pathways. DFT calculations performed on the model 2-CF3-DMBA substrate show that cyclopalladation on the dimer occurring via two sequential CMD steps is both kinetically and thermodynamically favored over the monomeric pathway. Similar to the work of Granel, Martínez et al.15 the first cyclopalladation is a rate-limiting step and facilitates the second cyclopalladation.
 | ||
| Scheme 3 C–H activation and functionalization of dimethylbenzylamines in the presence of MPAA. | ||
Finally, Henry experimentally showed that contrary to the mononuclear Na2Pd(OAc)4 the binuclear Na2Pd2(OAc)6 is the active precatalyst in the C–H activation of olefins.17
Trinuclear complexes
Over time, several complexes containing linear trinuclear palladium carboxylate motifs together with C–H activated ligands have been observed and characterized.11,18 These C(sp2)H and C(sp3)H activated complexes are summarized in Fig. 2. Analysis of the reaction conditions and stability of the compounds points to several common features. | ||
| Fig. 2 List of known types of linear trinuclear complexes. | ||
In all cases, the reaction conditions require a sub-stoichiometric or stoichiometric amount of substrate as compared to the amount of palladium carboxylate. Next, with the exception of olefins, all these complexes are relatively unstable. Finally, in many cases these complexes are found in combination with their binuclear analogues. There are only few studies reporting these complexes as reaction intermediates during the C–H activation.
Brown et al. observed in the reaction of olefins with palladium acetate in acetic acid solutions the formation of linear trinuclear complexes 11 that are gradually transformed to the binuclear ones.18a This transformation is reversible and treating binuclear complexes with palladium acetate leads to the formation of trinuclear complexes 11.
Analogous results were obtained by Váňa et al. in a detailed mechanistic study of C–H activation reaction of acetanilides with the palladium acetate/TFA system in DCM (Scheme 4).11 The NMR kinetics shows that linear trinuclear complex 12 is an intermediate in the reaction pathway leading to a stable binuclear palladacycle 13. The observed changes of abundance of trinuclear complex 12 caused by the change in the ratio of the mixed reactants lead to the suggestion that the transformation of trinuclear 12 to binuclear complex 13 is induced by the molecule of the nonactivated substrate. This could explain the fact that trinuclear complexes are isolated only rarely in C–H activations. Next, in a typical catalytic arrangement the palladium loading is only a few molar percent related to the substrate. Thus the unreacted substrate causes fast transformation of trinuclear complexes to the binuclear ones.
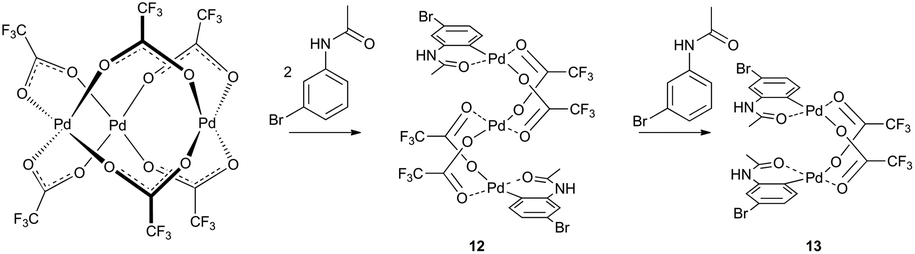 | ||
| Scheme 4 A simplified reaction scheme considering the reaction pathway proceeding via trifluoroacetate palladium complexes. Reproduced from ref. 11 with permission from the Royal Society of Chemistry. | ||
Another view on the whole issue is brought by the work of Houk, Yu and coworkers who prepared an acetate-bridged trinuclear complex 14 by the reaction of substituted oxazoline with palladium acetate in DCM.18c The comparison of the calculated reaction mechanisms proceeding via mononuclear and trinuclear species shows the mononuclear reaction pathway to be energetically favored (Scheme 5). The mononuclear reaction pathway leading to the trinuclear complex could be in principle described as follows: the trinuclear cyclic palladium acetate is attacked by molecules of oxazoline to give Pd(OAc)(μ-OAc)(oxazoline) 16. Next, the C–H activation step proceeds via the mononuclear transition state 17 and the monomeric palladacycle 18 is formed. Finally, after the release of acetic acid the stable trinuclear species 14 is formed. The alternative reaction pathway involving C–H activation directly from the trinuclear precomplexes 15 is less favorable than the mononuclear one due to the necessity of breaking of one of the bridged acetato groups in the trimer to generate a free coordination site on palladium 19. Finally, Vázquez-García et al. observed transformation of trinuclear complex 20 to binuclear one upon heating in toluene.18t
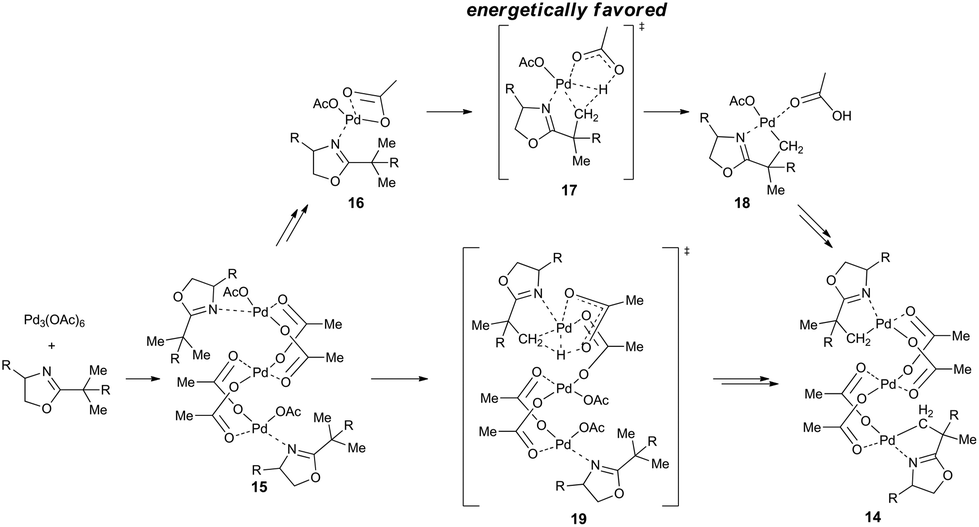 | ||
| Scheme 5 Mononuclear and trinuclear reaction pathways for the C–H activation of oxazolines. | ||
Conclusions and outlook
The presented results show that there is no comprehensive view on the whole issue of the precise nature of the key reaction species involved in the C–H activation reactions. Exactly the opposite, in many cases the results are contradictory. With no doubt, it can be concluded that in non-coordinating solvents and in stoichiometric amounts of palladium and substrate the polynuclear species are involved during the C–H activation processes. However, many important questions must be answered or clarified: is the nuclearity of the species involved in the C–H activation step dependent on the nature of the substrate, precatalyst, carboxylate ligand, or solvent or a combination of these factors? Is there any cooperation between palladium atoms in polynuclear species during the C–H activation step? Can C–H activation involving polynuclear complexes lead to different regioselectivities in comparison with the mononuclear pathway?19 How are reactivity and nuclearity influenced by the addition of carboxylates of different metals?20 What is the role of polynuclear complexes in the ongoing catalytic cycles of C–H functionalization reactions, where their formation is less probable? Answering all these and other related questions will require a lot of effort. However, we believe that this effort is crucial and will be reaped in the design of new catalysts and catalytic reactions and in many other ways.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Project GAČR 17-08499S.References
- Review: V. K. Jain and L. Jain, Coord. Chem. Rev., 2010, 254, 2848 CrossRef CAS.
- Review: D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed . Recent article: J. Váňa, V. Petrović, T. Terencio, O. Tischler, Z. Novák and J. Roithová, Organometallics, 2017, 36, 2072 CrossRef.
- Review: (a) T. A. Stromnova, Russ. J. Inorg. Chem., 2008, 53, 2019 CrossRef. Reactivities of binuclear complexes in further functionalization reactions were explored mainly by Sanford and Ritter. For an illustration see: (b) D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840 CrossRef CAS PubMed; (c) D. C. Powers, E. Lee, A. Ariafard, M. S. Sanford, B. F. Yates, A. J. Canty and T. Ritter, J. Am. Chem. Soc., 2012, 134, 12002 CrossRef CAS PubMed; (d) I. A. Sanhueza, A. M. Wagner, M. S. Sanford and F. Schoenebeck, Chem. Sci., 2013, 4, 2767 RSC.
- T. A. Stephenson, S. M. Morehouse, A. R. Powell, J. P. Heffer and G. Wilkinson, J. Chem. Soc., 1965, 3632 RSC.
- (a) A. C. Skapski and M. L. Smart, J. Chem. Soc., Chem. Commun., 1970, 658 RSC; (b) A. S. Batsanov, G. A. Timko, Y. T. Struchkov, N. V. Gerbeleu, K. M. Indrichan and G. A. Popovich, Koord. Khim., 1989, 15, 688 CAS.
- E. S. Stoyanov, J. Struct. Chem., 2000, 41, 440 CrossRef CAS.
- V. I. Bakhmutov, J. F. Berry, F. A. Cotton, S. Ibragimov and C. A. Murillo, Dalton Trans., 2005, 1989 RSC.
- (a) W. A. Carole and T. J. Colacot, Chem. – Eur. J., 2016, 22, 7686 CrossRef CAS PubMed; (b) W. A. Carole, J. Bradley, M. Sarwar and T. J. Colacot, Org. Lett., 2015, 17, 5472 CrossRef CAS PubMed; (c) R. B. Bedford, J. G. Bowen, R. B. Davidson, M. F. Haddow, E. A. Seymour-Julen, H. A. Sparkes and R. L. Webster, Angew. Chem., Int. Ed., 2015, 54, 6591 CrossRef CAS PubMed; (d) V. V. Tatarchuk, A. P. Sergievskaya, N. V. Kuratieva, I. V. Korol'kov, L. A. Sheludyakova and S. A. Gromilov, J. Struct. Chem., 2014, 55, 142 CrossRef CAS; (e) V. M. Nosova, Y. A. Ustynyuk, L. G. Bruk, O. N. Temkin, A. V. Kisin and P. A. Storozhenko, Inorg. Chem., 2011, 50, 9300 CrossRef CAS PubMed; (f) S. E. Bajwa, T. E. Storr, L. E. Hatcher, T. J. Williams, C. G. Baumann, A. C. Whitwood, D. R. Allan, S. J. Teat, P. R. Raithby and I. J. S. Fairlamb, Chem. Sci., 2012, 3, 1656 RSC.
- C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS PubMed.
- (a) O. Swang, R. Blom, O. B. Ryan and K. Fægri, J. Phys. Chem., 1996, 100, 17334 CrossRef CAS; (b) J. A. Tunge and L. N. Foresee, Organometallics, 2005, 24, 6440 CrossRef CAS.
- J. Váňa, J. Lang, M. Šoltésová, J. Hanusek, A. Růžička, M. Sedlák and J. Roithová, Dalton Trans., 2017, 46, 16269 RSC.
- T. Diao, P. White, I. Guzei and S. S. Stahl, Inorg. Chem., 2012, 51, 11898 CrossRef CAS PubMed.
- B. E. Haines, J. F. Berry, J.-Q. Yu and D. G. Musaev, ACS Catal., 2016, 6, 829 CrossRef CAS.
- J. Vicente, I. Saura-Llamas, M. G. Palin, P. G. Jones and M. C. R. de Arellano, Organometallics, 1997, 16, 826 CrossRef CAS.
- H. Font, M. Font-Bardia, K. Gómez, G. González, J. Granell, I. Machod and M. Martínez, Dalton Trans., 2014, 43, 13525 RSC.
- J. J. Gair, B. E. Haines, A. S. Filatov, D. G. Musaev and J. C. Lewis, Chem. Sci., 2017, 8, 5746 RSC.
- S. Winstein, J. McCaskie, H.-B. Lee and P. M. Henry, J. Am. Chem. Soc., 1976, 98, 6913 CrossRef CAS.
- (a) R. G. Brown, R. V. Chaudhari and J. M. Davidson, J. Chem. Soc., Dalton Trans., 1977, 176 RSC; (b) L. Y. Ukhin, N. A. Dologopolova, L. G. Kuzmina and Y. T. Struchkov, J. Organomet. Chem., 1981, 210, 263 CrossRef CAS; (c) R. Giri, Y. Lan, P. Liu, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 14118 CrossRef CAS PubMed; (d) R. Giri, X. Chen and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 2112 CrossRef CAS PubMed; (e) R. Giri, J. Liang, J.-G. Lei, J.-J. Li, D.-H. Wang, X. Chen, I. C. Naggar, C. Guo, B. M. Foxman and J.-Q. Yu, Angew. Chem., Int. Ed., 2005, 44, 7420 CrossRef CAS PubMed; (f) K. Guo, X. Chen, M. Guan and Y. Zhao, Org. Lett., 2015, 17, 1802 CrossRef CAS PubMed; (g) M. J. Tredwell, M. Gulias, N. G. Bremeyer, C. C. C. Johansson, B. S. L. Collins and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 1076 CrossRef CAS PubMed; (h) J. M. Chitanda, D. E. Prokopchuk, J. W. Quail and S. R. Foley, Organometallics, 2008, 27, 2337 CrossRef CAS; (i) R. E. Podobedov, T. A. Stromnova, A. V. Churakov, L. G. Kuzmina and I. A. Efimenko, J. Organomet. Chem., 2010, 695, 2083 CrossRef CAS; (j) M. T. Alonso, O. Juanes, J. de Mendoza and J. C. Rodriguez-Ubis, J. Organomet. Chem., 1992, 430, 349 CrossRef CAS; (k) F. K. Friedlein, K. Kromm, F. Hampel and J. A. Gladysz, Chem. – Eur. J., 2006, 12, 5267 CrossRef CAS PubMed; (l) F. K. Friedlein, F. Hampel and J. A. Gladysz, Organometallics, 2005, 24, 4103 CrossRef CAS; (m) A. Moyano, M. Rosol, R. M. Moreno, C. López and M. A. Maestro, Angew. Chem., Int. Ed., 2005, 44, 1865 CrossRef CAS PubMed; (n) Y. Fuchita, K. Hiraki and Y. Matsumoto, J. Organomet. Chem., 1985, 280, C51 CrossRef CAS; (o) M. Pfeffer, J. Organomet. Chem., 1985, 282, 127 CrossRef CAS; (p) G. Balavoine and J. C. Clinet, J. Organomet. Chem., 1990, 390, C84 CrossRef CAS; (q) D. H. R. Barton, J. Khamsi, N. Ozbalik and J. Reibenspies, Tetrahedron, 1990, 46, 3111 CrossRef CAS; (r) Y. Fuchita, M. Kawakami and K. Shimoke, Polyhedron, 1991, 10, 2037 CrossRef CAS; (s) J. E. Bercaw, M. W. Day, S. R. Golisz, N. Hazari, L. M. Henling, J. A. Labinger, S. J. Schofer and S. Virgil, Organometallics, 2009, 28, 5017 CrossRef CAS; (t) D. Vázquez-García, A. Fernández, M. López-Torres, A. Rodríguez, N. Gómez-Blanco, C. Viader, J. M. Vila and J. J. Fernández, Organometallics, 2010, 29, 3303 CrossRef.
- J. Milani, N. E. Pridmore, A. C. Whitwood, I. J. S. Fairlamb and R. N. Perutz, Organometallics, 2015, 34, 4376 CrossRef CAS.
- Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |