
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Induced chirality of cage metal complexes switched by their supramolecular and covalent binding†
Vladyslava B.
Kovalska
 ab,
Serhii V.
Vakarov
c,
Marina V.
Kuperman
a,
Mykhaylo Y.
Losytskyy
a,
Elzbieta
Gumienna-Kontecka
d,
Yan Z.
Voloshin
ef and
Oleg A.
Varzatskii
*bc
ab,
Serhii V.
Vakarov
c,
Marina V.
Kuperman
a,
Mykhaylo Y.
Losytskyy
a,
Elzbieta
Gumienna-Kontecka
d,
Yan Z.
Voloshin
ef and
Oleg A.
Varzatskii
*bc
aInstitute of Molecular Biology and Genetics NASU, 150 Zabolotnogo St., 03143 Kiev, Ukraine
bSC Princeton Biomolecular Research Labs, Saperne pole st., 26A, 01042, Kyiv, Ukraine. E-mail: connector9@gmail.com
cVernadskii Institute of General and Inorganic Chemistry NASU, 32/34 Palladin Av., 03080 Kiev, Ukraine
dFaculty of Chemistry, University of Wroclaw, 14 F. Joliot-Curie St., 50-383 Wroclaw, Poland
eNesmeyanov Institute of Organoelement Compounds RAS, 28 Vavilova St., 119991 Moscow, Russia
fKurnakov Institute of General and Inorganic Chemistry RAS, 31 Leninsky Prosp., 119991 Moscow, Russia
First published on 19th December 2017
Abstract
An ability of the ribbed-functionalized iron(II) clathrochelates to induce a CD output in interactions with a protein, covalent bonding or supramolecular interactions with a low-molecular-weight chiral inductor, was discovered. The interactions of CD inactive, carboxyl-terminated iron(II) clathrochelates with serum albumin induced their molecular asymmetry, causing an appearance of strong CD signals in the range of 350–600 nm, whereas methyl ester and amide clathrochelate derivatives remained almost CD inactive. The CD spectra of carboxyl-terminated clathrochelates on supramolecular interactions or covalent bonding with (R)-(+)-1-phenylethylamine gave a substantially lower CD output than with albumin, affected by both the solvent polarity and the isomerism of clathrochelate's ribbed substituents. In supramolecular assemblies, the bands were most intensive for ortho-substituted carboxyl-terminated clathrochelates. The ortho- and meta-phenylethylamide cage complexes in tetrachloromethane inverted the signs of their CD bands compared with those in acetonitrile. It was suggested that the tris-dioximate metal clathrochelates possess a Russian doll-like molecular system. Because of the distorted TP–TAP geometry, their coordination polyhedron had no inversion centre and possessed an inherent chirality together with the equiprobability of its left(Λ)- and right(Δ)-handle twists. The selective fixation of one of these C3-distorted conformations resulted in the appearance of the CD signal in the range of their visible metal-to-ligand charge transfer bands. Calculations by DFT methods were used to illustrate the possible conformations of the macrobicyclic molecules, as well as the intramolecular interactions between the cage framework and optically active distal substituents responsible for the chirality induction of the metal-centred coordination polyhedra.
1. Introduction
Circular dichroism (CD) spectroscopy is considered to be among the most efficient techniques for both stereochemical analysis and the study of chiral biopolymers.1 It has evoked an interest in the development of chirooptical probes, the parent molecules of which are CD inactive due to either their achirality or their existence as a racemic mixture of enantiomers. When binding of such molecules to a biotarget is accompanied by asymmetry induction, the molecules show the pronounced CD signal(s). The most intensive, characteristic CD output is described1 to be induced by the molecules undergoing rapid backbone reorganization toward formation of a distinct chiral isomer upon their interactions with target biopolymers. Metal complexes have been earlier reported among the potential stereodynamic probes responding to the binding to biomolecules. In particular, metal porphyrins have been used2 as CD sensors of secondary DNA structure, as well as for stereochemical assignment of various chiral substrates and peptides.3–8Clathrochelates9–11 are three-dimensional macropolycyclic complexes with an encapsulated metal ion, and previously, we have suggested that these polyazomethines have the ability of to give a CD response upon their supramolecular binding with chiral inductors, such as biomacromolecules, because of the specific geometry of MN6-coordination polyhedra, which is an intermediate between a trigonal prism (TP) and a trigonal antiprism (TAP).11 Due to such distorted TP–TAP geometry, the polyhedra have no inversion centre and possess an inherent chirality. An equiprobability of their left(Λ)- and right(Δ)-handed distortions and a rapid transition between them cause the absence of optical activity. Hence, a selective fixation of one of the C3-distorted conformations may result in an appearance of a CD signal in the spectra of these clathrochelates in the range of their visible metal-to-ligand charge transfer (MLCT) bands (400–600 nm).
Biological activity of the clathrochelates has been widely studied previously. In particular, the dioximate iron(II) mono- and bis-clathrochelates are reported12–16 to be very efficient (including submicromolar) inhibitors in in vitro transcription system of T7 RNA polymerase. Moreover, such compounds are found17 to affect the pathway of protein fibrilization reaction, leading to changes in its kinetics as well as in the morphology of the aggregates formed.18 Formation of the supramolecular assemblies of the ribbed-functionalized iron(II) complexes with globular proteins (in particular, those with serum albumins) has been studied19,20 using various spectral techniques. These interactions are strongly affected by the nature of the substituents in a clathrochelate framework: the iron(II) tris-dioximates with carboxyphenylsulfide functionalizing groups are reported19 to possess the strongest binding to the albumins.
Here, for the first time, we report an ability of the ribbed-functionalized iron(II) clathrochelates to induce a CD output upon their interaction with proteins and upon their covalent bonding or supramolecular interactions with a low-molecular-weight chiral inductor. A series of iron(II) clathrochelates 1–14 with one or two carboxyphenylsulfide ribbed substituents (Scheme 1), as well as their methyl esters and amide derivatives, were prepared. A chirooptical response of the cage complexes 1–9 upon their interactions with bovine serum albumin (BSA) was characterized by CD spectroscopy and protein fluorescence quenching method as a complimentary assay. To observe the effect of a low-molecular-weight chiral inductor, we recorded the CD spectra of the carboxyphenylsulfide iron(II) clathrochelates upon their ionic or covalent bonding with (R)-(+)-1-phenylethylamine. The ab initio calculations with DFT functionals (PBE and PBE0) were conducted to illustrate the possible conformations of these macrobicyclic molecules, as well as the intramolecular interactions between the cage framework and the optically active distal substituents, which are responsible for chirality induction of the metal-centred coordination polyhedra.
 | ||
| Scheme 1 Chemical drawings of the iron(II) clathrochelates under study. | ||
2. Materials and methods
2.1. Materials
BSA was obtained commercially (Sigma-Aldrich ®). Commercial DMSO was dried with molecular sieves before the experiments. 0.05 M Tris-HCl aqueous buffer with pH 7.9 was used for the preparation of BSA solution with cBSA = 0.2 mg cm−1; an aliquot of the freshly prepared 2 mM DMSO solution of a clathrochelate under study was added to this BSA buffer solution. Since the amount of the above DMSO solution was rather small (up to 2.5% of the total volume), the BSA concentration practically persisted, while the concentration of the clathrochelate under study changed from 0.5 or 1 to 20 μM.The preparative synthetic protocols and spectral and analytical data for the compounds 1–12 are presented in the ESI section.† The mono- and difunctionalized carboxyphenylsulfide iron(II) clathrochelates 1–6 were synthesized using nucleophilic substitution of the corresponding mono- and dichloroclathrochelate precursors shown in Schemes 2 and 3 as reported;21 the alkylsulfide clathrochelate 7 was prepared analogously to the arylsulfide clathrochelates 4–6 as shown in Scheme 3. The synthesis of the cage compounds 8 and 9 (the amide derivative of the macrobicyclic complex 5 and its methyl ester, respectively) was performed as shown in Scheme 4 as described elsewhere.20,22 The clathrochelate 10 was prepared as reported;23 the complexes 11 and 12 were synthesized using the analogous synthetic procedures (see ESI†).
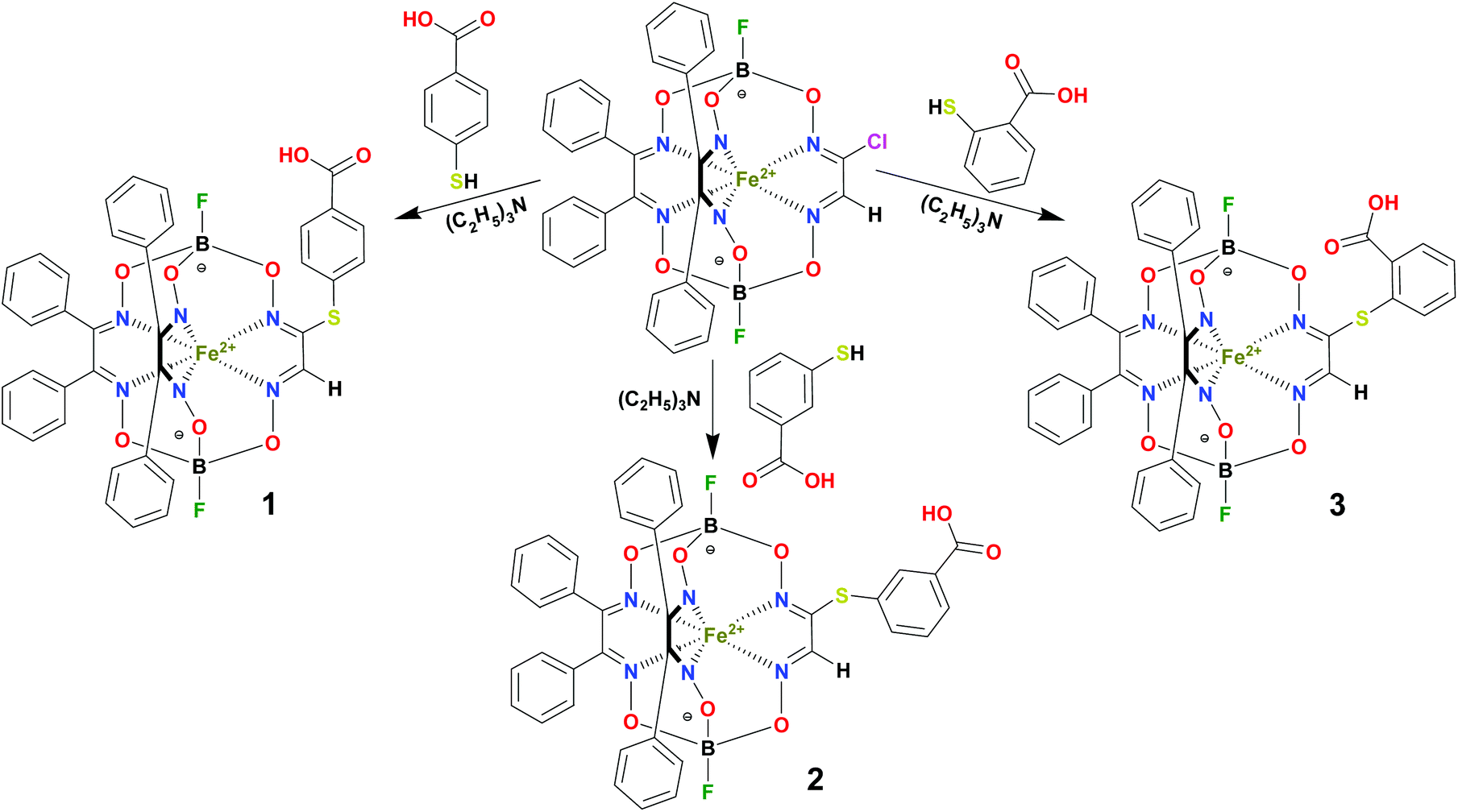 | ||
| Scheme 2 Preparation of the monofunctionalized carboxyphenylsulfide iron(II) clathrochelates 1–3. | ||
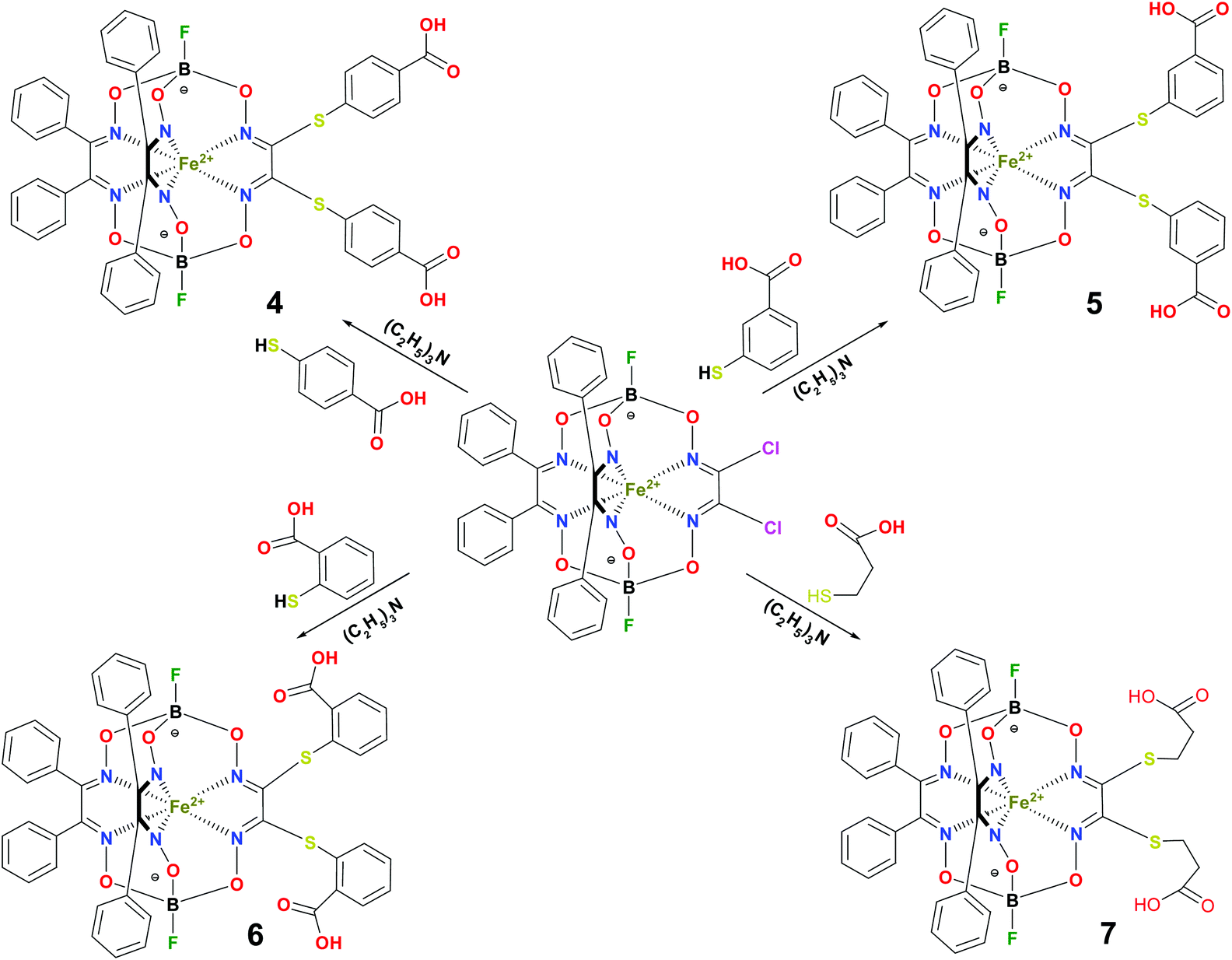 | ||
| Scheme 3 Synthesis of the dicarboxyl-terminated iron(II) clathcrochelates 4–7. | ||
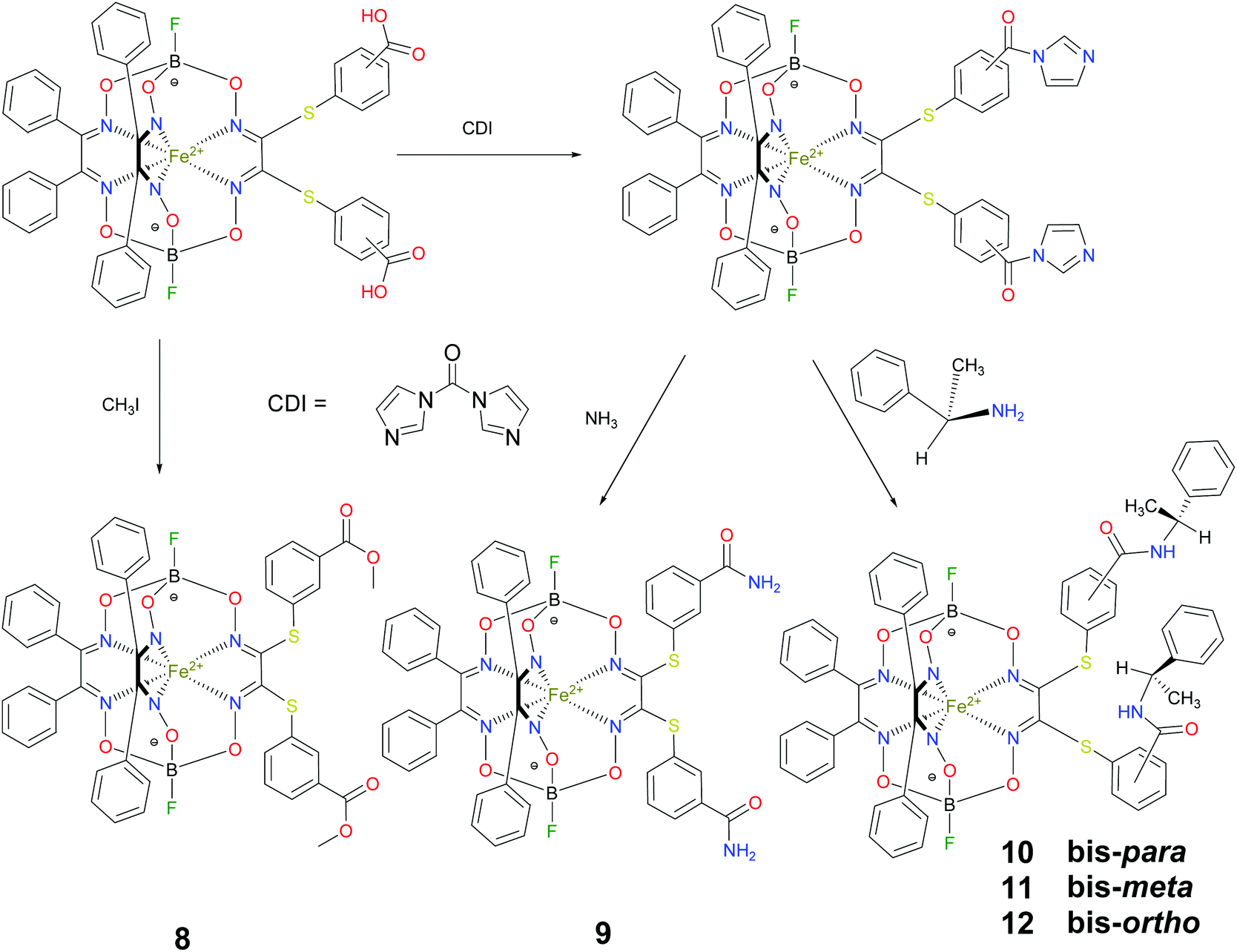 | ||
| Scheme 4 Synthesis of the difunctionalized amide and carboxymethyl iron(II) clathrochelates 8–12. | ||
2.2. Protein fluorescence quenching studies
The fluorescence spectra were obtained at room temperature on a Cary Eclipse fluorescence spectrophotometer (Varian) with a quartz cell (1 × 1 cm). To observe the quenching of BSA fluorescence by the iron(II) clathrochelates under study, the fluorescence spectra of the initial protein and those upon addition of the DMSO solution were recorded. Fluorescence of BSA was excited at 280 nm and its intensity was registered at a maximum of the corresponding emission spectrum. An increase in the concentration of clathrochelates led to a simultaneous increase in the optical density at the maxima of BSA excitation and/or emission, thus causing a decrease in protein fluorescence due to the so-called “inner filter effect” (IFE), as well as because of the reabsorption process.24,25 Such effects should be taken into account for correct interpretation of the spectral data.25–27 So, the intensities of all the fluorescence spectra measured were corrected using the following equation: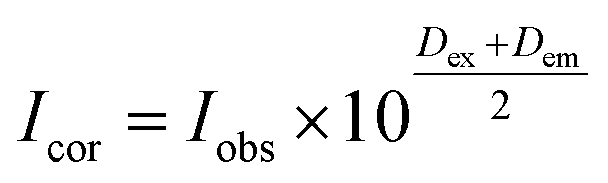 , where Icor and Iobs are the corrected and observed fluorescence intensities, respectively, and Dex and Dem are the total optical densities of the corresponding BSA–clathrochelate system at the maxima of its excitation and emission, respectively.
, where Icor and Iobs are the corrected and observed fluorescence intensities, respectively, and Dex and Dem are the total optical densities of the corresponding BSA–clathrochelate system at the maxima of its excitation and emission, respectively.
The solutions of the clathrochelates under study in 0.05 M Tris-HCl aqueous buffer system at pH 7.9 with their concentrations from 0.5 to 20 μM were used for the correction of IFE.
2.3. UV-vis spectra
UV-vis spectra of the BSA–clathrochelate systems were recorded at room temperature on a Specord M-40 spectrophotometer (Carl Zeiss, Germany).2.4. CD spectra
CD spectra were recorded in the range of 300–600 nm at room temperature on a Jasco – J-715 spectropolarimeter in 1 × 1 cm quartz cell. The data obtained were represented as ellipticity in mdeg, obtained directly from the spectra. Tris-HCl aqueous buffer solutions of the clathrochelates under study at pH 7.55 with BSA-to-clathrochelate molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (cBSA = 8 × 10−5 M; cClt = 4 × 10−5 M) were used for the experiments. Deconvolution of CD spectra of the mono- and difunctionalized iron(II) clathrochelates on their Gaussian components was performed using Origin 7.0 program and the equation: y = ∑[Ai × exp(−0.5 × ((ν − νi)/Δνi)2)] with i = 3; the values of Ai, νi and Δνi were obtained as the fitting parameters; the parameters were the band amplitude, maximum wavelength and parameter, characterizing its half-width and equal to 2.35 × Δνi, respectively. Because the spectra of the above clathrochelates were similar, fitting was performed only for the complex 5, the spectrum of which was found to be the most suitable for this procedure.
:1 (cBSA = 8 × 10−5 M; cClt = 4 × 10−5 M) were used for the experiments. Deconvolution of CD spectra of the mono- and difunctionalized iron(II) clathrochelates on their Gaussian components was performed using Origin 7.0 program and the equation: y = ∑[Ai × exp(−0.5 × ((ν − νi)/Δνi)2)] with i = 3; the values of Ai, νi and Δνi were obtained as the fitting parameters; the parameters were the band amplitude, maximum wavelength and parameter, characterizing its half-width and equal to 2.35 × Δνi, respectively. Because the spectra of the above clathrochelates were similar, fitting was performed only for the complex 5, the spectrum of which was found to be the most suitable for this procedure.
2.5. Computer simulation
The details of the performed quantum chemical calculations are presented in the ESI section.† All chemical calculations described below were carried out using an ORCA package with at least PBE (def2-SVP) level of accuracy. Further details are presented in the ESI section.†3. Results and discussion
3.1. Protein fluorescence quenching studies
The protein fluorescence quenching experiments (Fig. 1) were performed to observe the formation of the supramolecular assemblies between BSA and the clathrochelates 1; these cage complexes do not possess their own intrinsic fluorescence in the UV-vis range. Earlier, this method has been successfully used19 to study the interaction of globular proteins (in particular, BSA) with difunctionalized iron(II) clathrochelates. BSA has an intensive inherent emission with a maximum at 348 nm (λex = 280 nm), assigned to its two “bright” tryptophan fluorophores (Trp 134 and Trp 213). Exposed Trp-134 residue can be easily affected by the solvent molecules or by a close binding of various guest molecules, whereas Trp-213 residue of BSA is hidden within its protein globule, so is less accessible.28 The molecule of BSA has three domains, each of which is divided into two subdomains. This protein contains two main drug binding sites 1 and 2; the first of them is located in a pocket of subdomain IIA, while the subdomain IIIA contains its second drug binding site.29–32 Each of these cavities is mostly hydrophobic, but includes clusters of polar residues (mainly, positively charged Lys and Arg residues29). One Trp residue (Trp-134) is located in proximity to the drug binding site 1; another Trp residue (Trp-213) is located inside the drug binding site 1,19,29 so the binding of a guest cage complex to these sites can cause substantial changes in BSA fluorescence.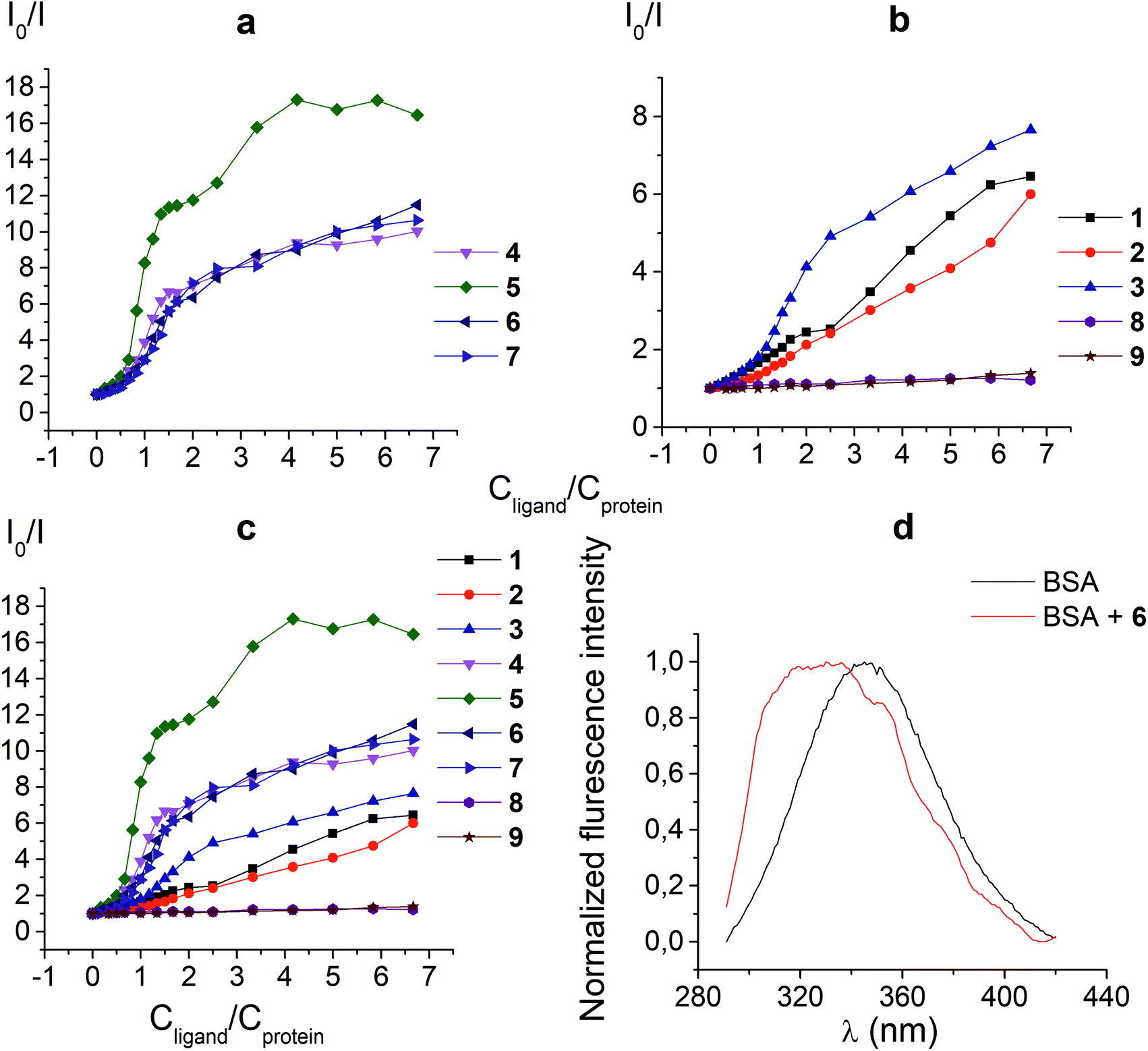 | ||
| Fig. 1 Stern–Volmer plots (a, b and c) of the fluorescence quenching of BSA by the clathrochelates under study. I0 and I are the protein fluorescence intensities in the absence and presence of these cage complexes, cBSA = 0.2 g l−1 (3 μM), cClt is varied from 0 to 20 μM; 6 (d) fluorescent spectra of 0.2 g l−1 BSA (cBSA = 3 × 10−6 M) in the absence and presence of 15 μM of the clathrochelate 6; these spectra were normalized to the same maximal intensity (BSA to clathrochelate molar ratio of 1:5). | ||
An interaction of the clathrochelate guests with BSA caused a substantial quenching of its fluorescence (mainly quenching of Trp fluorescence), as well as a longwave shift of the maximum of the emission band (Fig. 1d), thus suggesting the formation of the BSA–clathrochelate supramolecular assemblies. This quenching may be explained by the changes in the environment close to Trp or by FRET between Trp residue and the cage framework of a clathrochelate guest. The Stern–Volmer plots of the intrinsic fluorescence intensity of BSA versus the concentrations of the clathrochelates are shown in Fig. 1a–c. Quenching of BSA fluorescence with an excess of protein (BSA to clathrochelate molar ratio of 2:1) was rather small, being up to two-fold for the clathrochelate 5 and even less for other carboxyl-terminated cage compounds. In the case of equimolar amounts of BSA and clathrochelates, such quenching was substantially higher for the di-meta-substituted compound 5 than that for other difunctionalized clathrochelates 4, 6 and 7, as well as the quenching for their monofunctionalized analogues 1–3. With an excess in the cage complex (BSA to clathrochelate molar ratio is approximately 1:6.7), the difunctionalized carboxyaryl- and carboxyalkylsulfide clathrochelates 4–7 caused a stronger (approximately 10–17-fold) decrease in BSA fluorescence, reaching its maximum for the meta-substituted cage complex 5 (Table 1). Quenching in the presence of the monofunctionalized complexes 1–3 was substantially lower (in 6–7.5 times), whereas the iron(II) clathrochelates 8 and 9, with the terminal methyl ester and amide groups, respectively, caused almost no changes in the intensity of BSA fluorescence.
| Parameter | BSA + 1 | BSA + 2 | BSA + 3 | BSA + 4 | BSA + 5 | BSA + 6 | BSA + 7 | BSA + 8 | BSA + 9 |
|---|---|---|---|---|---|---|---|---|---|
| The molar ratio BSA to clathrochelate is 1:6.67. |
|||||||||
| Degree of quenching | 6.5 | 6.0 | 7.7 | 10.0 | 16.5 | 11.5 | 10.6 | 1.2 | 1.4 |
Thus, a quenching of the protein fluorescence showed the supramolecular binding of the carboxyl-terminated iron(II) clathrochelates to BSA. The dicarboxyl-terminated cage complexes 4–7 affected this fluorescence more substantially than their monocarboxyl-containing analogues 1–3. The efficiency of such quenching is also affected by the chemical constitution of a clathrochelate molecule (in particular, by the positions of the terminal carboxyl groups in its functionalizing ribbed substituents). At the same time, the amide and methyl ester derivatives of the carboxyl-terminated clathrochelates 8 and 9 affected the BSA fluorescence only slightly, if at all. Hence, we suggest the high impact of electrostatic interactions of the clathochelate's terminal carboxyl groups with the complimentary Lys or Arg residues of BSA in the formation of their host–guest assemblies.
3.2. CD spectra of the BSA–clathrochelate supramolecular assemblies
The ability of the functionalized iron(II) clathrochelates to induce a chirooptical response upon their supramolecular binding to BSA was studied using the CD method; the spectra were recorded in the range of the MLCT Fed → Lπ bands (400–600 nm, Fig. 2), characteristic of cage metal complexes of this type.9 The visible bands for the complexes under study with maxima from 465 to 490 nm are rather wide.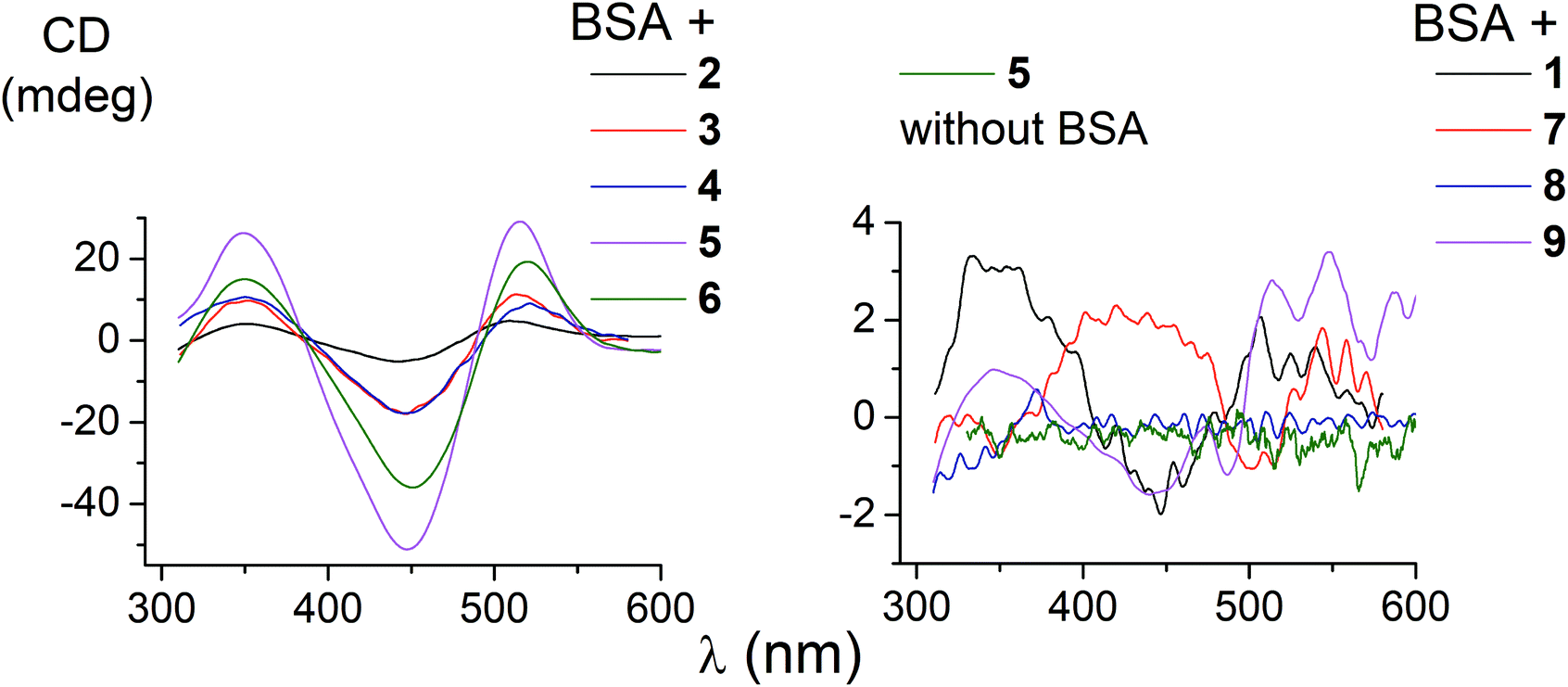 | ||
| Fig. 2 CD spectra of the supramolecular BSA–clathrochelate assemblies. | ||
Because the initial clathrochelates are inherently CD-inactive, their buffer solutions only exhibited a deviation in the baseline (Table 2). Their interaction with the above protein was found to induce a CD response with an intensity of optical output affected by the nature and number of their terminal functional groups. The data presented in Table 2 and Fig. 2 were obtained at the BSA to clathrochelate molar ratio of 2:1, wherein the assemblies gave the most intensive CD signals. Changing the molar ratio to 1:3 mainly affected the intensity of the corresponding CD band, whereas its shape and maximum wavelength remained unchanged. The binding of clathrochelate with one or two carboxyphenylsulfide functionalizing ribbed substituent(s) to BSA induced an appearance of intensive CD bands with maxima at approximately 350, 450 and 515 nm, having similar shapes (Fig. 2). We characterized the intensity of this CD output as the sum of modulus of maximum-to-minimum band intensities (ΔCD) in the range of 300–600 nm (Table 2 and Fig. 2).
| Parameter | 1 + BSA | 2 + BSA | 3 + BSA | 4 + BSA | 5 + BSA | 6 + BSA | 7 + BSA | 8 + BSA | 9 + BSA | 5 (without BSA) |
|---|---|---|---|---|---|---|---|---|---|---|
| a ΔCD = |CDmax| + |CDmin| is the intensity of CD output in the presence of BSA. b The intensity of CD signal for the initial clathrochelate 5. | ||||||||||
| ΔCDa (mdeg) | 5.3 | 7.4 | 27 | 28 | 86 | 45 | 3.3 | 0.8 | 2.4 | 1.6b |
| ε × 10−4 (mol−1 L cm−1) | 3.5 | 2.6 | 2.6 | 2.1 | 4.0 | 3.3 | 2.9 | 2.1 | 2.6 | |
| λ max (nm) | 465 | 470 | 472 | 480 | 480 | 480 | 480 | 490 | 485 | |
The most intensive chirooptical output (86 mdeg) was observed upon the supramolecular binding of BSA with the clathrochelate 5 bearing two meta-carboxyphenylsulfide substituents. The intensities of CD output for the cage compounds 6 and 4 with two ortho- and para-carboxyphenylsulfide ribbed substituents were also pronounced (45 and 28 mdeg, respectively). Those for the clathrochelates with a single terminal carboxyl group were substantially lower (27, 7.4 and 5.3 mdeg for the ortho-, meta- and para-substituted cage complexes 3, 2 and 1, respectively) than the outputs for their difunctionalized analogues. In contrast to the parent dicarboxyl-terminated clathrochelates, the methyl ester 8 and the amide derivative 9 showed only very slight, if at all, changes in the CD spectra in the presence of BSA (0.8 and 2.4 mdeg, respectively). The spectra of host–guest assemblies of the mono- and difunctionalized iron(II) clathrochelates with BSA had similar shapes, thus suggesting a similarity in the asymmetric TP–TAP distortions of their cage frameworks resulting from the supramolecular self-assembly.
The iron(II) clathrochelate 7 bearing two flexible carboxyalkylsulfide groups induced a very weak CD signal (3.3 mdeg) in the presence of BSA. At the same time, a substantial quenching of albumin fluorescence by this cage complex was observed (see above), thus evidencing its binding to BSA. So, a conformational lability of the linking moieties of the ribbed substituents in a cage framework affects the capability of a clathrochelate molecule to induce chirooptical response upon its binding with a protein. The rigidity of the whole macrobicyclic system seems to be very important for chiral recognition and the transmission of chirality to its metal-centered coordination polyhedron.
Therefore, the formation of supramolecular assemblies of the clathrochelates with proteins may induce an optical activity in these metal-centered macrobicyclic compounds, thus causing an appearance of the CD signal in the range of characteristic visible MLCT bands. The intensity of the signal and its shape are affected by the nature and the number of clathrochelate ribbed functionalizing substituents, as well as by the position of the terminal functionalizing groups in these substituents.
3.3. Deconvolution of CD spectra on Gaussian components
CD bands, induced by all the carboxyphenylsulfide iron(II) clathrochelates after their binding with BSA, have similar shapes and maxima. So, their deconvolution on the Gaussian components was performed only for few mono- and difunctionalized cage complexes (Table 3). Such deconvolution (Fig. 3) gave the Gaussian components with maxima at approximately 350, 450 and 510 nm. The components of all the above spectra have the same signs, which are positive for the bands at approximately 350 and 510 nm, and negative for those at approximately 450 nm. Therefore, the following conclusions can be made: (i) three electronic transitions are observed in CD spectra of the mono- and difunctionalized cage complexes, (ii) their macrobicyclic frameworks undergo similar TP–TAP distortions upon supramolecular binding with BSA, (iii) the iron(II) clathrochelates with different number and position of terminal carboxyl groups have a similar asymmetry as their metal-centred frameworks in the host–guest assemblies.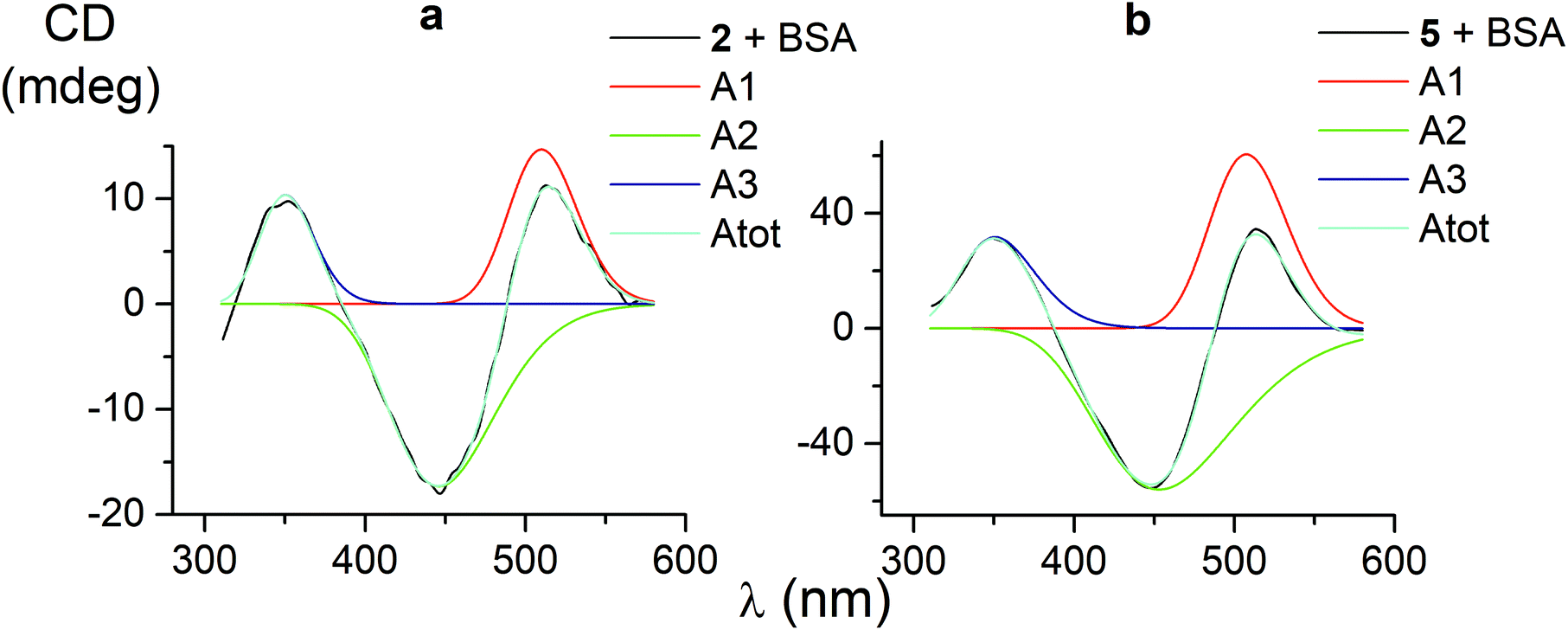 | ||
| Fig. 3 CD spectra of the mono- and difunctionalized iron(II) clathrochelates 2 and 5 (a and b, respectively), deconvoluted on the three Gaussian components. | ||
| Compound |
A
1c |
Δλ1b nm |
λ
1a nm |
A
2c |
Δλ2b nm |
λ
2a nm |
A
3c |
Δλ3b nm |
λ
3a nm |
R
2d |
|---|---|---|---|---|---|---|---|---|---|---|
| a Maxima of the component bands. b Half-widths of these bands. c Amplitudes of the bands. d Fitting parameter. | ||||||||||
| 1 | 1.5 | 133 | 511 | −1.7 | 122 | 446 | 3.3 | 132 | 349 | 0.95697 |
| 2 | 14.7 | 102 | 510 | −17.3 | 157 | 446 | 10.4 | 80 | 350 | 0.99603 |
| 5 | 60.5 | 115 | 508 | −56.1 | 212 | 453 | 31.7 | 112 | 351 | 0.99884 |
| 4 | 11.3 | 108 | 515 | −16.6 | 155 | 446 | 12.6 | 93 | 348 | 0.98904 |
| Average value | 114 | 511 | 161 | 448 | 105 | 350 | ||||
Using the obtained Gaussian components of the CD spectrum of the di-meta-substituted iron(II) clathrochelate 5, the deconvolution of its UV-vis spectrum was also performed (Fig. 4). Three components with their fixed maxima and half-widths but varying intensities were included in the calculation, and two additional bands in the range of 300–600 nm with maxima at 385 and 472 nm, as well as one more short-wavelength UV band with maximum at approximately 290 nm, were obtained. Thus, the results of the fitting proved the existence of three electronic transitions for the clathrochelate molecule 5 in the range of 300–600 nm; these transitions are associated with CD activity. The above results also suggest the presence of at least two electronic transitions, which are not related to the above chirooptical properties.
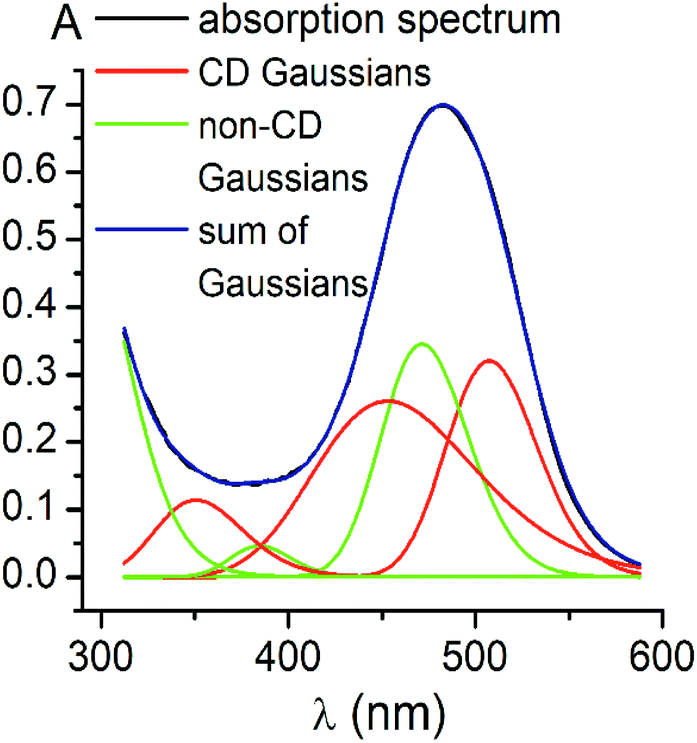 | ||
| Fig. 4 Deconvolution of the absorbance UV-vis spectrum of the clathrochelate 5 into its Gaussian components: the bands obtained by Gaussian fitting of its CD spectrum (in red); the additional bands (in green); the sum of all the Gaussian components (in blue); the experimental spectrum (cClt = 25 μM) in 50 mM Tris-HCl aqueous buffer at pH 7.9 (in black). | ||
3.4. Binding of the iron(II) clathrochelates with a low-molecular-weight optically active organic molecule
We also studied the effects of both the supramolecular binding of a low-molecular-weight optically active compound, (R)-(+)-1-phenylethylamine, with dicarboxyphenylsulfide iron(II) clathrochelates (the compounds 4–6) and that of its covalent bonding (the cage complexes 10–12) on the chirooptical properties of such macrobicyclic molecules.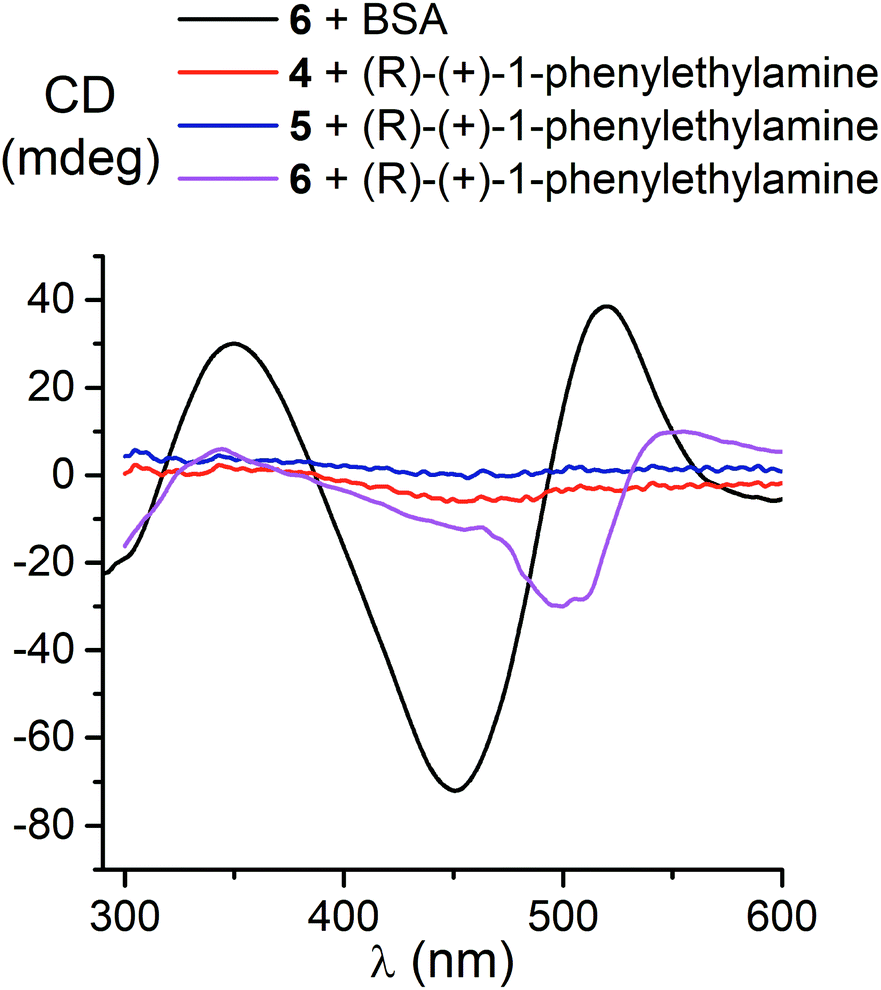 | ||
| Fig. 5 CD spectra of the dicarboxyphenylsulfide iron(II) clathrochelates 4–6 in the presence of (R)-(+)-1-phenylethylamine and BSA. | ||
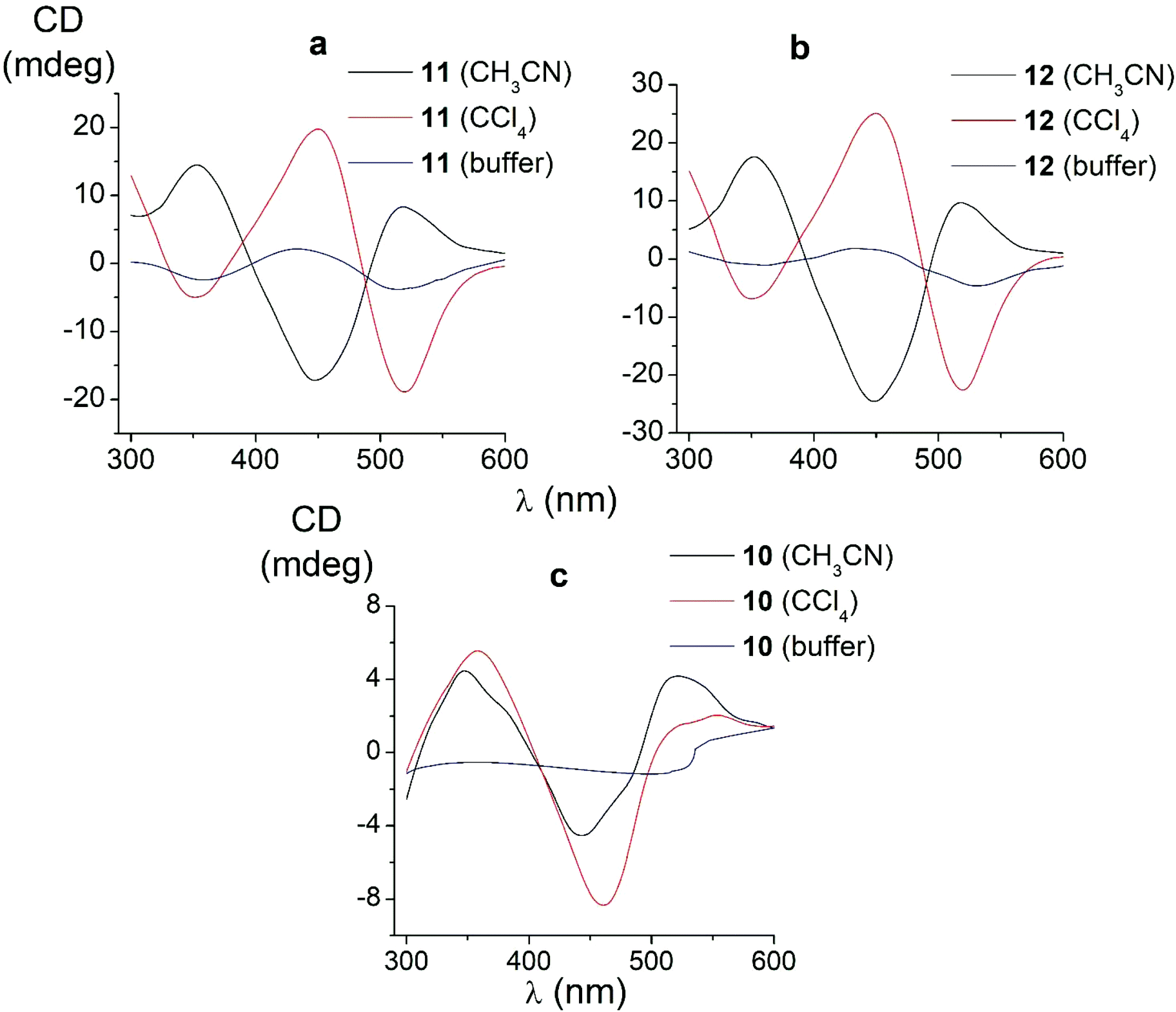 | ||
| Fig. 6 CD spectra of the amide iron(II) clathrochelates 10–12 in the Tris-HCl aqueous buffer at pH 7.55, acetonitrile and tetrachloromethane solutions. | ||
In non-polar tetrachloromethane medium, the ortho- and meta-substituted cage complexes 11 and 12 had the inverted signs of the CD bands as compared with those in more polar acetonitrile as the solvent. On the other hand, both the intensities and maxima of the bands in the tetrachloromethane and acetonitrile solutions were similar. CD bands of the para-substituted clathrochelate 10 possessed a low intensity and showed the same signs in both acetonitrile and tetrachloromethane solutions. The above results suggest that the solvent polarity substantially affects a dominant, optically active conformation of the cage framework of the ortho- and meta-substituted amide iron(II) clathrochelates 11 and 12 (i.e., it determines the stabilization of Δ or Λ conformations of their FeN6-coordination polyhedra). This effect may be caused by the difference in the dipole moments of these conformers and therefore by their different stabilization in solvents of different polarity. The predominant conformations can be additionally stabilized via the formation of supramolecular assemblies with the solvent molecules.
Thus, we suggest that the intramolecular interactions of the optically active terminal substituents with a macrobicyclic framework can determine the preferable stabilization of a given conformation of the corresponding clathrochelate molecule. This stabilization is also found to be affected by the nature of the solvent: in several cases, the use of solvents of different polarity cause the stabilization of either Δ or Λ conformation, thus resulting in the induction of CD signals with opposite signs.
Based on our preliminary experiments, an addition of BSA to the solutions of all the constitutional isomers 10–12 of an amide iron(II) clathrochelate with optically active terminal substituents only slightly affects their initial CD spectra. Such spectral behaviour of these optically active clathrochelate amides is similar to that of their regular meta-amide-terminated clathrochelate analogue 9. This result can be explained by the absence of the terminal carboxyl groups in the macrobicyclic molecules, which are responsible for electrostatic (polar) interactions between the clathrochelates and the charged or polar groups of a protein macromolecule, causing the formation and stabilization of host–guest assemblies.
3.5. Structural peculiarities of macrobicyclic metal tris-dioximates: the pathways of an inversion of their coordination polyhedra and the plausible scheme of the chirality induction for these clathrochelates
All the cage metal tris-dioximates possess the geometry of their MN6-coordination polyhedra, that is an intermediate between a TP (the distortion angle φ = 0°) and TAP (φ = 60°) with φ in the range of approximately 1 to 50°, depending on the nature of the encapsulated metal ion and that of the capping atoms (groups).9,11 TP clathrochelates are achiral complexes, whereas an Δ ↔ Λ inversion of the optically active forms of the TAP macrobicyclic molecules is accompanied by substantial conformational changes; therefore, such inversions have some energy barriers. The iron(II) clathrochelates under study possess an intermediate TP–TAP geometry with φ of approximately 25° and the calculated inversion barriers for the methine-containing cage compounds, the derivative of glyoxime, and the aromatic macrobicyclic complexes, the derivative of α-benzildioxime, are relatively low (5 and 6 kcal mol−1, respectively). So, the dynamic equilibrium between the Δ and Λ conformers (Fig. 7) is observed in solution at room temperature. An encapsulated metallocenter of the above metal clathrochelates is almost completely isolated from external factors by a rigid three-dimensional polyazomethine caging ligand, and therefore, it cannot directly interact with the chirality inductors, thus causing the Cotton effect. On the other hand, we observe a substantial chirooptical response that results from even weak non-covalent bonding of the cage complexes with chiral protein structures, providing supramolecular assemblies (i.e. an intermolecular chirality induction), as well as that from the intramolecular interactions of the terminal chiral center in a clathrochelate molecule with its cage framework (i.e. an intramolecular chirality induction). The above optical effects can be explained by the structural peculiarities of their molecules with rigid macrobicyclic frameworks, allowing dissymmetric supramolecular interactions and by efficient allosteric transmission of chirooptical effect of the terminal optically active groups as distal effectors on the encapsulated metallocenter as well (vide infra).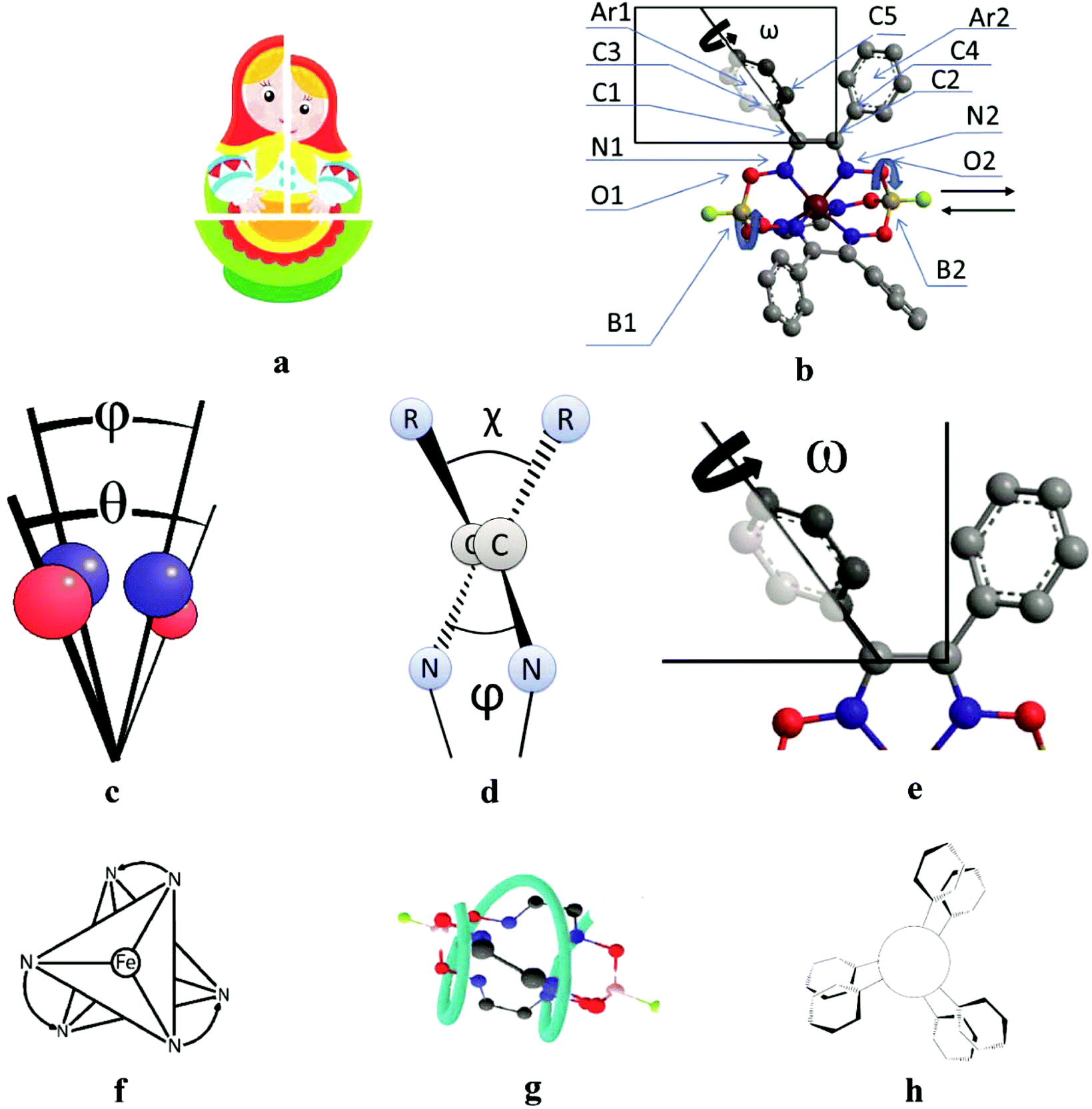 | ||
| Fig. 7 The clathrochelate molecule 8, possessing a Russian doll-like molecular structure (a), with a known X-ray structure (b) and its transformations caused by an inversion of FeN6-coordination polyhedron; the distortion angle φ of this TP–TAP polyhedron and the distortion angle θ of the O3BF capping group (c); the dihedral chelate angle ψ and the twisting angle χ of the ribbed α-dioximate fragment (d); the twisting angle ω of the ribbed aromatic substituents (e); the Δ ↔ Λ structural transitions of the above coordination polyhedron, causing a formation of enantiomeric Δ,Λ-pair of a chiral metallocenter (a point chirality, f); the P-to-M structural rearrangement of the cage framework at a Δ-to-Λ inversion of the FeN6-polyhedron (a helical chirality, g); the inherent aromatic substituents forming a molecular propeller (their flipping forms propeller chirality, h). | ||
| Angle | Structural moiety | Dissymmetry element |
|---|---|---|
| φ | N1–B1–B2–N2 | TP–TAP distortion of MN6-coordination polyhedron |
| θ | O1–B1–B2–O2 | Helical twisting of the macrobicyclic framework |
| χ | C3–C1–C2–C4 | Angle between the vic-substituents in the same chelate cycle |
| ψ | N1–C1–C2–N2 | Dihedral angle in the chelate fragment |
| ω | C5–C3–C1–C2 | Distortion of a propeller system of the inherent aromatic substituents |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CN fragments (approximately 2–5°), as well as in the ω (the dihedral angle between the inherent ribbed aromatic vic-substituents of the same α-benzildioximate fragment, Fig. 7e) from 60 to 140°. As a result, the system of the substituents at the clathrochelate framework undergoes an inversion in its configuration via a flipping process to minimize the sterical clashes between them and to increase their conjugation with the glyoximate chelate fragment. This also causes an inversion in the chelate angle ω with the simultaneous rotation of the above aromatic substituents by 80°. Hence, the conformational transition forms the so-called “hanged gear” between the twisting (the distortions) of the different structural subparts of a clathrochelate molecule in various planes and directions.
C–CN fragments (approximately 2–5°), as well as in the ω (the dihedral angle between the inherent ribbed aromatic vic-substituents of the same α-benzildioximate fragment, Fig. 7e) from 60 to 140°. As a result, the system of the substituents at the clathrochelate framework undergoes an inversion in its configuration via a flipping process to minimize the sterical clashes between them and to increase their conjugation with the glyoximate chelate fragment. This also causes an inversion in the chelate angle ω with the simultaneous rotation of the above aromatic substituents by 80°. Hence, the conformational transition forms the so-called “hanged gear” between the twisting (the distortions) of the different structural subparts of a clathrochelate molecule in various planes and directions.
For the computational study of the above transition, we performed quantum chemical calculations (PBE0, def2-TZVP, see ESI†) of the potential energies of the clathrochelate molecules 13 and 14versus the values of φ, ψ and ω (Fig. 8a, b and c, respectively).
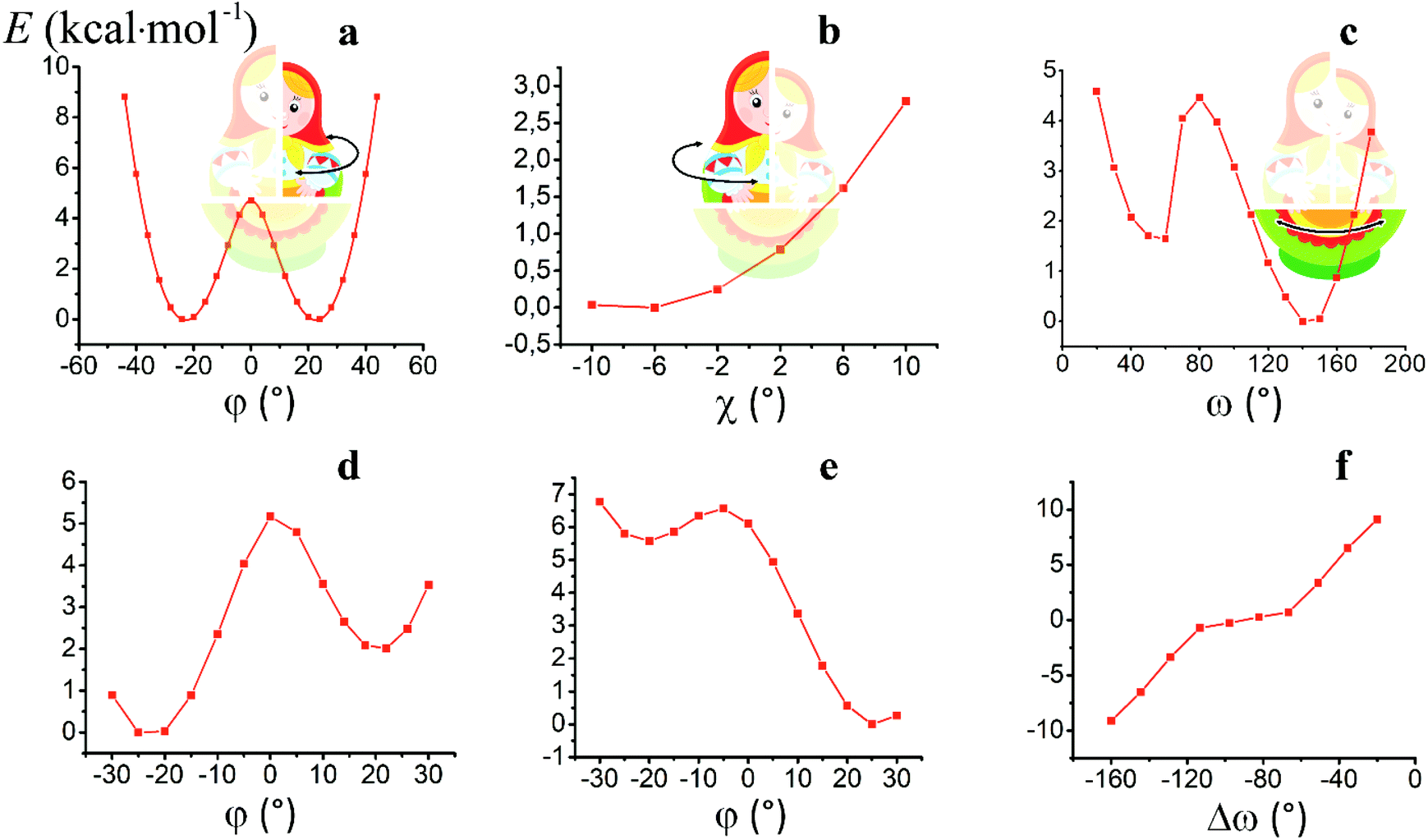 | ||
| Fig. 8 Plots of the energy of the clathrochelate molecule 13versus the values of φ and ψ at a fixed Λ-conformation of its FeN6-coordination polyhedron (a and b, respectively) and that of molecule 14versus the value of ω at a fixed conformation of its coordination polyhedron (c); versus the distortion angle φ at a given ω value (d); versus the distortion angle φ at a given ψ value (e); plot of the difference in energy between the Λ- and Δ-conformations of the molecule 14versus value of ω (f). | ||
Plot of the potential energy of the molecule 13versus the value of φ has an ω-shape character with an energy barrier between the above conformations of approximately 5 kcal mol−1 (Fig. 8a); this is 13's Δ-to-Λ racemization energy. Fig. 8b illustrates the variation in this energy in the course of a hypothetical inversion of the molecule 13, possessing a Λ-conformation of the FeN6-coordination polyhedron, versus its other structural characteristic, the angle ψ. This plot showed only one minimum corresponding to the same absolute configurations of both the above polyhedron and the macrobicyclic framework of this molecule. So, any conformational changes of 13's FeN6-coordination polyhedron induce corresponding and simultaneous changes in the whole cage molecule of 13, and therefore, its symmetry and conformation.
The calculated plot of the energy of the molecule 14versus the value of ω of the single ribbed pair (Fig. 8c) showed an even more intriguing picture: the twisting angle of the phenyl ribbed substituents in the cage framework described a propeller chirality of the macrobicycle 14, and therefore, this Fig. 8c illustrates the changes in 14's energy during the course of an inversion of a propeller system of the phenyl substituents. The calculated plot contains two minima with different energies, as well as a single maximum that corresponds to the barrier of the flipping transition of this system. Unequal energies of the above local minima suggest the presence of an energetically favourable orientation of these aromatic ribbed substituents in a given enantiomeric form that is in agreement with the conformations of both the FeN6-coordination polyhedron and the macrobicyclic framework of 14. In the case of the absence of an agreement between these three structural subparts, the calculated energy of this molecule, having a relaxation barrier of 5 kcal mol−1, is higher by approximately 2 kcal mol−1. Therefore, the above conformational transition of the hexaphenyl-substituted molecule 14 can be regarded as a simultaneous changing of at least two structural elements, described by the angles φ and ω. The corresponding 3D plot of the potential energy of the molecule 14versus its φ and ω values (simultaneous for all three phenyl ribbed substituents) is shown in Fig. 9. The calculated surface of this energy suggests the presence of two general and two local minima, corresponding to the stable and metastable states of this molecule, respectively, while the barrier of the D-to-L inversion of its FeN6-coordination polyhedron is affected by the pathway of this inversion. The above energy barrier is approximately 14 kcal mol−1 for the pathway I and is approximately 10 kcal mol−1 for the stepwise process (Pathways II + III) in which the inversions of the clathrochelate proceed via the corresponding metastable state. The above conformational transitions of the rigid three-dimensional clathrochelate structure as the so-called “molecular machine” can be virtually regarded as a mechanical gear in a gearbox, which transmits forces between the axes located in different planes; these interconnected structural subparts transfer the twisting of the ribbed aromatic substituents to the distortion angle φ of the FeN6-coordination polyhedron of the molecule 14. So, the dissymmetric supramolecular interactions with these inherent substituents (and, therefore, those with the whole cage molecule 14) can cause a splitting in energy of its Λ- and Δ-conformers, thus inducing the chirality of this macrobicycle. Fig. 8d illustrates the plot of energy of 14versus the angle φ at a given value of ω of 60°; the orientations of its ribbed phenyl substituents were fixed in this calculation. The calculated splitting in energy between these conformers of the molecule 14, having an inversion energy barrier of 2.5 kcal mol−1, was approximately 5 kcal mol−1. A similar effect was also observed in the case of the dissymmetric supramolecular interactions of a macrobicyclic framework of 14. The plot of its energy versus the angle φ at a given angle χ, corresponding to the average φ value for a Λ-conformation of this macrobicycle, is shown in Fig. 8e.
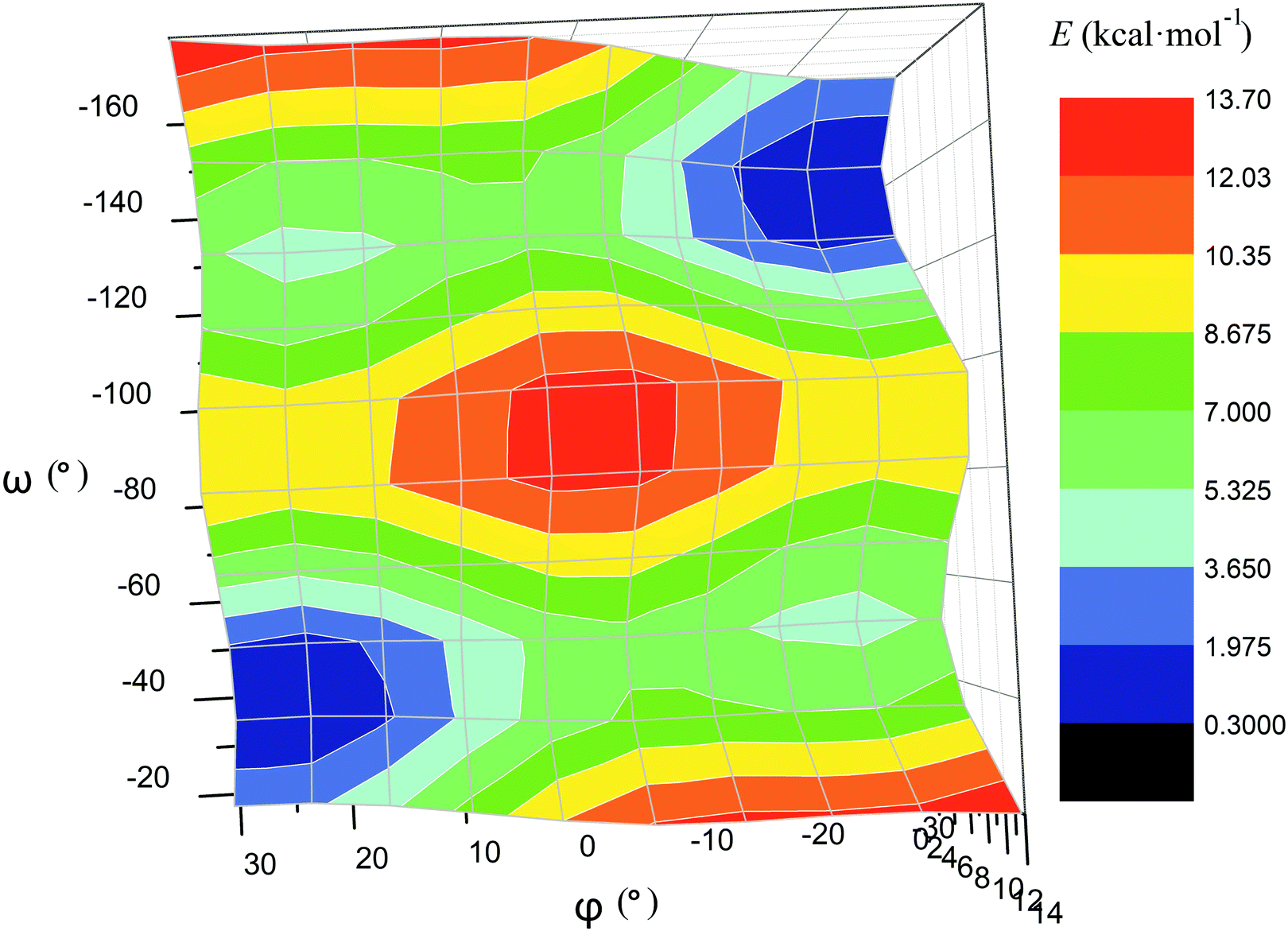 | ||
| Fig. 9 Potential energy surface of the clathrochelate molecule 14versus its φ and ω values. | ||
Thus, it is clear that the weak but dissymmetric supramolecular interactions between the chiral inductor and the peripheral groups of a clathrochelate molecule, such as their stacking interactions and/or hydrogen bonding, can also cause a difference in energy of the Δ- and Λ-conformations of the macrobicyclic molecule (the plot of this difference versus the value of ω is shown in Fig. 8f) and cause the inversion in the sign of the Cotton bands.
In the initial stage, we analyzed the different conformations of the clathrochelates 10–12 and main types A–C of their intramolecular interactions (Fig. 10), such as
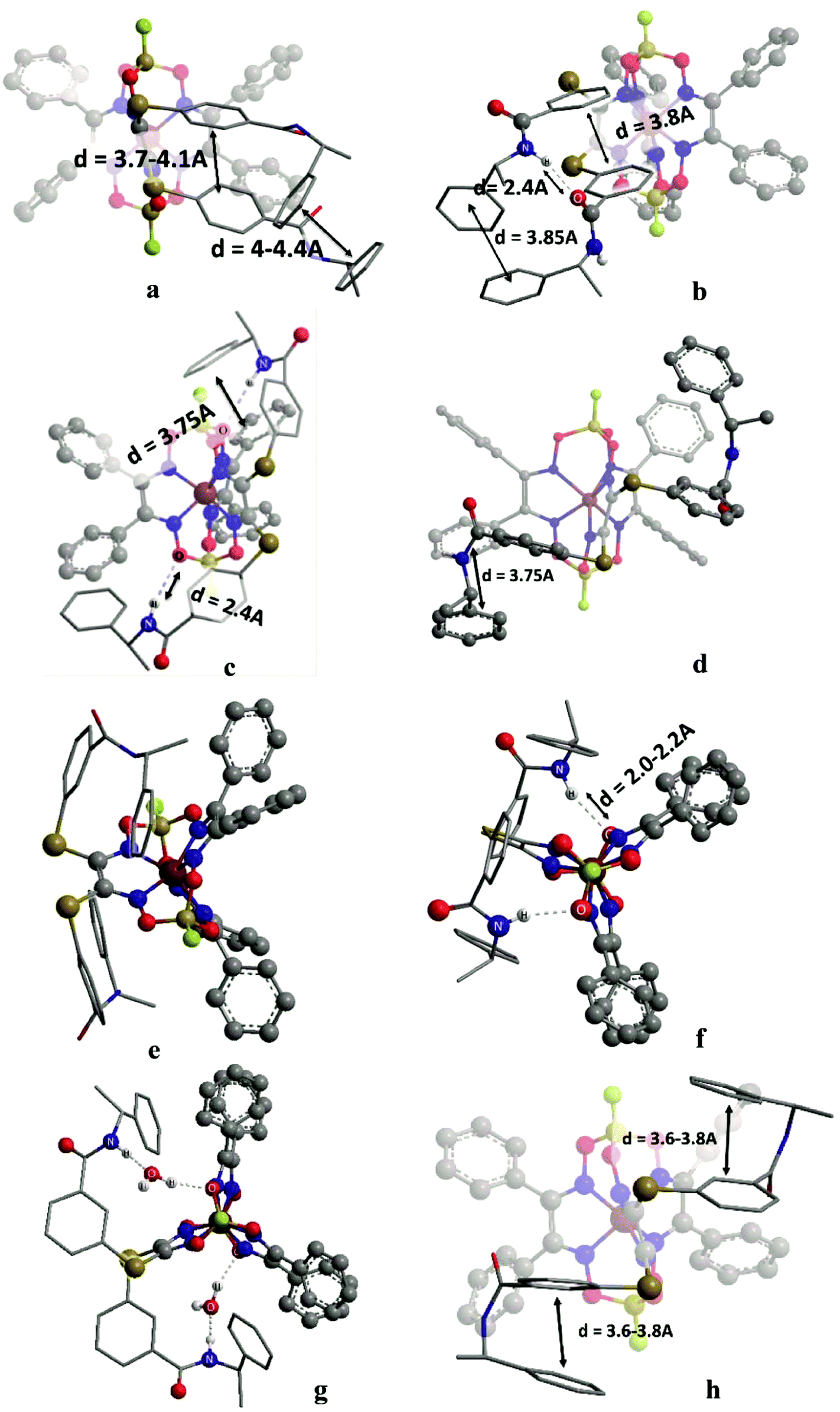 | ||
| Fig. 10 Main types of weak intramolecular interactions in the clathrochelate molecules 10–12. | ||
(i) intra-ribbed interactions (type A)
(ii) interactions between the functionalized ribbed substituents and the capping groups (type B)
(iii) inter-ribbed interactions (type C).
Main types of these interactions are as follows:
(i) aromatic interactions (Fig. 10a and b)
(ii) dispersion interactions
(iii) hydrogen bonding without a solvent molecule (Fig. 10c–e)
(iv) formation of the bridging hydrogen bond via the solvate water molecule (Fig. 10g)
(v) intra-ribbed stacking interactions (i.e. those in the same ribbed chelate fragment of the clathrochelate framework).
We calculated the difference in energy of the D and L conformers for three clathrochelate molecules 10–12, the constitutional isomers. This allowed evaluating their conformations in which the intramolecular stacking interactions and hydrogen bonding of the types A–C are formed; the most interesting results are shown in Fig. 10 and summarized in Table S3 (see ESI†).
3.6. Reasons of induced chirality
Conformations of type A are able to transmit chirality by a change in the angle ψ in the functionalized ribbed chelate fragment of a macrobicyclic ligand. Such conformations were found for the clathrochelates 10 and 11 (Fig. 10a, b and f). Disregarding some of the chiral interactions, an inversion of the FeN6-coordination polyhedron should change only the sign of ψ, while its magnitude should persist. In the optimized molecular structures of the clathrochelates under study, the value of the angle is in the range 8–18° for the clathrochelate's diastereomeric pairs. The calculated energies of these conformations are relatively high, while the differences between them are low. Due to their “prolonged” molecular shape, such conformations display high dipole moments. In non-polar media, some of these conformations are negligible, but their population can be higher in more polar solvents (see ESI, Table S3†).Conformations of type B (Fig. 10f and g) were found for the clathrochelate 12, as well as for the monohydrated form of the complex 11 (i.e. accounting for one solvent water molecule in the calculations). These conformations have the lowest calculated energies, high dipole moments and substantial differences in energy between their diastereomeric pairs. Conformation of this type allows the chiral inductor to directly affect the conformation of the clathrochelate framework by changing the angle θ via the formation of hydrogen bond(s).
Conformations of type C were found for the clathrochelates 10 and 11 (Fig. 10d and e). Their molecular geometries can be affected by a change in the angle χ both in the functionalized ribbed fragment of a macrobicyclic ligand and/or in its neighbouring α-benzildioximate moieties due to the aromatic and dispersion intramolecular interactions. As a result, the difference in energy between these conformations is higher, and the substitutional variations of their energies and dipole moments are observed.
B-conformation of 10 (Fig. 10c) is an interesting example of a clathrochelate molecule in which both the inter-ribbed aromatic interactions and hydrogen bonding with the apical fragments were found. This conformation only slightly differed in energy from the A-conformation of the same macrobicycle 10. This configuration was calculated to have the lowest difference in energy ΔE for its diastereomers but a substantial difference in their dipole moments, thus suggesting an increase in ΔE in polar media.
For all the above calculated conformations of the clathrochelates under study, substantial changes both in energy and dipole moments of their diastereomeric forms are observed. These effects can cause chirality induction of their mirror isomers, thus resulting in appearance of Cotton bands having different signs. A difference in energy of these diastereomers can be affected by the nature of the solvent used due to a difference in their dipole moments.
Because we experimentally observed an inversion in the signs of CD bands for the ortho- and meta-substituted constitutional isomers 11 and 12 caused by change in solvent polarity, while those for the para-substituted clathrochelate 10 was substantially less intensive and did not undergo any solvent-induced inversion, we suggested that the clathrochelate molecules 11 and 12 exhibited chirality in their conformations e, g and f, showing high ΔE values (see ESI, Table S3†). On the other hand, the above interactions for the para-substituted complex 10 could be realized in its C-conformation with a low ΔE value. Slight chirality induction was observed in the case of supramolecular interactions of the terminal (R)-(+)-1-phenylethylamine residues only with the cage framework of the ortho-substituted clathrochelate 12. We suggest that this chirality inductor forms the intramolecular bridging moiety between the fragments of its macrobicyclic ligand via electrostatic (Coulombic) interactions, including their carboxylic and amino groups, aromatic interactions of this aromatic amine residue with the neighbouring α-benzildioximate ribbed fragments of 12, and via its N–H⋯O hydrogen bonding with the apical O3BF fragment of 12. This intramolecular assembly resembles the conformation f (Fig. 10). The formation of the above intramolecular interactions and a bridging moiety for its meta- and para-substituted constitutional isomers 10 and 11 is hindered due to geometrical restrictions.
3.7. Reasons of the formation of CD-active protein–clathrochelate assemblies
Formation of these supramolecular host–guest assemblies, causing a fixation of the optically active TP–TAP distorted configuration of a clathrochelate framework, may be explained by intermolecular non-covalent interactions of different types. Selectivity of the fixation of a given clathrochelate conformer (Δ or Λ) in a protein binding site (and therefore, the appearance of the strong chirooptical output) can be caused by the asymmetry of the arrangement of this binding site, adopting a conformer that possesses a “complimentary geometry”. The electrostatic (polar) interactions were shown to play a key role in clathrochelate–BSA binding. Those can be formed by the terminal carboxyl groups of the corresponding clathrochelate molecule and the charged side groups of Lys or Arg residues of this protein macromolecule. In general, the recognition and stabilization of a given clatrochelate stereoisomer can be due to weak supramolecular bonds, such as aromatic interactions, hydrogen bonds and dispersion interactions. The aromatic (stacking) interactions may be formed by the inherent ribbed phenyl groups of the suitable clathrochelate guest and the aromatic side groups of the amino acid residues Phe, Tyr or Trp. The rigidity of the linking moieties of the functionalizing ribbed substituents, bearing the terminal carboxyl groups, also substantially affects the above chirality recognition and its transmission to the encapsulated metal-centred polyhedron {vide supra comparison of CD-active assemblies of the clathrochelates 4, 5, and 6 and CD-inactive assembly of the complex 7. All of them possess a similar strong binding to BSA (as follows from the fluorescence quenching data)}. The molecular docking of the clathrochelate molecules–leaders to the protein macromolecules with known X-ray structures, as well as the computer simulations of their structural changes caused by clathrochelate-to-protein binding (in particular, to the drug site 1 of BSA), are in progress in our research team.4. Conclusions
In this pioneering work, we discovered that the ribbed-functionalized iron(II) tris-dioximate clathrochelates are able to induce a CD response because of their supramolecular interaction either with a protein as a macromolecular chirality inductor or with an optically active amine as a low-molecular-weight chirality inductor, or by the covalent attachment of this amine. The induced CD signals are observed in the spectral range characteristic to the clathrochelate MLCT absorption bands (400–600 nm). The chirality induction is explained by the predominant stabilization of a rigid clathrochelate framework in one of its “mirror” TP–TAP-distorted configurations. The binding to a protein is found to be determined by the nature of clathrochelate terminal groups; the electrostatic (polar) interactions between its carboxyl groups and complimentary binding groups of a protein are suggested to play a key role for clathrochelate-to-protein assembling. Intensity of the CD output upon binding depends on the constitutional isomerism of the clathrochelate ribbed substituents. The precise intermolecular interactions are also shown to be affected by the conformational lability of the ribbed substituents. All these factors cause the stabilization of a given conformer (Δ or Λ) of a cage framework in the corresponding protein–clathrochelate assembly. Such supramolecular self-assemblies are studied in this work using optical methods; the corresponding isothermal titration calorimetry data will be published in due time. The covalent binding of the carboxyl-terminated clathrochelates with (R)-(+)-1-phenylethylamine caused the induction of CD bands, the shape, intensity and sign of which are affected by both the constitutional isomerism of a clathrochelate molecule and the nature of the solvent used. The inversion of the sign of the Cotton bands in solvents of different polarities confirms the strong solvato-chirooptic effects and importance of supramolecular interactions for the stabilization of certain diastereomeric conformations.We suggest that each of the clathrochelate molecules can be divided into three flexible interrelated subparts that form a Russian-doll-like system and this interrelation is a key element of the induction of chirality of the metal-centered coordination polyhedron of these cage complexes. This suggestion is supported by the ab initio DFT calculations of an energy barrier of their racemisation. The simulations of the intramolecular interactions between the distal chiral groups of a clathrochelate molecule and its macrobicyclic framework is impacted by supramolecular interactions such as hydrogen bonds and stacking and dispersion interactions in the stabilization of a given optically active conformer. Therefore, a macrobicyclic framework of these clathrochelate molecules can be regarded as a suitable molecular scaffold for the design of chirooptical probes for proteins (in particular, for sensing of their conformational changes and transitions). To design CD probes for proteins of different classes and types, a wide range of ribbed-functionalized metal clathrochelates with a given nature, number and position of their terminal groups should be obtained; their synthesis and characterization are in progress.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project leading to these results has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 778245. The synthesis of cage complexes was supported by the Russian Science Foundation (grant 16-13-10475). The contribution of the Center for Molecule Composition Studies of INEOS RAS is also gratefully acknowledged. Y. Z. V. thanks RFBR (grant 15-03-07509) for financial support.References
- C. Wolf and K. W. Bentley, Chem. Soc. Rev., 2013, 42, 5408–5424 RSC.
- M. Balaz, M. De Napoli, A. E. Holmes, A. Mammana, K. Nakanishi, N. Berova and R. Purrello, Angew. Chem., Int. Ed., 2005, 44, 4006–4009 CrossRef CAS PubMed.
- S. Matile, N. Berova, K. Nakanishi, S. Novkova, I. Philipova and B. Blagoev, J. Am. Chem. Soc., 1995, 117, 7021–7022 CrossRef CAS.
- T. Mizutani, T. Ema, T. Yoshida, Y. Kuroda and H. Ogoshi, Inorg. Chem., 1993, 32, 2072–2077 CrossRef CAS.
- M. Takeuchi, H. Kijima, I. Hamachi and S. Shinkai, Bull. Chem. Soc. Jpn., 1997, 70, 699–705 CrossRef CAS.
- Y. Furusho, T. Kimura, Y. Mizuno and T. Aida, J. Am. Chem. Soc., 1997, 119, 5267–5268 CrossRef CAS.
- M. Sakamoto, A. Ueno and H. Mihara, Chem. – Eur. J., 2001, 7, 2449–2458 CrossRef CAS.
- H. Tsukube and S. Shinoda, Chem. Rev., 2002, 102, 2389–2404 CrossRef CAS PubMed.
- Y. Voloshin, N. Kostromina and R. Krämer, Clathrochelates: synthesis, structure and properties, Elsevier, 2002 Search PubMed.
- Y. Voloshin, I. Belaya and R. Krämer, The Encapsulation Phenomenon: Synthesis, Reactivity and Applications of Caged Ions and Molecules, Springer, 2016 Search PubMed.
- Y. Voloshin, I. Belaya and R. Krämer, Cage Metal Complexes clathrochelates revisited, Springer, 2017 Search PubMed.
- V. Novikov, O. Varzatskii, V. Negrutska, Y. Bubnov, L. Palchykovska, I. Dubey and Y. Voloshin, J. Inorg. Biochem., 2013, 124, 42–45 CrossRef CAS PubMed.
- O. Varzatskii, V. Novikov, S. Shulga, A. Belov, A. Vologzhanina, V. Negrutska, I. Dubey, Y. Bubnov and Y. Voloshin, Chem. Commun., 2014, 50, 3166–3168 RSC.
- O. Varzatskii, S. Shul'ga, A. Belov, V. Novikov, A. Dolganov, A. Vologzhanina and Y. Voloshin, Dalton Trans., 2014, 43, 17934–17948 RSC.
- A. Belov, A. Vologzhanina, V. Novikov, V. Negrutska, I. Dubey, Z. Mikhailova, E. Lebed and Y. Voloshin, Inorg. Chim. Acta, 2014, 421, 300–306 CrossRef CAS.
- Y. Voloshin, V. Novikov and Y. Nelyubina, RSC Adv., 2015, 5, 72621–72637 RSC.
- V. Kovalska, M. Losytskyy, O. Varzatskii, V. Cherepanov, Y. Voloshin, A. Mokhir, S. Yarmoluk and S. Volkov, Bioorg. Med. Chem., 2014, 22, 1883–1888 CrossRef CAS PubMed.
- V. Kovalska, S. Chernii, V. Cherepanov, M. Losytskyy, V. Chernii, O. Varzatskii, A. Naumovets and S. Yarmoluk, J. Mol. Recognit., 2017, 30, e2622 CrossRef PubMed.
- M. Losytskyy, V. Kovalska, O. Varzatskii, A. Sergeev, S. Yarmoluk and Y. Voloshin, J. Fluoresc., 2013, 23, 889–895 CrossRef CAS PubMed.
- S. Vakarov, M. Kuperman, V. Kovalska and O. Varzatskii, Ukr. Chem. J., 2015, 81, 116–120 CAS.
- I. Belaya, G. Zelinskii, A. Belov, O. Varzatskii, V. Novikov, A. Dolganov, H. Kozlowski, Ł. Szyrwiel, Y. Bubnov and Y. Voloshin, Polyhedron, 2012, 40, 32–39 CrossRef CAS.
- O. Varzatskii, S. Vakarov, A. Belov, E. Lebed, A. Vologzhanina and Y. Voloshin, J. Coord. Chem., 2017 DOI:10.1080/00958972.2017.1407923.
- S. Vakarov, V. Kovalska and O. Varzatskii, Ukr. Chem. J., 2017, 83, 123–128 Search PubMed.
- J. Lakowicz, Principles of fluorescence spectroscopy, Springer, 1999 Search PubMed.
- Q. Gu and J. E. Kenny, Anal. Chem., 2008, 81, 420–426 CrossRef PubMed.
- B. Birdsall, R. W. King, M. R. Wheeler, C. A. Lewis, S. R. Goode, R. B. Dunlap and G. C. K. Roberts, Anal. Biochem., 1983, 132, 353–361 CrossRef CAS PubMed.
- M. Kubista, R. Sjöback, S. Eriksson and B. Albinsson, Analyst, 1994, 119, 417–419 RSC.
- Y. Moriyama, D. Ohta, K. Hachiya, Y. Mitsui and K. Takeda, J. Protein Chem., 1996, 15, 265–272 CrossRef CAS PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- D. Venturini, A. R. de Souza, I. Caracelli, N. H. Morgon, L. C. da Silva-Filho and V. F. Ximenes, PLoS One, 2017, 12, e0178597 Search PubMed.
- F. Graciani and V. Ximenes, PLoS One, 2013, 8, e76849 CAS.
- V. Ximenes, L. da Fonseca and A. de Almeida, Arch. Biochem. Biophys., 2011, 507, 315–322 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7dt03731g |
| This journal is © The Royal Society of Chemistry 2018 |