
Dynamic equilibria in supported ionic liquid phase (SILP) catalysis: in situ IR spectroscopy identifies [Ru(CO)xCly]n species in water gas shift catalysis†
Tanja
Bauer
 a,
Robert
Stepic
b,
Patrick
Wolf
c,
Fabian
Kollhoff
a,
Weronika
Karawacka
a,
Christian R.
Wick
d,
Marco
Haumann
c,
Peter
Wasserscheid
cf,
David M.
Smith
de,
Ana-Sunčana
Smith
b and
Jörg
Libuda
*af
a,
Robert
Stepic
b,
Patrick
Wolf
c,
Fabian
Kollhoff
a,
Weronika
Karawacka
a,
Christian R.
Wick
d,
Marco
Haumann
c,
Peter
Wasserscheid
cf,
David M.
Smith
de,
Ana-Sunčana
Smith
b and
Jörg
Libuda
*af
aLehrstuhl für Physikalische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, D-91058 Erlangen, Germany. E-mail: joerg.libuda@fau.de
bPULS Gruppe, Lehrstuhl für Theoretische Physik I, Friedrich-Alexander-Universität Erlangen-Nürnberg, Nägelsbachstraße 49b, D-91052 Erlangen, Germany
cLehrstuhl für Chemische Reaktionstechnik, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, D-91058 Erlangen, Germany
dRuđer Bošković Institute, Bijenička cesta 54, P.P. 180, HR-10002 Zagreb, Croatia
eComputer Chemie Centrum, Friedrich-Alexander-Universität Erlangen-Nürnberg, Nägelsbachstraße 25, 91052 Erlangen, Germany
fErlangen Catalysis Resource Center and Interdisciplinary Center Interface-Controlled Processes, Friedrich-Alexander-Universität Erlangen-Nürnberg, 91058 Erlangen, Germany
First published on 28th November 2017
Abstract
Ru-based supported ionic liquid phase (SILP) systems efficiently catalyze the low-temperature water-gas shift reaction (WGSR). While previous studies suggest that Ru-carbonyl species play an important role in the mechanism, detailed knowledge on the catalytically active species is still missing. To identify these carbonyl complexes, we apply in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) in combination with density functional theory (DFT). Investigations of an as-prepared [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 catalyst indicate splitting of the dimer induced by Cl−. Subsequently, an equilibrium between several [Ru(CO)xCly]n species is established, in which the IL serves as an effectively infinite Cl− reservoir. We find that the major species in the system freshly-prepared from [Ru(CO)3Cl2]2 is indeed [Ru(CO)3Cl3]−. A smaller amount of [Ru(CO)2Cl3]− and chloride-rich species [Ru(CO)2Cl4]2− or [RuCOCl4]2− are also found in the SILP. Similar Ru-carbonyl species are observed during carbonylation of RuCl3/[C4C1C1Im]Cl/Al2O3, another potential WGSR catalyst. The response of [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 to heating and/or CO dosing was probed in situ and the results confirm the presence of the equilibrium proposed above.
1. Introduction
The increasing importance of hydrogen-based technologies leads to an increasing demand for H2 production from hydrocarbon sources such as syngas and methanol, which may serve as energy storage media.1–2 To maximize the H2 release from these sources, one important step is the water-gas shift reaction (WGSR), i.e. the exothermic conversion of CO and H2O to CO2 and H2, which decreases the amount of CO and increases the H2 concentration in the product gas.1–3 Ru-based systems, especially Ru-carbonyls, are common catalysts to promote the WGSR at low temperature, where the forward reaction is thermodynamically favored.1,3,4 Werner et al. screened a series of homogeneous Ru catalysts in supported ionic liquid phase (SILP) systems.5 In this study, the SILP concept is applied to immobilize the active catalyst on a porous support using a thin coating of ionic liquid (IL).6,7 The negligible vapor pressure of ILs prevents stripping of the IL layer and leads to a very high long-term stability of the SILP catalysts.6–8 In their work, the authors showed that a SILP system prepared from the simple RuCl3 precursor, with 1-butyl-2,3-dimethylimidazolium trifluoromethanesulfonate ([C4C1C1Im][OTf]) as the IL, formed the most active catalyst. However, the full activity of this system was only reached after an induction period of approximately 60 h.9Interestingly, spectroscopic ex situ analysis of a used RuCl3/[C4C1C1Im][OTf]/Al2O3 sample indicated the presence of Ru-carbonyl species. This finding suggests that new [Ru(CO)xCly]n complexes with enhanced catalytic activity are formed during the induction period of the WGS reaction itself.9 Indeed, the desired activity and selectivity was achieved without an induction period when [Ru(CO)3Cl2]2/[C4C1C1Im][OTf]/Al2O3 was used as a catalyst.10 However, these studies did not reveal the exact nature of the catalytically active carbonyl complex. Thus, the following questions still remain unanswered to date: a) what is the exact nature and concentration of the carbonyl species present in the SILP WGSR system? b) Which of the present species shows the highest catalytic activity (in terms of TON and TOF)? It is obvious that only if the most active carbonyl complex is also the most abundant one in the SILP system under reaction conditions, the activity of the resulting SILP catalyst system it at its maximum.
The present study addresses mainly the first question and elucidates the existence and amount of different [Ru(CO)xCly]n species in two representative SILP systems: RuCl3/[C4C1C1Im]Cl/Al2O3 and [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3. For this purpose, we use in situ infrared (IR) spectroscopy, a particularly powerful tool in metal carbonyl chemistry. In combination with density functional theory (DFT) calculations, this approach allows us to identify the nature of the carbonyl complexes by careful analysis of the CO stretching frequency region. Previously, even the nature of intermediates could be identified by transient IR spectroscopy.11 In this work, we aim at a detailed understanding of the species which are present during the reaction. In contrast to earlier studies,10 we utilize here 1-butyl-2,3-dimethylimidazolium chloride ([C4C1C1Im]Cl) as the IL, because the enhanced chloride content has been shown to further enhance the activity. In particular, we focus on the influence of the IL upon preparation of the active catalyst, as well as on the effects of temperature and CO on the speciation in the active catalysts.
2. Experimental procedures, setups and materials
2.1. Reactor setup for infrared measurements
All IR experiments were performed using a Bruker Vertex 80v FTIR spectrometer with KBr beam splitter and a liquid nitrogen cooled mercury–cadmium–telluride (LN-MCT) detector.Transmission infrared spectroscopy (TIRS) measurements were either conducted with KBr pellets or using a KBr window as IR transparent support. The solutions were drop-cast onto a KBr window which, after evaporation of the solvent, was mounted in the sample compartment on a sample holder (Pike). Evacuation of the sample compartment leads to complete removal of the solvent. The acquisition time for TIR spectra was 1 min with a spectral resolution of 2 cm−1.
All diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) experiments were carried out in a home-built reactor system, which is described in detail elsewhere.12 Briefly, a high temperature reactor chamber and praying mantis diffuse reflectance accessory (both from Harrick) are placed in the sample compartment of the spectrometer. A home-built extension of the sample compartment, equipped with all necessary electrical and gas dosing feedthroughs, allows for keeping the complete optical path evacuated during the measurements. This maximizes the stability of the system, especially for long-term experiments. The reactor is equipped with CaF2 windows and a type K thermocouple directly contacting the examined sample, which provides excellent temperature control. Currently, the setup can be operated at pressures adjustable from 1 mbar to 20 bar using three pressure controllers (PCs). Up to five different gases can be dosed via mass flow controllers (Bronkhorst, MFCs). The setup can be operated in an automated way, which allows accurate programming of temperature, pressure and gas composition. In the CO (Linde, 4.7) dosing experiments the gas dosing (30 min, 1 bar, 20 mLN min−1) was followed by evacuation to 1 mbar for 1 min and, finally, by purging with Ar (Linde 5.0, 15 min, 1 bar, 20 mLN min−1) at 1 bar. The two last steps result in efficient removal of the CO gas phase from the reactor. This procedure was carried out repeatedly, if required, at different temperatures. All DRIFT spectra were recorded using an acquisition time of 5 min and a spectral resolution of 2 cm−1.
2.2. Preparation of the SILP systems
A typical SILP catalyst is made from a metal complex which is dissolved in an IL and, subsequently, dispersed onto the surface of a porous solid support. For the current study aluminum oxide (γ-Al2O3) was used as a support, which was purchased from Sasol Germany GmbH (LOT: B39598) and sieved to a fraction of 200 μm < dP < 500 μm. [Ru(CO)3Cl2]2 which was used as precursor complex was purchased from Alfa Aesar (LOT: T20C0136B). Two ILs were used, [C4C1C1Im]Cl and [C4C1C1Im][OTf], which were purchased from Merck KGaA (LOT: 99/818 and LOT: 99/902 respectively). The SILP materials were prepared with high purity dichloromethane (DCM) as solvent in the preparation procedure (Sigma-Aldrich, LOT: SZBG073AV). RuCl3 was purchased from Sigma Aldrich (LOT: MKBH0927V).In a first step, [C4C1C1Im]Cl was dissolved in high purity DCM (typically 30 mL) in a round-bottom Schlenk flask under Ar atmosphere. After stirring till complete dissolution of the IL (10–15 min at 25 °C), the Ru catalyst (either RuCl3 or [Ru(CO)2Cl3]2) was added followed by another stirring for 10 min. Subsequently, the γ-Al2O3 support was added and stirred quickly for 2–5 min. To avoid crushing of the particles due to mechanical stress, the stirrer bar was taken out before removal of the solvent in two steps (step 1: 3 h, 40 °C, 900 mbar; step 2: 1 h, 40 °C, <3 mbar) in a rotary evaporator. After solvent removal, free flowing SILP catalyst particles were obtained. The resulting catalyst has a Ru loading of wRu = 0.04 gRU gsupport−1 while the pore filling grade with IL is α = 0.34. The pore filling grade describes the IL volume related to the total pore volume of the support material (VILVsupport−1).
2.3. Computational details
The DFT calculations were carried out using the TURBOMOLE13,14 software package. The BP8615,16 functional was used in combination with the def2-TZVPP17 basis set, which was augmented by diffuse functions for C, Cl, H and O atoms taken from the aug-cc-PVTZ18,19 basis set. This approach has been successfully applied to similar systems.20 Optimizations were carried out without any constraints and vibrational frequencies were determined using the analytical derivatives of the energy as implemented in the AOFORCE module within TURBOMOLE.3. Results and discussion
In the following, experimentally observed spectral features are mainly compared to the respective DFT results. For sake of clarity, a complete summary and comparison of DFT and experimental spectra of all Ru-species is given in Table 1.| Ru species | Configuration | Peak center DFT [cm−1] | Peak center exp. [cm−1] | Δ(DFT–exp) [cm−1] | E rel [kJ mol−1] |
|---|---|---|---|---|---|
| [RuII(CO)3Cl2]2 | trans | 2107, 2040 | 2122, ∼2045 | −15, −5 | 0 |
| [RuII(CO)3Cl2] | s.-pyr. cis | 2101, 2033 + 2025 | 2122, 2055 | −21, −26 | 0 |
| [RuII(CO)3Cl2] | s.-pyr. trans | 2054, 2015 | 2040, 2002 | 14, 13 | 44 |
| [RuII(CO)3Cl3]− | facial | 2063, 1983 | ∼2030, ∼1950 | ∼33, ∼33 | 0 |
| [RuII(CO)3Cl3]− | meridial | 1972, 2003, 2091 | — | — | 39 |
| [RuII(CO)2Cl3]− | s.-pyr. cis | 2010, 1939 | 2008, 1922 | 2, 17 | 0 |
| [RuII(CO)2Cl3]− | s.-pyr. trans | 1943 | — | — | 123 |
| [RuII(CO)2Cl4]2− | cis | 1969, 1893 | —, ∼1900 | —, ∼−7 | 0 |
| [RuII(CO)2Cl4]2− | trans | 1894 | ∼1900 | ∼6 | 88 |
| [RuIICOCl4]2− | s.-pyr. | 1862 | ∼1900 | ∼38 | |
| [RuII(CO)4Cl2] | cis | (2149), 2086 + 2066, 2035 | 2140, 2078, — | 9, 8, — | 0 |
| [RuII(CO)4Cl2] | trans | 2060 | — | — | 40 |
| [Ru0(CO)3Cl2]2− | trig.-bipyr. cis | 1910, 1803, 1787 | — | — | 25 |
| [Ru0(CO)3Cl2]2− | trig.-bipyr. trans | 1777 | — | — | 0 |
| [RuII(CO)2(CO–OH)HCl3] | 2025, 1953, 1704, 1678 | (1780/1730) | — | 42 | |
| [RuII(CO)2(CO–OH)Cl2] + HCl | 2117, 2013, 1944, 1728 | — | — | 0 |
3.1. Influence of the IL on [Ru(CO)3Cl2]2 during catalyst preparation
The TIR spectrum of a freshly-prepared SILP catalyst in a KBr disc is shown in Fig. 1a. The spectrum can be divided into regions dominated by IL features (<1600 cm−1 and >2500 cm−1) and carbonyl features from the Ru-species (2200–1700 cm−1). The spectral region below ∼1000 cm−1 cannot be probed due to total absorption of Al2O3.21 [C4C1C1Im]Cl gives rise to asymmetric and symmetric C–H stretching features νas/νs at 3115, 2963 and 2875 cm−1, respectively.22 Furthermore, peaks in the fingerprint region can be assigned to C–N stretching ν (1588 and 1540 cm−1), C–H rocking δas (1540 and 1254 cm−1) and C–H scissor δs (1469 and 1422 cm−1) vibrations.22 The carbonyl region shows very intense C–O stretching features corresponding to CO-ligands of [Ru(CO)xCly]n. Strikingly, the CO-features at 2033 and 1951 cm−1 obtained from the [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 SILP system differ strongly from what was found by us and others in previous studies with [Ru(CO)3Cl2]2 in other Cl-free ILs.20,23,24 We attribute the occurrence of the new peaks to a transformation of the Ru complex in the chloride IL phase. The most likely scenario is a splitting of the dimer by addition of Cl− and formation of [Ru(CO)3Cl3]−, a reaction which was proven previously by experimental and theoretical studies.25 Furthermore, the calculated IR spectrum of the energetically favorable facial (fac-)[Ru(CO)3Cl3]− isomer fits the experimentally obtained peaks well (see Table 1). With respect to this finding, the IL may be regarded as an “infinite” reservoir of Cl− ligands. Dimer splitting was previously also observed in an ion-free environment by Motterlini et al., who showed that, in DMSO, solvent molecules may serve as ligands as well, breaking the dimer into di- and tricarbonyl monomers.26 | ||
| Fig. 1 Reference TIR spectra for; a) the as-obtained [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 SILP system. The assignment is given for IL bands; b) [Ru(CO)3Cl2]2/[C4C1C1Im]Cl mixtures prepared with DCM. | ||
To further prove this hypothesis, we prepared [C4C1C1Im]Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Ru(CO)3Cl2]2 mixtures (weight ratio 2:1 and 5:1) in DCM. The TIRS spectra are displayed in Fig. 1b. All spectra are normalized to the peak at 2030–2040 cm−1, therefore no scale bar is given. The freshly prepared 2:1 mixture (black curve) shows two features at 2122 and 2043 cm−1. Based on our DFT calculation we attribute these peaks to intact [Ru(CO)3Cl2]2.23,26 Previously, Tsuchiya et al. assigned these peaks to [Ru(CO)3Cl3]−,25 which, however, would not agree with our present DFT data. The more pronounced red-shift with respect to previously published IRAS data is attributed to the solvent effects due to the increased amount of IL.20 With increasing IL concentration in the 5:1 mixture (red curve), a small feature appears at 1950 cm−1 and the intensity of the peak at 2122 cm−1 decreases. Presumably, part of the [Ru(CO)3Cl2]2 dimer is at this stage already converted to [Ru(CO)3Cl3]−. The spectrum taken of the 5:1 mixture after 5 days (blue curve) follows this trend. No peak can be seen at 2122 cm−1, which indicates that the dimer species is completely consumed. In addition to the expected features of [Ru(CO)3Cl3]− (2032 and 1950 cm−1) a new peak is visible at 1908 cm−1. It most likely corresponds to chloride-rich, di-anionic species like, i.e., [Ru(CO)2Cl4]2− or [Ru(CO)Cl4]2−, which are formed by further CO/Cl− ligand-exchange reactions. Comparison with DFT data shows that the peak matches very well the calculated position for trans-[Ru(CO)2Cl4]2−, but this complex is energetically unfavorable (88 kJ mol−1). Its corresponding cis-isomer on the other hand should lead to two peaks. The calculated spectrum of [RuCOCl4]2− shows a single peak (1862 cm−1) and could therefore also be responsible for the feature at 1908 cm−1. This would then mean that a direct ligand substitution reaction from [Ru(CO)2Cl3]− to [RuCOCl4]2− takes place. Although no unambiguous assignment of all features can be made at the moment, we summarize that chloride-rich [Ru(CO)2Cl4]2− or [RuCOCl4]2− di-anions are formed from the dimer by sequential ligand substitution reactions. The two small features at 2008 and 1922 cm−1 can be assigned to an intermediate species cis-[Ru(CO)2Cl3]−.
:[Ru(CO)3Cl2]2 mixtures (weight ratio 2:1 and 5:1) in DCM. The TIRS spectra are displayed in Fig. 1b. All spectra are normalized to the peak at 2030–2040 cm−1, therefore no scale bar is given. The freshly prepared 2:1 mixture (black curve) shows two features at 2122 and 2043 cm−1. Based on our DFT calculation we attribute these peaks to intact [Ru(CO)3Cl2]2.23,26 Previously, Tsuchiya et al. assigned these peaks to [Ru(CO)3Cl3]−,25 which, however, would not agree with our present DFT data. The more pronounced red-shift with respect to previously published IRAS data is attributed to the solvent effects due to the increased amount of IL.20 With increasing IL concentration in the 5:1 mixture (red curve), a small feature appears at 1950 cm−1 and the intensity of the peak at 2122 cm−1 decreases. Presumably, part of the [Ru(CO)3Cl2]2 dimer is at this stage already converted to [Ru(CO)3Cl3]−. The spectrum taken of the 5:1 mixture after 5 days (blue curve) follows this trend. No peak can be seen at 2122 cm−1, which indicates that the dimer species is completely consumed. In addition to the expected features of [Ru(CO)3Cl3]− (2032 and 1950 cm−1) a new peak is visible at 1908 cm−1. It most likely corresponds to chloride-rich, di-anionic species like, i.e., [Ru(CO)2Cl4]2− or [Ru(CO)Cl4]2−, which are formed by further CO/Cl− ligand-exchange reactions. Comparison with DFT data shows that the peak matches very well the calculated position for trans-[Ru(CO)2Cl4]2−, but this complex is energetically unfavorable (88 kJ mol−1). Its corresponding cis-isomer on the other hand should lead to two peaks. The calculated spectrum of [RuCOCl4]2− shows a single peak (1862 cm−1) and could therefore also be responsible for the feature at 1908 cm−1. This would then mean that a direct ligand substitution reaction from [Ru(CO)2Cl3]− to [RuCOCl4]2− takes place. Although no unambiguous assignment of all features can be made at the moment, we summarize that chloride-rich [Ru(CO)2Cl4]2− or [RuCOCl4]2− di-anions are formed from the dimer by sequential ligand substitution reactions. The two small features at 2008 and 1922 cm−1 can be assigned to an intermediate species cis-[Ru(CO)2Cl3]−.
For comparison, we extracted the Ru-species also from the fresh catalyst using DCM. The corresponding TIR spectra are included in Fig. 1b (in green). As expected, the spectrum obtained is similar to both the TIR spectra of the “intact” SILP (Fig. 1a) and the one obtained from the 5:1 mixture after 5 days in solution. This proves that the Cl−-induced splitting of the Ru-dimer observed without support material in solution also takes place during preparation of the SILP catalyst. We assume that the formation of monomers is a function of the IL concentration and time. As shown by the spectra of the catalyst after extraction, the Ru-carbonyls can be completely removed from the as-prepared SILP with the non-ligating solvent DCM. Therefore, one can conclude that all species are physically dissolved in the IL phase in the system.23
More detailed insight into the conversion of the dimer species to a variety of monomers in the chloride-containing IL can be gained by studying the transformation in situ. To this end, the 2:1 [C4C1C1Im]Cl:[Ru(CO)3Cl2]2 mixture was deposited on a KBr window and IR spectra were taken in vacuo every 30 min (∼1 mbar, air). The results are displayed in Fig. 2. As the features at 2047 and 2036 cm−1 lie quite close, they cannot be resolved for integration. The peak assigned to the dimer at 2121 cm−1 decreases linearly in time, while the peak related to [Ru(CO)3Cl3]− at 1951 cm−1 increases simultaneously. The integrated peak areas of the intermediate species [Ru(CO)2Cl3]− with peaks at 2009 and 1919 cm−1 increase more steeply in the first 480 min indicating that this complex forms more rapidly at the beginning. As previously observed, its amount is rather small throughout the experiment. The experiment shows the first stage of dimer splitting, therefore, no peak related to the di-anionic species is observed.
 | ||
| Fig. 2 Top: Offset, time-resolved transmission spectra of a [Ru(CO)3Cl2]2:[C4C1C1Im]Cl (1:2) mixture deposited on a KBr window taken every 30 min in vacuum; bottom: normalized peak areas (all lines are guides to the eye). | ||
In order to prove that Cl− ions in the IL are responsible for the transformation of the dimer, we repeated the TIRS measurements with IL solutions providing lower Cl− concentration. First, a 1:1.2 mixture of [C4C1C1Im][OTf] and [C4C1C1Im]Cl was applied to prepare the solutions as described above. Second [C4C1C1Im][OTf] with no additional Cl−, apart from the intrinsic amount from [Ru(CO)3Cl2]2 was used. In contrast to the pure Cl− containing IL, the spectrum of the [OTf]−/Cl− mixture after 5 days (see Fig. 3a) shows a peak at 2125 cm−1 indicating that the dimer is indeed only partly split. In the spectra obtained from the pure [C4C1C1Im][OTf] solution no monomer peak at 1950 cm−1 is formed and the peaks of the dimer species around 2130 and 2056 cm−1 are present even after 5 days (see Fig. 3b). This proves that a smaller Cl− content is associated with a higher amount of intact dimer in the DCM solution. In the case of a chloride-free IL no transformation of [Ru(CO)3Cl2]2 takes place. Nevertheless, spectral changes are visible for the high frequency peak, which forms a shoulder at higher IL concentration. A closer look at the IL region of the spectra helps to understand these changes. Apart from the expected peaks at 1265, 1226, 1156 and 1036 cm−1, which were observed previously, additional weaker features form at 1203 and 1015 cm−1 over time. We attribute this behavior to ligand exchange with the [OTf]− anions themselves. In previous work it was already reported that in DCM solution Cl− at the RuII metal center can be substituted by [OTf]− ligands.27 Although the amount of [OTf]−-species in this previous study was very low (∼1%), the authors could obtain a crystal structure of this complex. Moreover, we were able to spectroscopically identify [OTf]− bound in a ligand-like fashion to a metal support in recent ultra-high vacuum studies with 1-ethyl-2-methylimidazolium trifluoromethanesulfonate ([C2C1Im][OTf]).28 In the [C2C1Im][OTf] monolayer, the anions interact with a Pd support via the SO3-group, leading to red-shifted νs(S–O) and νas(S–O) signals at ∼1010 and 1216 cm−1, respectively. Similar features located at 1204 and 1016 cm−1 can also be found in the TIR spectra recorded from [C4C1C1Im][OTf]:[Ru(CO)3Cl2]2 mixtures as seen in the spectrum recorded after 5 days. The band intensity of the [OTf]− ligand species is lower compared to its solvent counterpart, but its temporal evolution shows that the concentration of the RuII[OTf]− complex increases with time.
 | ||
| Fig. 3 Offset and normalized reference TIRS spectra for a) [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/[OTf] mixture and b) [Ru(CO)3Cl2]2/[C4C1C1Im][OTf] mixtures (IL region included) both prepared with DCM. | ||
Our findings support the assumption that there is a slow time- and concentration-dependent conversion of [Ru(CO)3Cl2]2 to a series of different monomers driven by Cl− from the supporting IL phase. The major species on the freshly prepared SILP catalyst using [Ru(CO)3Cl2]2 as a precursor is [Ru(CO)3Cl3]−. In addition, we find species with a larger number of Cl− ligands, formed at lower concentration. The formation and the nature of the [Ru(CO)xCly]n species clearly depends strongly on the concentration of Cl− ions. It is expected that the speciation should have a direct influence on the catalytic properties of the SILP system.
3.2. Influence of Cl−-content on overall catalytic behavior of the SILP system
To ascertain the dependence of speciation and catalytic activity on the Cl− concentration, a series of experiments with different sources and concentration of Cl− was performed. Apart from [C4C1C1Im]Cl, NaCl was also chosen as the Cl− source. All catalysts were prepared from [Ru(CO)3Cl2]2 as described in the experimental section, except that the additional Cl− source was dissolved in DCM as well. The water content (20 vol%) under experimental conditions enhances NaCl dissolution. Details on the apparatus used to conduct the activity measurements can be found in the ESI.† Specifically, the investigated systems were (from low to high Cl− content): [C4C1C1Im][OTf], a mixture of [C4C1C1Im][OTf] doped with [C4C1C1Im]Cl (equal volume with respect to the pore filling ratio of α = 0.34), [C4C1C1Im][OTf] doped with NaCl (equimolar), [C4C1C1Im]Cl as the benchmark system and an equimolar mixture of [C4C1C1Im]Cl and NaCl. In Table 2 we summarize the activity given as the maximum turn over frequency (TOF140, molCO2molRu−1 h−1 at 140 °C and steady) and the Cl− concentration given as molar fraction of the complete mixture normalized to the benchmark. Furthermore, the Cl−-source other than experimentally induced by the precursor is given. The experiments show that at low concentration, Cl− has a positive effect on the catalytic performance. A certain fraction of Cl− (xCl = 0.31) is present in each of the above-mentioned systems. This fraction originates from the precursor complex, which contains Cl− as well (see [C4C1C1Im][OTf] with xCl = 0.31). By comparison of the [C4C1C1Im][OTf] system (TOF = 26.1 h−1, xCl = 0.31) with the NaCl doped equivalent (TOF = 33.8 h−1, xCl = 0.84), a relative increase in performance of 29.5% is observed, which supports the assumption that Cl− ions enhance the WGSR by the formation of different Cl−-rich monomers, starting from the [Ru(CO)3Cl2]2 dimer. The effect becomes even more visible when comparing [C4C1C1Im][OTf] with the [C4C1C1Im][OTf]/Cl mixture (TOF = 61.4 h−1, xCl = 0.66, αi = 0.17; αtot = 0.34; n[C4C1C1Im][OTf]:n[C4C1C1Im]Cl = 1:1.3). A boost in performance of more than 230% is achieved by doubling the Cl−-content. The large change in performance resulting from a relatively small increase in Cl− content suggests that the counter ion of the doping agent plays an important role as well. This assumption is supported by comparing the [C4C1C1Im]Cl benchmark system (TOF = 72.5 h−1, xCl = 1.00) with the NaCl-doped equivalent (TOF = 33.8 h−1, xCl = 0.84), where a decrease in performance by 53,3% can be observed. Additionally, a comparison between the [C4C1C1Im]Cl benchmark system (TOF = 72.5 h−1, xCl = 1.00) with the NaCl doped benchmark system shows a decrease in performance of 15,5%, in spite of the fact that an increase of the activity would be expected due to the higher Cl− content. These results indicate that Na+ as counter ion has a hindering effect as compared to [C4C1C1Im]+. In addition, NaCl may form solid precipitates in the pores of the support leading to partial blocking of the pores.
| IL | Cl−-sourcea | x(Cl−)a [—] | TOF [h−1] |
|---|---|---|---|
| a Other than experimentally induced Cl− source respectively Cl− content by precursor. | |||
| [C4C1C1Im][OTf] | — | 0.31 | 26.1 |
| [C4C1C1Im][OTf] + [C4C1C1Im]Cl | [C4C1C1Im]Cl | 0.66 | 61.4 |
| [C4C1C1Im][OTf] + NaCl | NaCl | 0.84 | 33.8 |
| [C4C1C1Im]Cl | [C4C1C1Im]Cl | 1.00 | 72.5 |
| [C4C1C1Im]Cl + NaCl | [C4C1C1Im]Cl + NaCl | 1.70 | 61.2 |
The Ru-carbonyls identified so far can be interconverted simply by consecutive Cl−/CO ligand substitutions, therefore we conclude that an equilibrium is established between the various observed species during the catalytic reaction.
3.3. Carbonylation of RuCl3
As mentioned earlier, previous experiments suggested that [Ru(CO)xCly]n species are also produced from RuCl3 under WGSR conditions in a SILP system.9 In order to prove that similar species are formed on the RuCl3- and [Ru(CO)3Cl2]2-based catalysts we monitored the carbonylation of RuCl3/[C4C1C1Im]Cl/Al2O3 by in situ IR spectroscopy. As described in the experimental section, CO was dosed in steps interrupted by purging periods, to be able to resolve also those peaks which are otherwise hidden by the gas-phase CO bands. Fig. 4 shows the last spectrum recorded in purge mode for each CO dosing step. We observe the same bands as in the reference experiments discussed above. After short CO exposure, the spectra show prominent features at 2031 and 1953 cm−1, indicating that the major species in the RuCl3-based system is again [Ru(CO)3Cl3]−. The occurrence of a small peak at 1907 cm−1 indicates that the di-anionic species [Ru(CO)2Cl4]2− or [RuCOCl4]2− is also formed in lower concentrations. Interestingly, the feature of [Ru(CO)2Cl3]− at 2008 cm−1 is quite intense at the early stages of CO dosing. Indeed, it is almost as intense as the peaks of [Ru(CO)3Cl3]−. This is probably due to the fact that [Ru(CO)2Cl3]− is one of the first carbonylation products formed from RuCl3 by addition of two CO ligands. In fact, the amount of [Ru(CO)2Cl3]− decreases at later stages of the experiment indicating its consumption by a consecutive reaction. This temporal behavior supports our previous assignment of the peak. | ||
| Fig. 4 Top: Offset DRIFT spectra taken during CO treatment on RuCl3/[C4C1C1Im]Cl/Al2O3. Each spectrum is taken after the respective cycle of 30 min CO dosing, a subsequent 1 min evacuation and 15 min Ar purge; bottom: integrated, normalized and averaged (over the complete Ar purging time) peak areas (all lines are guides to the eye). | ||
The sharp feature at 2121 cm−1 which corresponds to the [Ru(CO)3Cl2]2 dimer is quite small and increases in intensity more slowly. Previous results with the [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 system suggested the formation of monomers, thus, the back-reaction and formation of [Ru(CO)3Cl2]2 in the IL is implausible. One possible explanation is that no dimerization takes place, but the [Ru(CO)3Cl2] monomer is formed in small amounts. The calculated spectrum of square pyramidal (s.-pyr.) cis-[Ru(CO)3Cl2] strongly resembles the spectrum of the dimer. Although unambiguous assignment of the bands at 2121 and 2155 cm−1 may, therefore, not be possible, it is clear that upon CO dosing several of the previously identified Ru-carbonyls are formed. Similar species like [Ru(CO)3Cl2]2, fac-[Ru(CO)3Cl3]− and [Ru(CO)2Cl4]2− were also found during carbonylation of RuCl3 in refluxing ethanol/ethoxyethanol.29,30 Thus, freshly prepared [Ru(CO)3Cl2]2 (in the presence of Cl−) and carbonylated RuCl3 SILP catalysts exhibit similar Ru-species, mostly [Ru(CO)3Cl3]−. In the case of RuCl3/[C4C1C1Im]Cl/Al2O3 such carbonylated species can only be formed under reaction conditions. This explains the induction period observed in the catalytic activity.9 The new results indicate that CO dosing at ambient conditions (1 bar, RT) should give rise to a similar effect as preparation of the SILP catalyst with [Ru(CO)3Cl2]2. This finding is interesting from an economic point of view, as RuCl3 is a cheaper starting material than [Ru(CO)3Cl2]2.
In the next step, we tested the response of the carbonylated RuCl3/[C4C1C1Im]Cl/Al2O3 system to a change in temperature. The experiments were performed in Ar atmosphere at 1 bar. A summary of the results is displayed in Fig. 5. Upon heating above 75 °C, the peak at 2121 cm−1 decreases in intensity until no band is visible anymore above 125 °C. Simultaneously, a peak with increasing intensity is observed at 1902 cm−1. This points to the formation of chlorine-rich Ru-carbonyls from CO-rich species. The loss of CO ligands is expected, as carbonylchlororuthenium complexes like [Ru(CO)3Cl2]2 are frequently used as CO releasing molecules (CO-RM) in, for example, medical applications.31 It was reported that up to 0.7 mole CO per mole [Ru(CO)3Cl2]2 can be released in DMSO.26 As both peaks of [Ru(CO)2Cl3]− appear rather as shoulders, it cannot be clearly deduced whether this species increases in intensity or the signal is suppressed by the increase of the more dominant features of [Ru(CO)3Cl3]− at 2032 cm−1 and [Ru(CO)2Cl4]2− or [RuCOCl4]2− at 1902 cm−1. Moreover, a new peak is formed at 1735 cm−1 upon heating which was not observed before. Our DFT calculations (see ESI†) suggest that features at such low wavenumbers are found only for carbonyl–carboxylate complexes and five-coordinated Ru0 carbonyls in trigonal-bipyramidal (trig.-bip.) configuration (see Table 1). The formation of a Ru0 species is relatively unlikely under the present conditions (see also section 3.5). The formation of carboxylates could result from the presence of water in the system, which we consider as unlikely or, potentially, from OH species arising from the Al2O3 support. However, formation of a carboxylate from one CO ligand species should influence the carbonyl peaks at higher wavenumbers, which is not observed. The definite assignment of this peak is, therefore, not possible at the present time.
 | ||
| Fig. 5 Top: Offset DRIFT spectra during heating the carbonylated RuCl3/[C4C1C1Im]Cl/Al2O3 sample; bottom: selected, normalized peak areas (all lines are guides to the eye). | ||
In general, we observe that only little peak intensity is lost while, at the same time, several peaks drastically increase in intensity. One explanation for this observation is that upon heating IL and Ru species diffuse in the porous system and – at the applied high IL loading – even some may diffuse out of the pores of the support. The change in peak intensity of the IL-related peaks, which should increase correspondingly, supports this hypothesis (see ESI†). In addition residual RuCl3 species in the catalyst pellets may react with CO and form additional [Ru(CO)3Cl3]−, which would also lead to an increase in the intensity of the respective features.
The experiments with RuCl3/[C4C1C1Im]Cl/Al2O3 confirm that an equilibrium is established between different [Ru(CO)xCly]n species present in the two SILP systems that are active for WGSR.
3.4. Carbonylation of [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3
With the above findings, we now return to the initially investigated [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 system and its response to CO dosing. In Fig. 6, the spectra obtained during CO dosing on [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 are shown over a total duration of 360 min. As the background spectrum was taken before starting the experiment, peaks pointing upwards correspond to loss of intensity or decrease of the respective species, whereas peaks pointing downwards show the formation of the corresponding species. In line with results presented above, we assume that the major species in the examined sample is [Ru(CO)3Cl3]− with a smaller amount of [Ru(CO)2Cl3]− and chloride-rich species such as [Ru(CO)2Cl4]2− or [RuCOCl4]2− also being present. Upon CO dosing, we observe the loss of peak intensity around 1900 cm−1, a spectral region attributed to the latter chloride-rich complexes. The decrease is linear throughout the experiment, and is accompanied by an increase in intensity of the peaks at 2121 and 2055 cm−1, which are assigned to [Ru(CO)3Cl2] or [Ru(CO)3Cl2]2. This shows that the equilibrium is shifted towards complexes containing more CO ligands. The peak at 2055 cm−1 is quite small, which can be explained by the fact that strong absorption of [Ru(CO)3Cl3]− around 2030 cm−1 leads to a decrease in sensitivity compared to the spectral region around 2121 cm−1. Thus, the peak at 2055 cm−1 observed in the difference spectra appears to be less intense. We suggest that in the case of five coordinated Ru complexes the observed ligand substitution occurs in an associative fashion via a six-fold coordinated 18 valence electron transition state.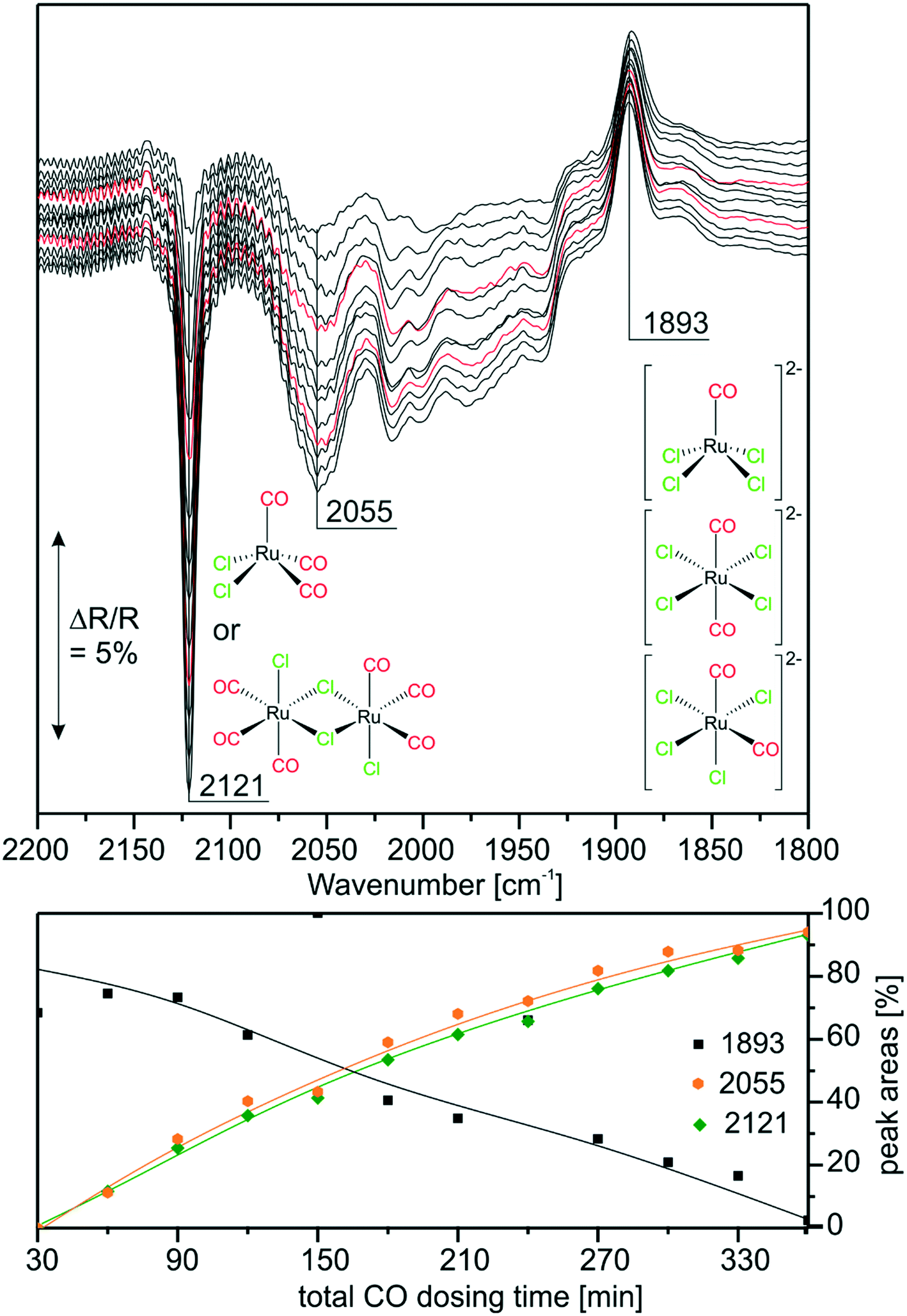 | ||
| Fig. 6 Top: Offset DRIFT spectra taken during CO treatment on the as-obtained SILP catalyst, each spectrum is taken after the respective cycle of 30 min CO dosing, a subsequent 1 min evacuation and 15 min Ar purge step (to remove the CO gas phase from the reactor); bottom: integrated, normalized peak areas (all lines are guides to the eye). | ||
Upon CO dosing the amount of chloride-rich species decreases and [Ru(CO)3Cl2] or [Ru(CO)3Cl2]2 species are formed with more CO ligands bound to the Ru metal center. This observation supports the presence of a dynamic equilibrium between these species.
3.5. Carbonylation of [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 at elevated temperatures
Next, we investigated the carbonylation at elevated temperatures from 30 to 140 °C. Fig. 7 shows the difference spectra recorded in Ar purge mode after CO dosing at each temperature step. We observe that the feature related to chloride-rich species at ∼1899 cm−1 decreases in intensity. These complexes undergo carbonylation to form the previously discussed (s.-pyr. cis-) [Ru(CO)3Cl2] or dimer species as indicated by the peaks at 2121 and 2060 cm−1. The overall increasing intensity of positive and negative peaks in the spectra with respect to the isothermal CO dosing experiment at RT indicates that, at elevated temperatures, the conversion to CO-rich species is enhanced. Moreover, new species can be identified. For example the two rather broad features at 2040 and 2002 cm−1 can be assigned to the s.-pyr. trans-[Ru(CO)3Cl2] monomer, which is less stable compared to the cis conformer. As the band at 2040 cm−1 lies in between two intense features at 2060 cm−1 and 2030 cm−1 ([Ru(CO)3Cl3]−), its quantification is difficult. Therefore, Fig. 7 only includes integrated and normalized areas of the peak at 2002 cm−1. The amount of s.-pyr. trans-[Ru(CO)3Cl2] (grey curve) increases quickly upon heating and CO dosing and it appears to be stable in CO atmosphere also during the cooling period. The behavior of the integrated peak areas of all observed [Ru(CO)3Cl2] monomer (s.-pry., cis/trans) and dimer complexes shows interconversion between the different isomers. At high temperatures, the less stable s.-pyr. trans structure is favored. | ||
| Fig. 7 Top: Offset DRIFT spectra taken during CO treatment upon heating on the as-obtained SILP catalyst, each spectrum is taken after a cycle of 30 min CO dosing, a subsequent 1 min evacuation and 15 min Ar purge (the two latter steps remove the CO gas phase from the reactor) at the respective temperature; bottom: integrated, normalized peak areas (all lines are guides to the eye). | ||
A closer look at the spectra reveals that a small feature at 2140 cm−1 in combination with a band at 2078 cm−1 (orange curve) develops at temperatures above ∼80 °C. As peaks located at such high wavenumbers point to CO-rich Ru-complexes, this signal pair can be assigned to [Ru(CO)4Cl2].32 The third peak expected from DFT at ∼2030 cm−1 coincides with bands from other species and, therefore, cannot easily be identified in the spectra. [Ru(CO)4Cl2] may indeed be formed from [Ru(CO)3Cl2] or the corresponding dimer in either configuration by carbonylation. It is stable above 90 °C in our experiment, whereas decreasing peaks areas of the [Ru(CO)3Cl2] conformers or dimer (green and grey curve) indicate consumption of the latter at high temperatures.
During cooling, [Ru(CO)4Cl2] re-converts to [Ru(CO)3Cl2] or [Ru(CO)3Cl2]2 complexes by loss of CO. The amount of s.-pyr. trans-[Ru(CO)3Cl2] at 2002 cm−1 decreases upon cooling, although the process seems not to be completed on the time scale of the present experiment.
Furthermore, the spectral region at lower wavenumbers shows an interesting behavior. Firstly, the peak at 1736 cm−1 develops upon heating, as expected from the heating experiment of carbonylated RuCl3. Secondly, the peak intensity decreases above 90 °C and a new peak at 1782 cm−1 is formed at the same time. The cooling ramp shows that this behavior is reversible. As discussed above the corresponding bands may be attributed to carbonyl–carboxylate or Ru0 species. We tentatively assign the low frequency peaks to carbonyl–carboxylate species formed with OH species from the Al2O3 support, as both the appreciable presence of water and reversible formation of Ru0 carbonyl species seem quite unlikely.
In the context of CO dosing, the question arises whether full carbonylation to Ru3(CO)12 is possible under the present conditions. The infrared spectrum of Ru3(CO)12 on Al2O3 shows a broad vibrational structure between 1900 and 2100 cm−1.32 On one hand, this could explain the broad smeared-out structure, which develops upon heating exactly in this spectral region. On the other hand, it has been reported that the presence of halide anions readily leads to CO displacement in Ru3(CO)12, especially in solvents which increase the nucleophilicity of Cl−.33 Still, these experiments have not been conducted in CO atmosphere.
In comparison with the CO dosing experiments performed at RT we conclude that at elevated temperatures carbonylation of chloride-rich species is possible even up to formation of [Ru(CO)4Cl2]. This species was previously not observed in the presented experiments and seems to be associated with appreciable concentration only at temperatures above 80 °C.
4. Conclusion
In this work, using a combination of DRIFTS and DFT, we investigated [Ru(CO)xCly]n based SILP catalysts, which are active for the low-temperature WGSR. From our results we are able to draw the following conclusions:1) It is shown that the [Ru(CO)xCly]n SILP is a dynamic system, with a speciation that responds to the reaction conditions. In particular, we investigated the role of the reaction temperature and the CO pressure.
2) With the help of DFT calculations, several species that are present under reaction conditions have been identified. Their nature is summarized in Fig. 8. Specifically, we identify the following species: fac-[Ru(CO)3Cl3]− (major species in the freshly prepared catalyst), cis-[Ru(CO)2Cl3]−, cis-/trans-[Ru(CO)2Cl4]2−, [Ru(CO)Cl4]2−, s.-pyr. cis-/trans-[Ru(CO)3Cl2] and cis-[Ru(CO)4Cl2].
 | ||
| Fig. 8 Overview of Ru complexes observed during the investigation of the different SILP systems and reference experiments. Peak positions taken from the measurements are included. Note that for the sake of clarity not all possible ligand exchange reactions are indicated. | ||
3) Successive elimination/addition steps lead to principle groups of intermediates. In this reaction sequence, the applied chloride IL acts as an effectively infinite reservoir of chloride ligands.
4) Upon preparation of [Ru(CO)3Cl2]2/[C4C1C1Im]Cl/Al2O3 by impregnation in DCM, the Ru dimer splits and a [Ru(CO)3Cl3]− monomer is formed, together with a small amount of [Ru(CO)2Cl3]−, [Ru(CO)2Cl4]2− and [RuCOCl4]2−.
5) The same species are found, if the catalyst is prepared from RuCl3/[C4C1C1Im]Cl/Al2O3 followed by subsequent CO dosing at room temperature.
6) Heating leads to CO release and an increasing fraction of chloride-rich di-anionic species (right-hand side in Fig. 8), whereas CO dosing drives the equilibrium to the CO-rich (left-hand) side. CO dosing at elevated temperatures enhances this transformation and leads even to formation of CO-rich species such as [Ru(CO)4Cl2].
7) In addition to Ru-complexes with varying numbers of Cl− and CO ligands, different conformers of the [Ru(CO)3Cl2] monomer have been identified. Some of the species observed are present only at elevated temperatures in CO atmosphere.
8) The beneficial effect of Cl− addition on the equilibria between different Ru-chloro-carbonyl species is also reflected in the catalytic activity. Higher Cl− concentrations in the IL layer result in more active WGS catalysts.
The presented experiments show that a variety of Ru-complexes can be identified by in situ DRIFTS in combination with DFT. Further experiments are planned involving the influence of water in operando mode. In this way, it should be feasible to assign catalytic activities in an even more reliable fashion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding by the German Research Council (DFG), which, within the framework of its “Excellence Initiative” supports the Excellence Cluster “Engineering of Advanced Materials” (http://www.eam.fau.de) at the Friedrich-Alexander-Universität Erlangen-Nürnberg” (project EXC 315) (Bridge Funding). T. B. gratefully acknowledges financial support from the “Fonds der Chemischen Industrie”. R. S., C. W., A.-S. S. and D. M. S. acknowledge the DAAD project Multiscale Modelling of Supported Ionic Liquid Phase Catalysis (2017–2018), as well as the NIS project 11311 at the Juelich Supercomputing facilities. P. Wa., M. H. and P. Wo. gratefully acknowledges financial support from the European Commission within the Horizon 2020-SPIRE project ROMEO (grand agreement number 680395).References
- S. R. J. Byron, L. Muruganandam and S. M. Shekhar, Int. J. Chem. React. Eng., 2010, 8 DOI:10.2202/1542-6580.2238.
- M. H. Halabi, M. H. J. M. de Croon, J. van der Schaaf, P. D. Cobden and J. C. Schouten, Appl. Catal., A, 2010, 389, 68–79 CrossRef CAS.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- G. Jacobs and B. H. Davis, in Catalysis: Volume 20, The Royal Society of Chemistry, 2007, vol. 20, pp. 122–285 Search PubMed.
- S. Werner, N. Szesni, R. W. Fischer, M. Haumann and P. Wasserscheid, Phys. Chem. Chem. Phys., 2009, 11, 10817–10819 RSC.
- A. Riisager, R. Fehrmann, M. Haumann and P. Wasserscheid, Eur. J. Inorg. Chem., 2006, 695–706, DOI:10.1002/ejic.200500872.
- C. P. Mehnert, Chem. – Eur. J., 2004, 11, 50–56 CrossRef CAS PubMed.
- H. P. Steinrück, J. Libuda, P. Wasserscheid, T. Cremer, C. Kolbeck, M. Laurin, F. Maier, M. Sobota, P. S. Schulz and M. Stark, Adv. Mater., 2011, 23, 2571–2587 CrossRef PubMed.
- S. Werner, N. Szesni, A. Bittermann, M. J. Schneider, P. Härter, M. Haumann and P. Wasserscheid, Appl. Catal., A, 2010, 377, 70–75 CrossRef CAS.
- S. Werner, N. Szesni, M. Kaiser, R. W. Fischer, M. Haumann and P. Wasserscheid, ChemCatChem, 2010, 2, 1399–1402 CrossRef CAS.
- J. M. Butler, M. W. George, J. R. Schoonover, D. M. Dattelbaum and T. J. Meyer, Coord. Chem. Rev., 2007, 251, 492–514 CrossRef CAS.
- T. Bauer, V. Hager, M. Williams, M. Laurin, T. Döpper, A. Görling, N. Szesni, P. Wasserscheid, M. Haumann and J. Libuda, ChemCatChem, 2017, 9, 109–133 CrossRef CAS.
- TURBOMOLE V6.6 2014, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com Search PubMed.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- M. Sobota, S. Schernich, H. Schulz, W. Hieringer, N. Paape, P. Wasserscheid, A. Görling, M. Laurin and J. Libuda, Phys. Chem. Chem. Phys., 2012, 14, 10603–10612 RSC.
- M. Kaltchev and W. T. Tysoe, Surf. Sci., 1999, 430, 29–36 CrossRef CAS.
- B. Haddad, D. Mokhtar, M. Goussem, E.-H. Belarbi, D. Villemin, S. Bresson, M. Rahmouni, N. R. Dhumal, H. J. Kim and J. Kiefer, J. Mol. Struct., 2017, 1134, 582–590 CrossRef CAS.
- L. Huang and Y. Xu, J. Mol. Catal. A: Chem., 2001, 176, 267–280 CrossRef CAS.
- D. Roberto, R. Psaro and R. Ugo, Organometallics, 1993, 12, 2292–2296 CrossRef CAS.
- K. Tsuchiya, J.-D. Huang and K.-i. Tominaga, ACS Catal., 2013, 3, 2865–2868 CrossRef CAS.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, E17–24 CrossRef CAS PubMed.
- G. Santiso-Quinones and R. E. Rodriguez-Lugo, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 859–861 CAS.
- T. Bauer, S. Mehl, O. Brummel, K. Pohako-Esko, P. Wasserscheid and J. Libuda, J. Phys. Chem. C, 2016, 120, 4453–4465 CAS.
- M. L. Berch and A. Davidson, J. Inorg. Nucl. Chem., 1973, 35, 3763–3767 CrossRef CAS.
- N. Lugan, G. Lavigne, J. M. Soulie, S. Fabre, P. Kalck, J. Y. Saillard and J. F. Halet, Organometallics, 1995, 14, 1712–1731 CrossRef CAS.
- A. A. Ahanger, S. Prawez, D. Kumar, R. Prasad, Amarpal, S. K. Tandan and D. Kumar, Naunyn-Schmiedeberg's Arch. Pharmacol., 2011, 384, 93–102 CrossRef CAS PubMed.
- V. L. Kuznetsov, A. T. Bell and Y. I. Yermakov, J. Catal., 1980, 65, 374–389 CrossRef CAS.
- J. Lillis, A. Rokicki, T. Chin and P. C. Ford, Inorg. Chem., 1993, 32, 5040–5043 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cy02199b |
| This journal is © The Royal Society of Chemistry 2018 |