
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction of tin(IV) phthalocyanine dichloride with decamethylmetallocenes (M = CrII and CoII). Strong magnetic coupling of spins in (Cp*2Co+){SnIVCl2(Pc˙3−)}˙−·2C6H4Cl2†
Dmitri V.
Konarev
 *a,
Sergey I.
Troyanov
b,
Alexander F.
Shestakov
a,
Evgeniya I.
Yudanova
a,
Akihiro
Otsuka
cd,
Hideki
Yamochi
cd,
Hiroshi
Kitagawa
c and
Rimma N.
Lyubovskaya
a
*a,
Sergey I.
Troyanov
b,
Alexander F.
Shestakov
a,
Evgeniya I.
Yudanova
a,
Akihiro
Otsuka
cd,
Hideki
Yamochi
cd,
Hiroshi
Kitagawa
c and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, Moscow Region, 142432 Russia. E-mail: konarev3@yandex.ru
bChemistry Department, Moscow State University, Leninskie Gory, 119991 Moscow, Russia
cDivision of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
dResearch Center for Low Temperature and Materials Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
First published on 13th December 2017
Abstract
The reaction of tin(IV) phthalocyanine dichloride {SnIVCl2(Pc2−)} with decamethylmetallocenes (Cp*2M, M = Co, Cr) has been studied. Decamethylcobaltocene reduces SnIVCl2(Pc2−) to form the (Cp*2Co+){SnIVCl2(Pc˙3−)}˙−·2C6H4Cl2 (1) complex. The negative charge of {SnIVCl2(Pc˙3−)}˙− is delocalized over the Pc macrocycle providing the alternation of the C–N(imine) bonds, the appearance of new bands in the NIR range and a strong blue shift of both the Soret and Q-bands in the spectrum of 1. The magnetic moment of 1 is equal to 1.68μB at 300 K, indicating the contribution of one S = 1/2 spin of the Pc˙3− macrocycles. These macrocycles form closely packed double stacks in 1 with effective π–π interactions providing strong antiferromagnetic coupling of spins at a Weiss temperature of −80 K. Decamethylchromocene initially also reduces SnIVCl2(Pc2−) to form the [(Cp*2Cr+){SnVICl2(Pc˙3−)}˙− complex but further reaction between the ions is observed. This reaction is accompanied by the substitution of one Cp* ligand of Cp*2Cr by chloride anions originating from {SnIVCl2(Pc˙3−)}˙− to form the complex {(Cp*CrCl2)(SnIV(μ-Cl)(Pc2−))}·C6H4Cl2 (2) in which the (Cp*CrCl2) and {SnIV(Pc2−)} species are bonded through the μ-bridged Cl− anion. According to the DFT calculations, this reaction proceeds via an intermediate [(Cp*2CrCl)(SnClPc)] complex.
Introduction
Metal phthalocyanines form compounds showing promising optical, conducting and magnetic properties.1–6 They are used as sensors, and materials for optical and electronic devices.1,2 Conducting compounds have been obtained either by the oxidation of phthalocyanines by iodine or by the electrochemical oxidation of the [MIIIPcL2]− anions (M = Co, Fe; L = CN, Cl, Br).3–5 Since metal phthalocyanines can contain paramagnetic metals, they are also used as active components in the design of magnetic compounds. For example, polymeric compounds with alternating (MnIIIPc)+ and tetracyanoethylene (TCNE˙−) ions show ferrimagnetic ordering of spins.6Reduced metal phthalocyanines can also potentially show promising conducting and magnetic properties. For example, metallic conductivity is predicted for electron doped non-transition metal phthalocyanines.7 A series of salts with the reduced metal phthalocyanines is obtained as single crystals some of which show strong magnetic coupling between spins.8–13 The reduction of iron(II) phthalocyanine by decamethylchromocene (Cp*2Cr) allows one to obtain a complex with π-stacks of alternating FeIPc (S = 1/2) and Cp*2Cr+ (S = 3/2) ions which shows ferrimagnetic ordering of spins below 4.5 K.14 This approach was also applied for free-base phthalocyanine (H2Pc). However, in this case no essential magnetic coupling was found in (Cp*2Cr+)(H2Pc˙−)·4C6H4Cl2 (Weiss temperature is −4 K) which has similar π-stacks of alternating Cp*2Cr+ and H2Pc˙− ions. Providing more effective π–π overlapping between Cp*2Co+ and H2Pc˙−, stronger magnetic coupling between H2Pc˙− spins is attained in (Cp*2Co+)(H2Pc˙−)·0.5C6H4Cl2·0.7C6H5CN·0.3C6H14 at a Weiss temperature of −23 K.15
Here we study the interaction of strong organometallic donors – decamethylcobaltocene (Cp*2Co) and decamethylchromocene (Cp*2Cr)16 – with tin(IV) phthalocyanine dichloride {SnIVCl2(Pc2−)} which tend to form closely packed one- or two-dimensional structures from the radical anion macrocycles.13,17 Cp*2Co reduces {SnIVCl2(Pc2−)} to form the crystalline charge transfer (Cp*2Co+){SnIVCl2(Pc˙3−)}˙−·2C6H4Cl2 (1) complex whose crystal structure, and optical and magnetic properties are discussed. Cp*2Cr also reduces {SnIVCl2(Pc2−)}, but in contrast to Cp*2Co, the substitution of one Cp* ligand of Cp*2Cr by chloride anions is observed. This provides an unusual coordination compound {(Cp*CrCl2)(SnIV(μ-Cl)(Pc2−))}·C6H4Cl2 (2) in which two ions are bound through the μ-bridged chloride anion.
Results and discussion
Synthesis
All investigations were carried out under strictly anaerobic conditions in a glove box. The addition of Cp*2Co to {SnIVCl2(Pc2−)} in o-dichlorobenzene and the stirring of the solution provides complete dissolution of phthalocyanine with the formation of a deep blue solution characteristic of the reduced Pc macrocycle. Slow mixing of the obtained solution with n-hexane affords the crystals of 1 as black needles in high yield. These crystals are of small size and are suitable for X-ray diffraction analysis using synchrotron radiation. The mixing of Cp*2Cr and {SnIVCl2(Pc2−)} in the same solvent is also accompanied by the formation of a deep blue solution indicating the reduction of the Pc macrocycle. However, after 4 hours of stirring at 80 °C, the color of the solution turned green which is characteristic of the dianionic Pc2− macrocycle. A similar color change from deep blue to green is observed in the reaction of the {SnIVCl2(Pc˙3−)}˙− radical anions with some transition metal complexes accompanied by chloride anion abstraction and the formation of coordination complexes with neutral SnII(Pc2−).9 Obviously, further reaction between the Cp*2Cr+ cations and the {SnIVCl2(Pc˙3−)}˙− radical anions occurs (Scheme 1, see also the theoretical part in the ESI†). The solution was stirred for additional 20 hours at 80 °C, preserving green color. Slow mixing with n-hexane precipitates green powder with a small amount of green plate-like crystals which were studied by X-ray diffraction on single crystals using synchrotron radiation. Thus, the composition of 1 and 2 was determined from X-ray diffraction on single crystals. The elemental analysis of green powder shows that its composition is close to that of single crystals. We also studied the optical properties of compound 2 in o-dichlorobenzene. On combining these data with X-ray diffraction data and DFT calculations, it is possible to evaluate the electronic states of the components in 2.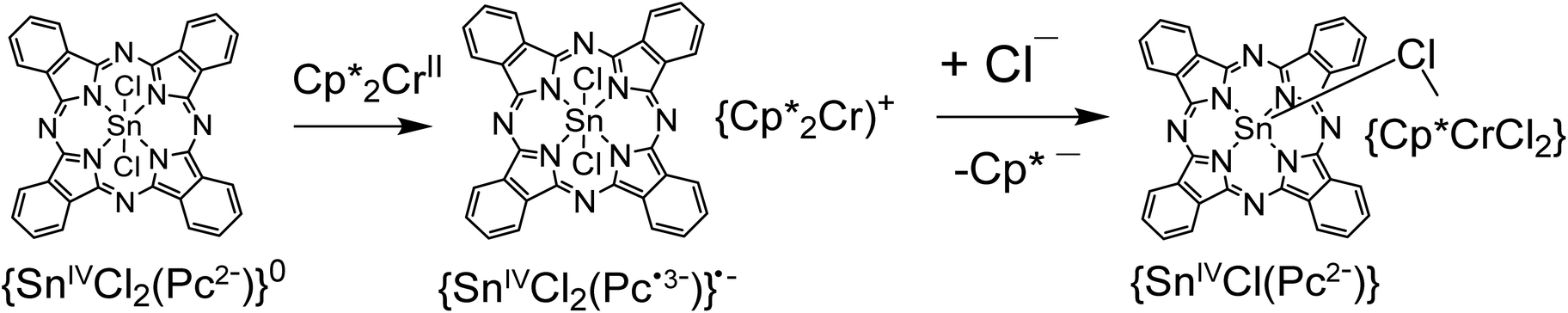 | ||
| Scheme 1 | ||
Crystal structures
Crystals of 1 and 2 were studied by X-ray diffraction at 100 K. Main structural blocks of these complexes except solvent C6H4Cl2 molecules are shown in Fig. 1. Complex 1 contains individual Cp*2Co+ cations and {SnIVCl2(Pc˙3−)}˙− radical anions. There is no π–π interaction between the Cp* ligand and the Pc˙3− macrocycle in 1 since the Cp*2Cr+ cations are oriented by methyl substituents towards the Pc˙3− plane. At the same time, these components are oriented in such a way that relatively close distances between the positively charged CoIII and negatively charged chloride anions of {SnIVCl2(Pc˙3−)}˙− are formed (5.6–5.8 Å) (Fig. 1a). The average length of the Co–C(Cp*) bonds for Cp*2Co in 1 is 2.046(3) Å. This length corresponds to the formation of the Cp*2Co+ cations which have the average Co–C(Cp*) bond length of 2.04–2.05 Å,18 whereas neutral Cp*2Co has longer Co–C(Cp*) bonds of 2.101(3) Å.19 The geometry of the Pc˙3− macrocycle is shown in Fig. 1a. There are two types of the C–N bonds with imine and pyrrole nitrogen atoms of Pc. Longer C–N(pyrrole) bonds with a length of 1.381(4) Å have no alternation. Shorter C–N(imine) bonds alternate in such a way that four bonds belonging to two oppositely located isoindole units are short (average length is 1.319(4) Å) and four other bonds are long (1.350(4) Å), and the difference is 0.031 Å. Such alternation is explained by the partial disruption of aromaticity of the Pc macrocycle in the formation of a less stable 19-π-electron system of Pc˙3−.20 The average length of the Sn–Cl bonds is 2.485(1) Å and that of the Sn–N(pyrrole) bonds is 2.049(3) Å. These lengths are rather close to those in pristine {SnIVCl2(Pc2−)}0 – 2.470(2) and 2.054(2) Å, respectively.21 The tin(IV) atom is positioned not exactly in the 24-atom Pc plane but is slightly displaced out of this plane by 0.034 Å. The reason for this is a slightly non-planar saddle-like shape of Pc˙3− with two phenylene groups located above the 24-atom Pc plane and two such groups located below this plane.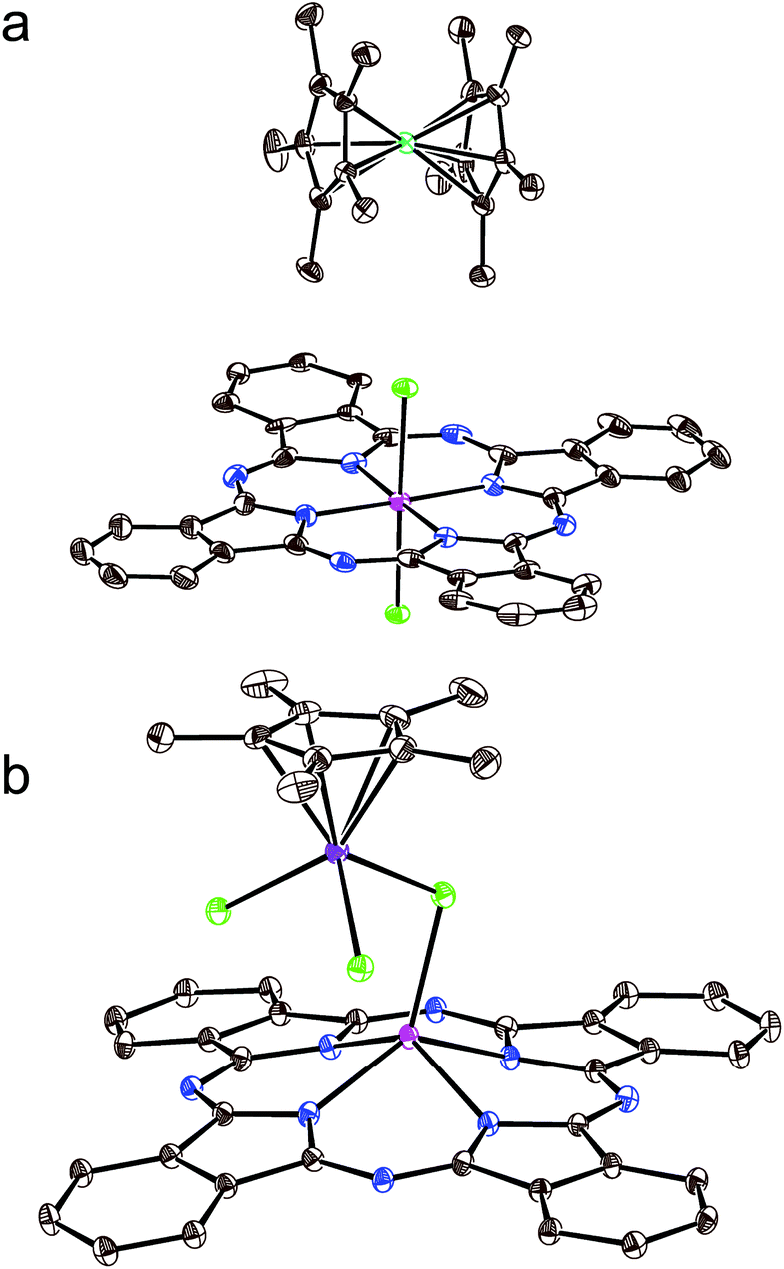 | ||
| Fig. 1 Molecular structures of the components for complexes 1 and 2 at 100 K: (a) the Cp*2Co+ cations and the {SnIVCl2(Pc˙3−)}˙− radical anions in 1; (b) the Cp*CrCl2 units in 2 bridged with SnIV(Pc2−) by the μ-chloride anion. Solvent C6H4Cl2 molecules are not shown for clarity. Ellipsoid probability is 25%. | ||
The crystal structure of 1 is shown in Fig. 2. It contains large channels occupied by the Cp*2Co+ cations (Fig. 2a). The walls of these channels are formed by double stacks from the closely packed Pc˙3− macrocycles (Fig. 2). There is an effective π–π interaction between Pc˙3− in these double chains since the phenylene group of one macrocycle is positioned over the phenylene group of the neighboring macrocycle. The planes of these groups are nearly parallel to each other with a dihedral angle of only 2.34°, and many short van der Waals C⋯C contacts are formed between the phenylene substituents of the Pc˙3− macrocycles in the 3.45–3.54 Å range (Fig. 2b). Such packing is observed for the first time for the radical anion salts with {SnIVCl2(Pc˙3−)}˙− since previously only closely packed single stacks17 and 2D phthalocyanine layers13 from {SnIVCl2(Pc˙3−)}˙− were observed.
 | ||
| Fig. 2 (a) View along the double phthalocyanine stacks and crystallographic a axis in 1. Pairs of {SnIVCl2(Pc˙3−)}˙− radical anions belonging to one stack are marked as orange ellipses; (b) view on the double stacks from {SnIVCl2(Pc˙3−)}˙− arranged in the ab plane. Short van der Waals C⋯C contacts between the Pc˙3− macrocycles are shown as green dashed lines. | ||
The molecular structure of the {(Cp*CrCl2){SnIV(μ-Cl)(Pc2−)}} units formed in 2 is shown in Fig. 1b. The chromium atom coordinates to one Cp* ligand by η5-type with an average length of the Cr–C(Cp*) bonds of 2.243(6) Å. These bonds are noticeably longer than the length of the Cr–C(Cp*) bonds in pristine Cp*2Cr (2.152(4) Å)22 and the Cp*2Cr+ cations (2.176(3)–2.198(3) Å).15,23,24 Moreover, three chloride anions are coordinated to the chromium atom. Two anions have the length of the Cr–Cl bonds of 2.313(1) and 2.318(1) Å but the third anion which is coordinated to both tin(IV) and chromium atoms as a μ-bridge has the length of the Cr–Cl bond of 2.393(1) Å. Since a shorter Sn–Cl bond between this chloride anion and {SnIV(Pc2−)} is observed (2.313(1) Å), it can formally be attributed to tin(IV) phthalocyanine {SnIV(μ-Cl)(Pc2−)}. Previously, the geometry of the (Cp*CrIICl)2 and (Cp*CrIIICl2)2 dimers bonded by μ-bridged chloride anions and that of the (Indigo-O,O)(Cp*CrIICl) complex are known.24–26 The average lengths of the Cr–Cl and Cr–C(Cp*) bonds are 2.331(2) (μ) and 2.251(2) Å for the first compound,24 2.396(3) (μ), 2.288(3) and 2.233(3) Å for the second compound,25 and 2.396(1) and 2.247(2) Å for the third compound,26 respectively. The lengths of these bonds in the (Cp*CrIIICl3)− anions in (Cp*2Cr)+(Cp*CrIIICl3)− are 2.324(3) and 2.253(3) Å,27 respectively. It is seen that the charged state of chromium does not noticeably affect the Cr–Cl and Cr–C(Cp*) bond lengths. Nevertheless, the presence of CrIII requires the formation of the {SnIV(μ-Cl)(Pc˙3−)}0 units but that is not agreed with the experimental data as we show below. Therefore, the (Cp*CrIICl2)− anions and the {SnIV(μ-Cl)(Pc2−)}+ cations are formed in 2.
There are also C–N bonds with imine and pyrrole nitrogen atoms in SnClPc in 2. The length of all eight C–N(pyrrole) bonds is centered at 1.381(7) Å. In contrast to 1, the alternation of the C–N(imine) bonds is absent in 2 since oppositely located short and long bonds have the length of 1.319(6) and 1.326(6) Å, respectively. The difference of 0.007 Å is comparable to the error for the length of these types of bonds and is essentially less than such a difference in 1 (0.031 Å). The absence of alternation justifies the formation of a dianionic Pc2− macrocycle in 2 and that corresponds well to the green color of compound 2. Thus, we suppose that coordination units in 2 consists of the (Cp*CrIICl2)− and {SnIV(μ-Cl)(Pc2−)}+ ions μ-bridged through the chloride anion. Since the chloride anion is coordinated to the tin(IV) atom only from one side, it displaces towards the chloride anion by 0.98 Å (relative to the 24-atom Pc plane), and the average Sn–N(Pc) bonds are also essentially elongated up to 2.141(4) Å. As a result, the Pc macrocycle has concave conformation with the deviation of all four phenylene groups of Pc to one side relative to the 24-atom Pc plane. The {SnIVCl(Pc2−)}+ cation observed in 2 is a rare example of pentacoordinated tin(IV) atoms in tin(IV) phthalocyanine which has some similarities with the {SnIVPh(Pc2−)}+ cations also containing dianionic Pc2− macrocycles.28
The {(Cp*CrCl2){SnIVCl(Pc2−)}} units in 2 form chains arranged along the c axis in which closely packed pairs of the Pc2− macrocycles with short van der Waals C⋯C contacts can be outlined (Fig. S5†).
Optical properties
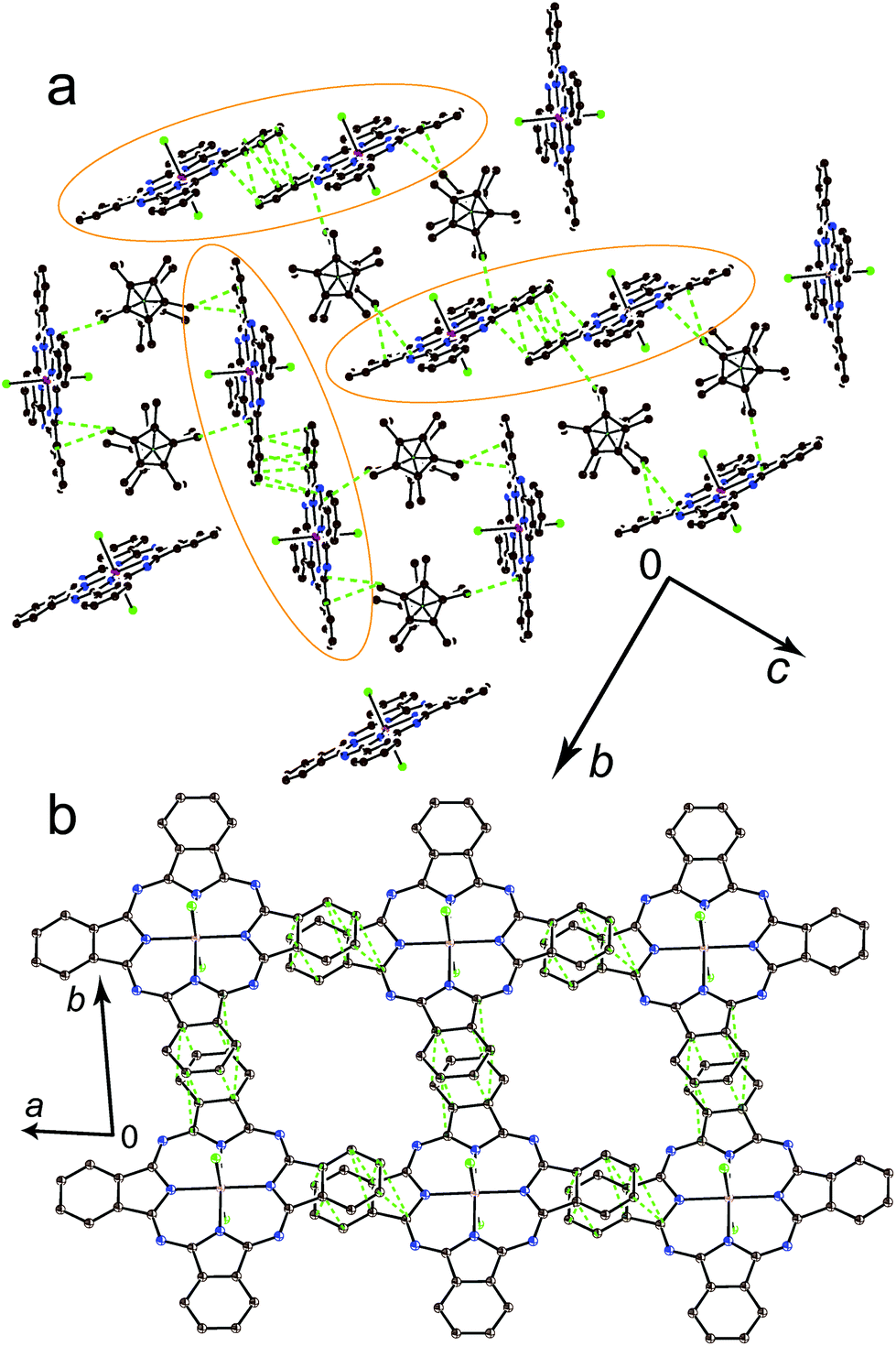
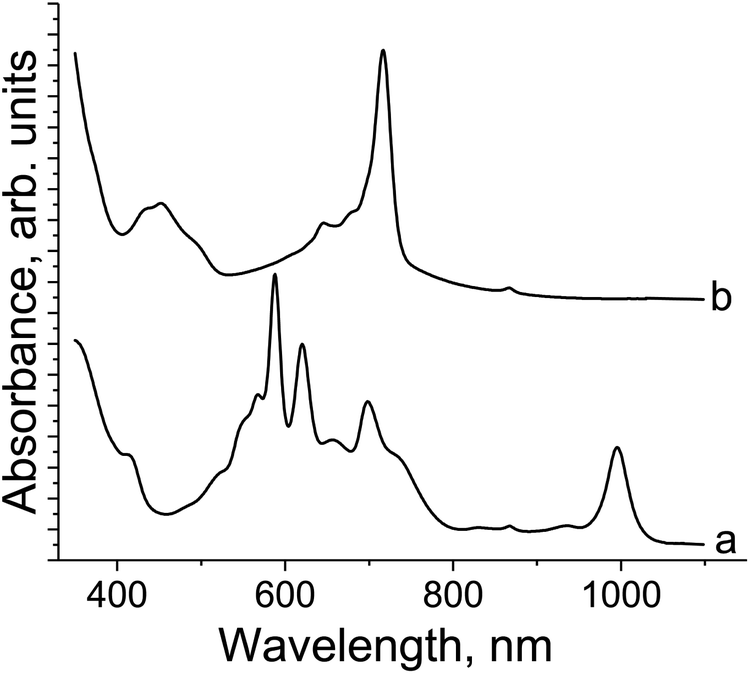
The spectrum of 1 in the UV-visible-NIR range is shown in Fig. 3. Pristine {SnIVCl2(Pc2−)}0 shows Soret and split Q-bands at 381 and 670, 740 (max), and 848 nm, respectively (Fig. 3, curve a). The formation of 1 is accompanied by the appearance of new bands in the NIR range at 1010 nm and a weaker band is observed at 840 nm. Both the Soret and Q-bands are noticeably blue shifted in the spectrum of 1 appearing at 338 nm and 712, 627 (max), and 600 nm, respectively (Fig. 3, curve b). Such behavior is generally observed in the formation of the {M(Pc˙3−)}˙− radical anions with the radical trianionic Pc˙3− macrocycles.8,9,11–13,15,17 | ||
| Fig. 3 UV-visible-NIR spectra of: (a) pristine {SnIVCl2(Pc2−)}0 and (b) the complex (Cp*2Co+){SnIVCl2(Pc˙3−)}˙−·2C6H4Cl2 (1) in KBr pellets prepared under anaerobic conditions. | ||
We also recorded the spectra in the o-dichlorobenzene solution of {SnIVCl2(Pc2−)}0 reduced by one equivalent of Cp*2Co (Fig. 4a) or Cp*2Cr (Fig. 4b) by the stirring of the solution at 80 °C for one day and filtering. The spectra show different states of complexes 1 and 2. Complex 1 has deep blue color in solution and manifests bands at 410, 588, 621, 657, 698 and 996 nm. The latter band in the NIR range and an essential blue shift of both the Soret and Q-bands are the signs of the formation of the Pc˙3− macrocycles. In contrast, the spectrum of the green solution of 2 contains bands only at 452 and 716 nm without any new bands in the NIR range. This spectrum indicates the formation of the Pc2− macrocycle in 2.
 | ||
| Fig. 4 UV-visible-NIR spectra in the o-dichlorobenzene solution of: {SnIVCl2(Pc2−)}0 reduced by one equivalent of Cp*2Co (a) and Cp*2Cr (b) after 1 day of stirring at 80 °C. | ||
Magnetic properties
The magnetic properties of 1 were studied by SQUID and EPR techniques. The effective magnetic moment of 1 is equal to 1.68μB at 300 K (Fig. 5a), indicating the contribution of one S = 1/2 spin of the Pc˙3− macrocycles (the calculated value for the system of one noninteracting S = 1/2 spin is 1.73μB). The reciprocal molar magnetic susceptibility is linear in the 130–300 K range allowing one to determine the Weiss temperature to be −80 K (Fig. 5b), indicating strong antiferromagnetic coupling of spins. Deviation towards the antiferromagnetic side from the Curie–Weiss law is observed below 130 K. However, long range antiferromagnetic ordering of spins is not observed down to 1.9 K. Strong antiferromagnetic coupling of spins is probably the reason for a lower magnetic moment of 1 in comparison with the calculated value and the decreases of the magnetic moment even below 300 K (Fig. 5a). Effective π–π interactions realized between the Pc˙3− macrocycles in the closely packed double chains of 1 can explain strong antiferromagnetic coupling between the spins.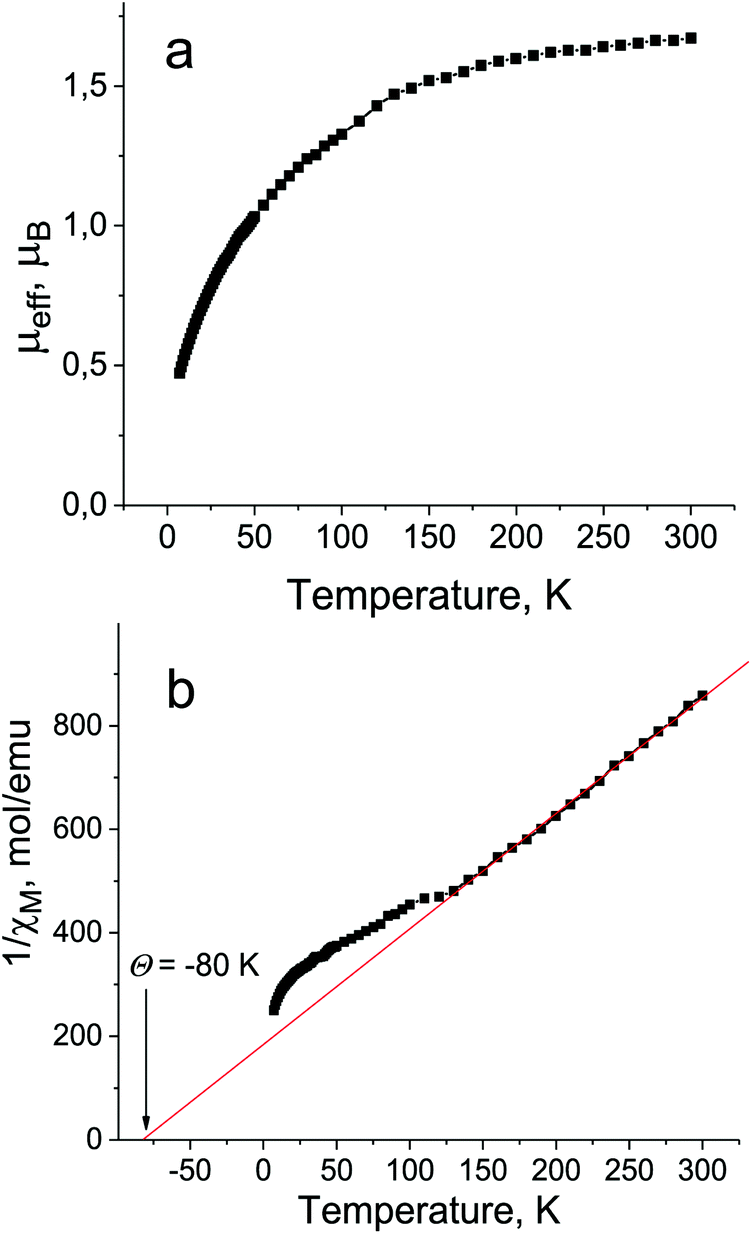 | ||
| Fig. 5 Temperature dependencies of the effective magnetic moment (a) and reciprocal molar magnetic susceptibility (b) for polycrystalline 1 in the 1.9–300 K range. | ||
The EPR spectra of 1 were studied in the 4–295 K range. The complex shows a broad single Lorentzian line with a g-factor of 1.9958 and a linewidth (ΔH) of 16.1 mT at 295 K. The signal can be attributed to the {SnIVCl2(Pc˙3−)}˙− radical anions. Similar broad EPR signals were previously observed for other salts with the {SnIVCl2(Pc˙3−)}˙− radical anions.13,17 The signal strongly narrows and the g-factor shifts to the smaller values with the temperature decrease (spectrum at 120 K is shown Fig. S6†). The signal splits into two lines below 30 K (Fig. 6a) which are broadened and shift strongly to the smaller and larger g-factors with the temperature decrease (Fig. 6b and c). The broadening of lines and shift of their g-factors can be attributed to strong antiferromagnetic coupling of spins. The split signal at 9 K is shown in Fig. 6a and the parameters of the lines are g1 = 1.9780, ΔH = 3.2 mT and g2 = 1.9912, ΔH = 2.6 mT. The EPR spectrum of {SnIVCl2(Pc˙3−)}˙− generated by Cp*2Co was studied in o-dichlorobenzene at 77 K (Fig. S7†), and the signals were not found at room temperature. The isolated {SnIVCl2(Pc˙3−)}˙− radical anions show an intense strongly asymmetric EPR signal at 77 K which can be fitted by three Lorentzian lines with g1 = 2.0050, ΔH = 0.482 mT; g2 = 2.0021, ΔH = 0.924 mT; and g3 = 1.9942, ΔH = 2.704 mT. The ratio of the integral intensities of the lines is 13/20/67% for g1, g2 and g3, respectively (Fig. S7†). It is seen that lines are essentially narrower at 77 K in solution in comparison with the solid state. These data also show that the asymmetry of the EPR signal of 1 is intrinsic and can be realized, for example, due to the static distortion of Pc˙3− at low temperatures. The behavior of the EPR signal in 1 correlates well with the behavior of salts with the radical anions of aluminium(III), gallium(III) and indium(III) phthalocyanines which show strong broadening of the EPR signal with the increasing size of the metal atom, and g-factors are noticeably lower than 2.000 and the signal shows strong asymmetry at low temperatures.12 The linewidth of the EPR signal in 1 at 295 K is comparable to that of the EPR signal from {InIIIBr(Pc˙3−)}˙− radical anions.12 The Cp*2Co+ cations with CoIII are diamagnetic and do not contribute to the EPR signal of 1.15,18
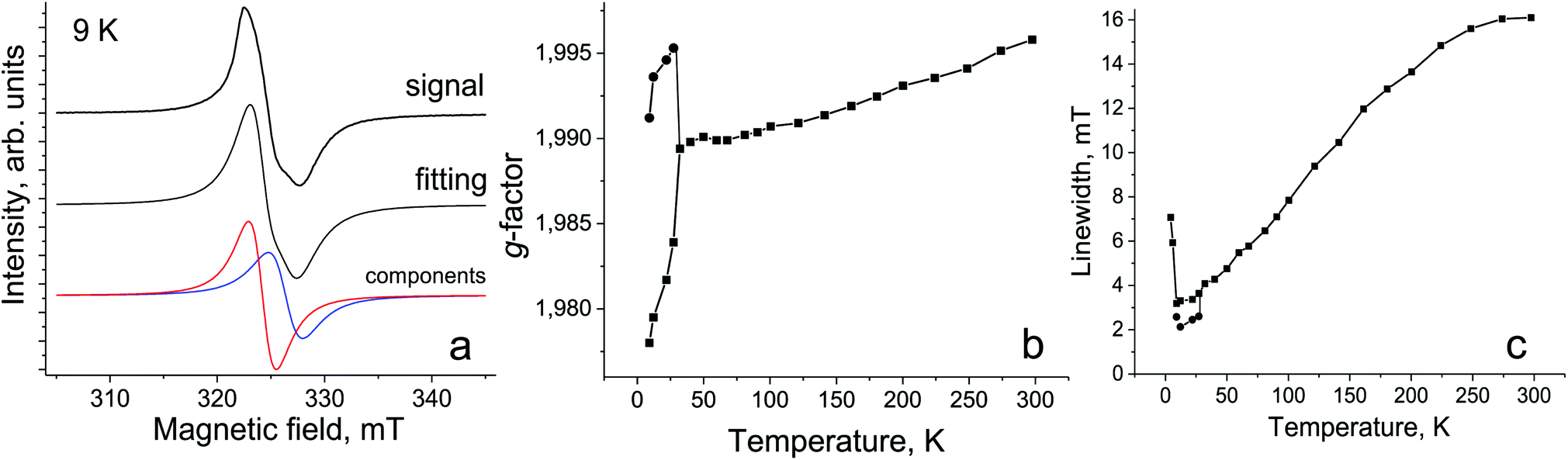 | ||
| Fig. 6 (a) EPR signal from polycrystalline 1 at 9 K. The fitting of the signal by two Lorentzian lines is shown below; temperature dependencies of the g-factor (b) and the linewidth (c) of the EPR signal in 1. The signal is split into two lines below 30 K. | ||
Experimental
Materials
Tin(IV) phthalocyanine dichloride {SnIVCl2(Pc2−)} was purchased from TCI. Decamethylchromocene (Cp*2Cr, >95%) was purchased from Strem and decamethylcobaltocene (Cp*2Co) was purchased from Aldrich. o-Dichlorobenzene (C6H4Cl2) was distilled over CaH2 under reduced pressure; n-hexane was distilled over Na/benzophenone. All operations on the synthesis of 1 and 2 and their storage were carried out in a MBraun 150B-G glove box with a controlled atmosphere and the water and oxygen content less than 1 ppm. The solvents were degassed and stored in the glove box. KBr pellets for IR- and UV-visible-NIR measurements were prepared in the glove box. The polycrystalline sample of 1 was placed in 2 mm quartz tubes under anaerobic conditions for EPR and SQUID measurements and sealed under 10−5 torr pressure.General
UV-visible-NIR spectra were recorded in KBr pellets on a PerkinElmer Lambda 1050 spectrometer in the 250–2500 nm range. FT-IR spectra (400–7800 cm−1) were recorded in KBr pellets with a PerkinElmer Spectrum 400 spectrometer. EPR spectra were recorded for polycrystalline samples of 1 with a JEOL JES-TE 200 X-band ESR spectrometer equipped with a JEOL ES-CT470 cryostat working between room and liquid helium temperatures. A Quantum Design MPMS-XL SQUID magnetometer was used to measure the static magnetic susceptibility of 1 at 100 mT magnetic field under cooling and heating conditions in the 300–1.9 K range. A sample holder contribution and core temperature independent diamagnetic susceptibility (χd) were subtracted from the experimental values. The χd values were estimated by the extrapolation of the data in the high-temperature range by fitting the data with the following expression: χM = C/(T − Θ) + χd, where C is the Curie constant and Θ is the Weiss temperature. The effective magnetic moment (μeff) was calculated with the following formula: μeff = (8·χMT)1/2.Synthesis
The crystals of (Cp*2Co+){SnIVCl2(Pc˙3−)}˙−·2C6H4Cl2 (1) were obtained by the following procedure. 29.2 mg of SnIVCl2(Pc2−) (0.042 mmol) was reduced by a slight excess of Cp*2Co (15 mg, 0.046 mmol) in 16 mL of o-dichlorobenzene upon stirring at 80 °C for 24 hours. Immediately after mixing the components, deep blue-colour of the solution formed which remained unchanged for 24 hours. Phthalocyanine was completely dissolved, the solution was cooled and filtered into a 50 mL glass tube of 1.8 cm diameter with a ground glass plug, and 30 mL of n-hexane was layered over the solution. The crystals were precipitated on the walls of the tube for 1.5 month. Then, the solvent was decanted from the crystals and they were washed with n-hexane to afford black needles in 76% yield. The composition of the crystals was determined from X-ray diffraction on single crystals using synchrotron radiation. Elemental analysis cannot be used to determine the composition of the obtained crystals due to their high air sensitivity (i.e., addition of oxygen to the samples during the procedure of elemental analysis).The crystals of {(Cp*CrCl2)(SnIVCl(Pc2−))}·C6H4Cl2 (2) were obtained similarly. 29.2 mg of SnIVCl2(Pc2−) (0.042 mmol) was reduced by a slight excess of Cp*2Cr (15 mg, 0.046 mmol) in 16 mL of o-dichlorobenzene upon stirring at 80 °C for 24 hours. Immediately after mixing of the components, deep blue-colour of the solution appeared indicating the reduction of the Pc macrocycle. However, after about 4 hours, the colour of the solution turned green and remained unchanged upon stirring at 80 °C for additional 20 hours. Phthalocyanine was completely dissolved, the solution was cooled and filtered into a 50 mL glass tube of 1.8 cm diameter with a ground glass plug, and 30 mL of n-hexane was layered over the solution. In 1 month, the solvent was decanted to yield green precipitates together with a small amount of dark green plates. They were washed with n-hexane. The composition of crystals was determined from X-ray diffraction on single crystals using synchrotron radiation and was confirmed by elemental analysis. Anal. calcd for C48H35Cl5CrN8Sn, Mr = 1071.78: C 53.74; H 3.27; Cl 16.56; N 10.45; found: C 53.08; H 3.18; N 10.34.
Theoretical calculations
The PBE density functional method30 and Λ2 basis31 of cc-pVTZ quality were used for theoretical calculations. See the ESI† for details.X-ray crystal structure determination
Crystal data for 1 at 100(2) K: C64H54Cl6CoN8Sn, F.W. 1325.47, black needle, 0.05 × 0.02 × 0.01 mm3, monoclinic, space group P21/c, a = 10.676(1), b = 28.916(3), c = 18.615(2) Å, β = 98.234(10)°, V = 5687.3(10) Å3, Z = 4, dcalcd = 1.548 M gm−3, μ = 1.680 mm−1, F(000) = 2692, λ = 0.84344 Å, 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 516 reflections collected, 13710 independent; R1 = 0.0529 for 12829 observed data [>2σ(F)] with 73 restraints and 946 parameters; wR2 = 0.1156 (all data); G.o.F. = 1.018. CCDC 1574909.†
516 reflections collected, 13710 independent; R1 = 0.0529 for 12829 observed data [>2σ(F)] with 73 restraints and 946 parameters; wR2 = 0.1156 (all data); G.o.F. = 1.018. CCDC 1574909.†
Crystal data for 2 at 100(2) K: C48H35Cl5CrN8Sn, F.W. 1071.78, dark-green plate, 0.03 × 0.02 × 0.005 mm3, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.086(1), b = 13.211(1), c = 16.647(1) Å, α = 82.580(6), β = 78.880(7), γ = 85.730(5)°, V = 2155.5(3) Å3, Z = 2, dcalcd = 1.651 M gm−3, μ = 2.070 mm−1, F(000) = 1076, λ = 0.87313 Å, 30393 reflections collected, 8157 independent; R1 = 0.0590 for 7316 observed data [>2σ(F)] with 573 parameters; wR2 = 0.1561 (all data); final G.o.F. = 1.028. CCDC 1574910.†
, a = 10.086(1), b = 13.211(1), c = 16.647(1) Å, α = 82.580(6), β = 78.880(7), γ = 85.730(5)°, V = 2155.5(3) Å3, Z = 2, dcalcd = 1.651 M gm−3, μ = 2.070 mm−1, F(000) = 1076, λ = 0.87313 Å, 30393 reflections collected, 8157 independent; R1 = 0.0590 for 7316 observed data [>2σ(F)] with 573 parameters; wR2 = 0.1561 (all data); final G.o.F. = 1.028. CCDC 1574910.†
The intensity data for 1 and 2 were collected on a MAR225 CCD detector using synchrotron radiation at the BESSY storage ring, BL 14.2 (PSF of the Free University of Berlin, Germany). The structures were solved by direct methods and refined by the full-matrix least-squares method against F2 using SHELX-2014/7 packages.32 All non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms were included into refinement in a riding model. The crystal structure of 1 contains disordered components. The Cp*2Co+ cation is rotationally disordered between two orientations with the 0.737(14)/0.262(14) occupancies. They are related to the rotation of the cation about the axis passing through the centres of Cp rings and the Co atom by about 30°. One phenylene group of Pc is disordered over two positions with the 0.64(5)/0.36(5) occupancies. o-Dichlorobenzene is statistically disordered between two orientations.
Conclusions
The study on the reaction of tin(IV) phthalocyanine dichloride with strong organometallic donors decamethylmetallocenes (M = Co, Cr) shows an essential difference between two decamethylmetallocenes. Decamethylcobaltocene reduces this phthalocyanine to the radical anion state forming a CT complex. Close packing of the radical trianionic Pc˙3− macrocycles in double stacks of 1 allows the observation of effective antiferromagnetic coupling of spins without their ordering down to 1.9 K. In contrast, decamethylchromocene can abstract the chloride anion from the {SnIVCl2(Pc˙3−)}˙− radical anions to form an unusual coordination compound in which the Cp*CrCl2 and SnIV(Pc2−) units are μ-bridged through the chloride anion. Previously, the substitution of the Cp* ligand in Cp*2Cr+ was demonstrated by indigo26 and thioindigo29 ligands, and the elimination of chloride anions from {SnIVCl2(Pc˙3−)}˙− is observed in the interaction of this radical anion with some transition metal complexes having metals in the low oxidation state, for example, for {CpMoI(CO)3}2.9 Compound 2 could show interesting optical and magnetic properties since in accordance with the DFT calculations it has the closely lying excited CT state in which the electron moves from Cr(II) to the Pc2− macrocycle of {SnIVCl(Pc2−)}+. The study on the optical and magnetic properties of 2 is in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by Russian Science Foundation grant no. 17-13-01215, and by JSPS KAKENHI Grant Number JP26288035, and the JST (ACCEL) 27 (100150500010) project.References
- C. G. Claessens, W. J. Blau, M. Cook, M. Hanack, R. J. M. Nolte, T. Torres and D. Wöhrle, Monatsh. Chem., 2001, 132, 3 CrossRef CAS.
- D. Wöhrle, G. Schnurpfeil, S. G. Makarov, A. Kazarin and O. N. Suvorova, Macroheterocycle, 2012, 5, 191 CrossRef.
- J. L. Petersen, C. S. Schramm, D. R. Stojakovic, B. M. Hoffman and T. J. Marks, J. Am. Chem. Soc., 1977, 99, 286 CrossRef CAS.
- H. Hasegawa, T. Naito, T. Inabe, T. Akutagawa and T. Nakamura, J. Mater. Chem., 1998, 8, 1567 RSC.
- T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503 CrossRef CAS PubMed.
- J. S. Miller, C. Vazquez, J. C. Calabrese, M. L. McLean and A. J. Epstein, Adv. Mater., 1994, 6, 217 CrossRef CAS.
- E. Tosatti, M. Fabrizio, J. Tóbik and G. E. Santoro, Phys. Rev. Lett., 2004, 93, 117002 CrossRef PubMed.
- D. V. Konarev, A. V. Kuzmin, M. A. Faraonov, M. Ishikawa, Y. Nakano, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Eur. J., 2015, 21, 1014 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, Y. Nakano, M. A. Faraonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2016, 55, 1390 CrossRef CAS PubMed.
- D. V. Konarev, L. V. Zorina, M. Ishikawa, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Cryst. Growth Des., 2013, 13, 4930 CAS.
- D. V. Konarev, S. S. Khasanov, A. V. Kuzmin, Y. Nakano, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Cryst. Growth Des., 2017, 17, 753 CAS.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Asian J., 2017, 12, 910 CrossRef CAS PubMed.
- D. V. Konarev, M. A. Faraonov, A. V. Kuzmin, S. S. Khasanov, Y. Nakano, M. S. Batov, S. I. Norko, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, New J. Chem., 2017, 41, 6866 RSC.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, E. U. Hakimova and R. N. Lyubovskaya, New J. Chem., 2012, 36, 48 RSC.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2017, 46, 3492 RSC.
- J. L. Robbins, N. Edelstein, B. Spencer and J. C. Smart, J. Am. Chem. Soc., 1982, 104, 1882 CrossRef CAS.
- D. V. Konarev, S. I. Troyanov, M. Ishikawa, M. A. Faraonov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, J. Porphyrins Phthalocyanines, 2014, 18, 1157 CrossRef CAS.
- (a) D. A. Dixon and J. S. Miller, J. Am. Chem. Soc., 1987, 109, 3656 CrossRef CAS; (b) D. Braga, O. Benedi, L. Maini and F. Grepioni, J. Chem. Soc., Dalton Trans., 1999, 2611 RSC; (c) D. V. Konarev, S. S. Khasanov, G. Saito, I. I. Vorontsov, A. Otsuka, R. N. Lyubovskaya and Y. M. Antipin, Inorg. Chem., 2003, 42, 3706 CrossRef CAS PubMed.
- M. M. Clark, W. W. Brennessel and P. L. Holland, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m391 CAS.
- J. A. Cissell, T. P. Vaid and A. L. Rheingold, Inorg. Chem., 2006, 45, 2367 CrossRef CAS PubMed.
- J. Janczak and R. Kubiak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m237 Search PubMed.
- J. Blümel, M. Herker, W. Hiller and F. H. Köhler, Organometallics, 1996, 15, 3474 CrossRef.
- (a) J. S. Miller, R. S. Mclean, C. Vazquez, J. C. Calabrese, F. Zuo and A. J. Epstein, J. Mater. Chem., 1993, 3, 215 RSC; (b) D. V. Konarev, S. S. Khasanov, A. Otsuka and G. Saito, J. Am. Chem. Soc., 2002, 124, 8520 CrossRef CAS PubMed.
- R. A. Heintz, R. L. Ostrander, A. L. Rheingold and K. H. Theopold, J. Am. Chem. Soc., 1994, 116, 11387 CrossRef CAS.
- F. H. Köhler, J. Lachmann, G. Müller and H. Zeh, J. Organomet. Chem., 1989, 365, C15 CrossRef.
- D. V. Konarev, S. S. Khasanov, A. V. Kuzmin, A. F. Shestakov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2016, 45, 17095 RSC.
- S. Aldridge, M. Shang and T. P. Fehlner, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 47 Search PubMed.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2016, 45, 10780 RSC.
- D. V. Konarev, S. S. Khasanov, A. F. Shestakov, A. M. Fatalov, A. Otsuka, H. Yamochi, H. Kitagawa and R. N. Lyubovskaya, Dalton Trans., 2017, 46, 14365 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- D. N. Laikov, Chem. Phys. Lett., 2005, 416, 116 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of theoretical calculations, IR spectra and crystal structure of 2. CCDC 1574909 and 1574910. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt03807k |
| This journal is © The Royal Society of Chemistry 2018 |