
Understanding the origins of Oyl–U–Oyl bending in the uranyl (UO22+) ion†

Abstract
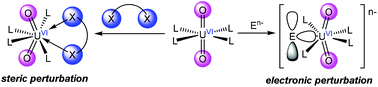
The structure of the uranyl ion (UO22+) has been the topic of investigation for almost a century. Since the first structural study of uranyl in 1935, over 4000 uranyl complexes have been characterized by X-ray crystallography. The vast majority of these complexes feature a linear uranyl group (e.g., Oyl–U–Oyl = 180°); however, there are a handful of complexes that feature much more acute Oyl–U–Oyl angles. In fact, the smallest experimentally observed Oyl–U–Oyl angles are ca. 161°. This Frontier Article catalogs every reported uranyl complex that features an Oyl–U–Oyl angle below 172°, and attempts to rationalize the origins of the observed Oyl–U–Oyl bending. In particular, I describe two distinct causes of Oyl–U–Oyl bending: (1) bending that occurs as a result of unfavourable steric interactions between the equatorial co-ligands and the uranyl oxo groups; and (2) bending that appears to have an electronic origin. In addition, I describe several possible avenues for future investigation. Understanding the effect that Oyl–U–Oyl bending has on uranyl electronic structure could ultimately provide insight into several unique aspects of the uranyl ion, such as the inverse trans influence and the involvement of the “pseudo-core” U6p orbitals in U–Oyl bonding.
- This article is part of the themed collections: Philip Power at 65: an icon of organometallic chemistry and 2018 Frontier and Perspective articles
 Please wait while we load your content...
Please wait while we load your content...

