
1,3,2-Diazaborole-derived carbene complexes of boron†

Abstract
Reaction of 2-bromo-1,3,2-diazaborole (1) with excess BBr3 induces 1,2-hydrogen migration, giving 1,3,2-diazaborole-derived carbene complexes of boron bromide (2). Compound 2 exists in a dynamic solution equilibrium with 1. The 1H NMR study shows that the equilibrium lies to the right side of the dissociation reaction of 2. Parallel reaction of 1 with excess BI3 gives the corresponding 1,3,2-diazaborole-derived carbene boron iodide complex (3). Notably, in contrast to 2, the dissociation reaction of 3 largely lies to the left side, favouring the formation of 3. The dynamic solution equilibrium behaviours of 2 and 3 are probed by both experimental and theoretical methods.
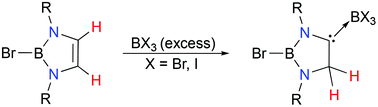
- This article is part of the themed collection: Philip Power at 65: an icon of organometallic chemistry
 Please wait while we load your content...
Please wait while we load your content...

