
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoencapsulation of phase change materials for advanced thermal energy storage systems
E. M.
Shchukina
 ,
M.
Graham
,
Z.
Zheng
and
D. G.
Shchukin
*
,
M.
Graham
,
Z.
Zheng
and
D. G.
Shchukin
*
Stephenson Institute for Renewable Energy, Department of Chemistry, University of Liverpool, Crown Street, L69 7ZD, Liverpool, UK. E-mail: d.shchukin@liverpool.ac.uk
First published on 16th April 2018
Abstract
Phase change materials (PCMs) allow the storage of large amounts of latent heat during phase transition. They have the potential to both increase the efficiency of renewable energies such as solar power through storage of excess energy, which can be used at times of peak demand; and to reduce overall energy demand through passive thermal regulation. 198.3 million tons of oil equivalent were used in the EU in 2013 for heating. However, bulk PCMs are not suitable for use without prior encapsulation. Encapsulation in a shell material provides benefits such as protection of the PCM from the external environment and increased specific surface area to improve heat transfer. This review highlights techniques for the encapsulation of both organic and inorganic PCMs, paying particular attention to nanoencapsulation (capsules with sizes <1 μm). We also provide insight on future research, which should focus on (i) the development of multifunctional shell materials to improve lifespan and thermal properties and (ii) advanced mass manufacturing techniques for the economically viable production of PCM capsules, making it possible to utilize waste heat in intelligent passive thermal regulation systems, employing controlled, “on demand” energy release/uptake.
 E. M. Shchukina | Dr Elena Shchukina received PhD in Organic Chemistry at the Belarussian Academy of Sciences, Belarus. She has gained experience in organic chemistry working in different industries. Since 2015, she is project leader in Stephenson Institute for Renewable Energy, University of Liverpool, working on the encapsulation of active materials. |
 M. Graham | Dr Michael Graham received his MChem degree in 2013 from the University of Liverpool. He completed his PhD in chemistry under the supervision of Prof. Dmitry Shchukin in 2017 at the Stephenson Institute for Renewable Energy, before continuing as a postdoctoral research associate. His current research is focused on the nanoencapsulation of crystallohydrate phase change materials for thermal energy storage and thermoregulation applications. |
 Z. Zheng | Dr Zhaoliang Zheng was PhD in Max-Planck Institute of Colloids and Interface (Germany) and Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences. After one-year postdoc experience in the German institute, he joined Prof. Shchukin's group in University of Liverpool (UK). Currently, he is working on (1) designing energy harvest/transfer device based on 2D material-encapsulated phase change material, (2) investigating functional carbonaceous hybrids and (3) novel 3D printed microfluidics for encapsulation of phase change materials. |
 D. G. Shchukin | Prof. Dmitry Shchukin was a group leader at the Department of Interfaces, Max Planck Institute of Colloids and Interfaces, Potsdam, Germany, in 2007–2012. He obtained his PhD (2002) in physical chemistry. Currently, he is Professor in Chemistry, Stephenson Institute for Renewable Energy, University of Liverpool. His main research expertise is encapsulation of energy materials and chemistry at cavitation interface. |
1. Introduction
With each passing year, energy becomes more crucial in modern society. A one third increase in demand is predicted by 2035.1 Fossil fuels have been humanity's greatest energy resource since the dawn of the Industrial Revolution. In 2001, global consumption of energy was 4.25 × 1020 J, of which 86% was produced by fossil fuels.2 However, oil, coal and natural gas reserves are not infinite, and have had an enormous impact on our environment. Large amounts of emitted greenhouse gases in the atmosphere have led to relatively rapid climate change and acidification of the oceans in the last 250 years. Environmental change can already be observed by the behaviour of wild plants and animals, with their geographical distribution being affected.3,4 Fossil fuels can also result in major political problems. Their uneven distribution can cause interdependencies between countries and may even lead to conflict.5Therefore, it is important to develop cleaner energy sources. The best possible energy sources are renewable energies. They are unlimited in the amount of energy they can supply, and often produce zero greenhouse gases. Unfortunately, due to the high demand for immediate power, renewable energies are not currently reliable or economically viable enough to fully replace oil, coal and natural gas. It is vital to develop energy storage systems to ensure clean energy can be provided round the clock.
Solar power is considered the most promising renewable energy due to its abundance, zero cost and lack of emissions.6 The US Department of Energy calculated that the worldwide consumption of energy in 2001 could be met with less than 1 and a half hours of sunlight.7,8
The major drawback of renewable energies such as solar power is their intermittency – when the sun is not shining, no energy can be produced. This is where thermal energy storage is of great importance. Excess of thermal energy can be stored using an energy storage media, which acts as energy sink. The energy can then be released during peak hours to meet demand, known as peak shifting. Factors involved in the selection of heat storage materials include cost, storage density and reliability.
Thermal energy storage approaches
There are three main approaches for thermal energy storage: sensible heat storage (SHS), latent heat storage (LHS) and thermochemical energy storage (TCS). Sensible heat refers to heat that can be detected (“sensed”) by a temperature change in a linear relationship with temperature (as seen in Fig. 1). The heat stored is dependent on the specific heat capacity of the material. SHS is the simplest and most developed form of heat storage, however, it suffers from low energy density and loss of thermal energy at any temperature.9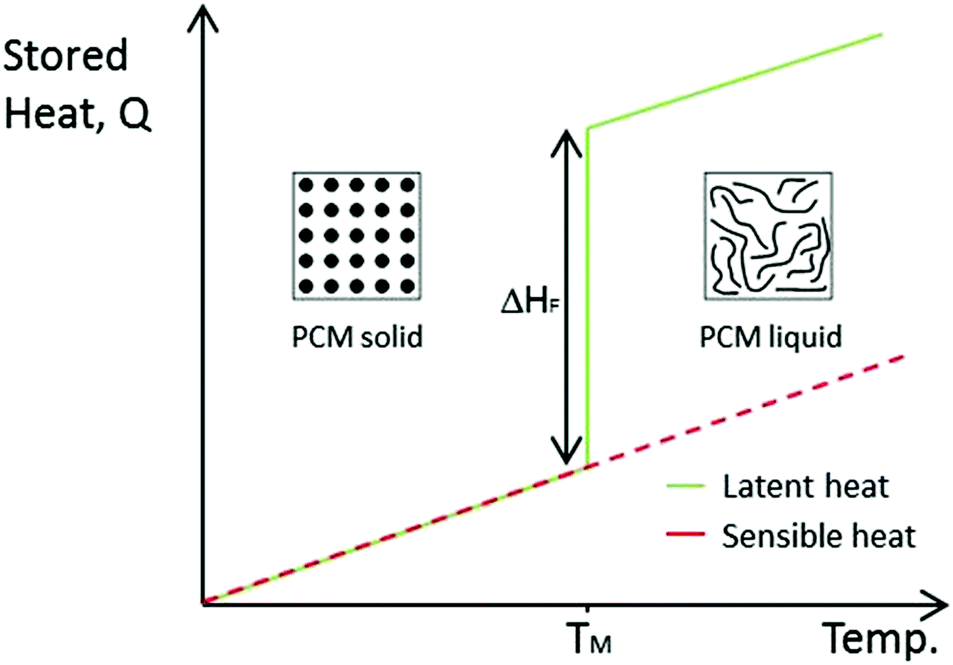 | ||
| Fig. 1 Comparison between SHS and LHS, ΔHF is the latent heat of fusion during melting. TM is the melting temperature. | ||
Latent heat storage refers to heat transfer associated with phase transitions, which cannot be detected with a thermometer. LHS is more efficient and has a far superior storage density than SHS.
Materials that utilise LHS are known as phase change materials (PCMs). Examples of phase transitions include melting and freezing (solid–liquid), evaporation and condensation (liquid–gas) or changes in crystalline structure (solid–solid). Essentially, the energy associated with these changes corresponds to the number of chemical bonds broken. Therefore, solid–gas transitions store the highest amount of energy. However, the large volume change of these transitions means pressurised containers are required. Solid–solid and solid–liquid PCMs have been researched since the oil crisis of the 1970s brought energy to the fore of scientific research. However, once the crisis was down, they were largely forgotten until the 2000s. With the current focus on clean energy sources, PCMs have become widely studied at an increasing rate. As can be seen from Fig. 1, an ideal LHS material stores a large amount of heat isothermally during melting. Once the material freezes, this energy is released. PCMs also store thermal energy sensibly whilst not undergoing phase transition. PCMs are far more efficient than SHS materials, especially over the small temperature range associated with their phase transition.
In 1983, Abhat10 outlined the ideal properties for a PCM:
1. Thermodynamic:
• Melting temperature (TM) in desired application range.
• High latent heat of fusion.
• High density.
• High specific heat for additional SHS.
• High thermal conductivity.
• Congruent melting.
• Small volume changes during phase transition.
• No supercooling.
2. Chemical:
• Chemically stable over long periods.
• Non-corrosive to container materials.
• Non-flammable, non-toxic and non-explosive.
3. Economic:
• Low cost.
• Available in large quantities.
However, there are no PCMs to date which fit all these criteria.
Thermochemical energy storage gives the highest energy density of all, around 5 to 10 times greater than LHS and SHS, respectively. TCS relates to energy stored and released during controlled reversible chemical reactions. Despite the progress made with TCS and its potential for high temperature applications, it has considerable issues with long-term stability. Reactions must have constant conversion efficiency without degradation of energy storage capacity over long periods of time.11 Essentially, the major problem for TCS is the lack of research and understanding. Currently, SHS has been developed to an industrial level, LHS to pilot plant scale, while TCS has only been tested on a laboratory scale.11 There is a recent review focused on TCS materials which we recommend to readers.12 TCS may be valuable in future, but LHS should be the focus for immediate research to solve energy storage issues.
PCMs have several applications, which can be grouped into two categories – thermal regulation and thermal energy storage. Thermal regulation is highly important, and PCMs with a TM in the desired application range can prevent temperature fluctuations with no energy input. To illustrate the vast potential of passive thermal regulation, PCMs with TMs in the human comfort range (10–25 °C) can be used to air condition buildings. Buildings account for approximately 40% of global energy usage, a large amount used for air conditioning.13–15 In 2013, 198.3 million tons oil equivalent were used for space heating in the EU alone. This is especially true of modern lightweight constructions, which suffer from large temperature swings. Other applications benefitting from thermal regulation include Li batteries,16–18 photovoltaics,19–21 spacecraft/spacesuits22 and textiles.23–25
PCMs can also be used as energy storage media for waste heat from industrial processes26 or fuel cells.27 The waste heat can then be reused, for example, it can be transported to off-site purchasers for applications such as greenhousing.28 Energy storage can also be employed to improve the efficiency of concentrated solar power, using high temperature PCMs with transition temperatures above 300 °C (salts or metals).29 Few attempts have been made to encapsulate high temperature PCMs on the macro30,31 and micro32 scale. This review (i) surveys the main encapsulation approaches for nanostructured and multifunctional capsule shells and (ii) focuses on the nanoencapsulation of low temperature paraffin waxes and salt hydrate. For a more detailed PCM loading into matrix-type cores or organic shells, we strongly recommend two recent papers.33,34
PCM classification
PCMs can be classified according to the specific phase transitions they undergo. As mentioned above, the sublimation and evaporation give the highest latent heat of fusion but are not practical due to the large volume change and need for specialised containment to prevent material loss. There are several categories of PCMs, as seen in Fig. 2. Solid–solid PCMs have low latent heat of fusion, and are not considered useful for practical applications. Solid–liquid PCMs give a good balance between high latent heat of fusion and manageable volume change. | ||
| Fig. 2 Classification of various solid–solid and solid–liquid PCMs. | ||
Solid–liquid PCMs can be divided into organic or inorganic materials (Fig. 2). Organic PCMs include paraffin wax, fatty acids and polyethylene glycol (PEG), whilst inorganic PCMs can be salt hydrates, salts or metallic.
Paraffin waxes are linear alkanes containing between 8–40 carbon atoms. Paraffins often display additional LHS in the form of solid–solid transitions associated with different crystalline phases. Their disadvantages include low thermal conductivity, bad odour, flammability and high cost.
Paraffin waxes are also non-renewable, as they are refined from petroleum with bleaching agents. Commercial paraffin contains formaldehyde and vinyl chloride as well as benzene, toluene, naphthalene and methyl ethyl ketone which are volatile and carcinogenic in nature, so care must be taken while using this materials in building applications.35
Fatty acids can be produced from vegetable based oils which are non-toxic. They have lower flash points and longer flame propagation than paraffins. However, their high cost (even higher than paraffin waxes) has rendered them unusable in practical applications. Due to the large volume change on melting, they must also be contained.
Salt hydrates (also known as crystallohydrates) are the major class of inorganic PCMs, and most promising PCMs overall due to their high latent heat, high energy storage density, low cost, abundance, reasonable thermal conductivity and wide variety of melting temperatures in the domestic application range (5–130 °C). Their favourable properties compared to paraffin waxes are displayed in Table 1. They have the general formula M·nH2O where M is a metal salt and n is the hydration number. Salt hydrates’ high energy density is especially attractive as less material is required, reducing the necessary volume of containers and reducing costs further. Salt hydrates have specific densities in the region of 1500–2000 kg m−3, whereas paraffin waxes have specific densities of around 900 kg m−3. Combined with their higher latent heats of around 200–250 J g−1 compared with 150–200 J g−1 for paraffins,36 their energy storage ability is far greater. Paraffin waxes have similar latent heat values to some salt hydrates. However, when the latent heat per unit volume is quoted, salt hydrates will demonstrate greater energy storage ability. Salt hydrates have energy densities of around 250–400 J dm−3 compared with around 125–200 J dm−3 for paraffin waxes.10,37,38 Data for energy density in J dm−3 is sparse in the literature, but is very useful when considering sizes of the macroscale thermal energy storage unit.
| Paraffin wax | Salt hydrate | |
|---|---|---|
| Energy density | 125–200 J dm−3 | 250–400 J dm−3 |
| Latent heat | 150–200 J g−1 | 150–250 J g−1 |
| T M range | −60 to 80 °C | 5–130 °C |
| Thermal conductivity (solid phase) | 0.2 W m−1 K−1 | 0.7–1 W m−1 K−1 |
| Supercooling | No | Yes |
| Congruent melting | Yes | No |
An advantageous property of salt hydrates that is not fully realised is the formation of mixtures and eutectics. When salt hydrates are mixed, their TM is lowered due to the inhibition of crystallisation of the components.39 The ratio which results in the lowest possible TM, always lower than both of the component compounds, is known as a crystallohydrate eutectic. Eutectics have a high latent heat due to the formation of a single phase, and often have reversible phase change without phase separation. Eutectic salts have relatively low thermal conductivity, the same as for single crystallohydrates, which reduces heat transfer.40 Research into eutectics is particularly useful as there are a few known pure crystallohydrates with a TM in the optimal range for applications such as air conditioning.41
Salt hydrates have several disadvantages. Incongruent melting is incomplete melting of the salt hydrate, leading to the irreversible formation of a salt of lower hydration number. This salt then precipitates at the bottom of the melt due to density difference, known as a phase separation. These effects reduce ΔH at the desired TM, and will eventually lead to zero latent heat, rendering salt hydrates chemically unstable, often after very few melting/freezing cycles. Supercooling (calculated from the difference between TM and freezing temperature (TF)) is also a major problem. It is a phenomenon where a material must be cooled far below its freezing point in order to freeze, and is caused by poor heat transfer. This can be as much as 40 °C. Salt hydrates also display corrosiveness towards container materials.42,43
Salt hydrates have varying levels of toxicity. They are generally non-toxic in nature but they can cause skin or eye irritation and respiratory problems. Most crystallohydrate salts have low prices (as cheap as 100 USD per ton for sodium sulphate decahydrate).41
Other major classes of inorganic PCMs are salts and metals, which are most suitable for high temperature applications. They have the widest range of melting temperatures, salts from −86 °C for a 24.8 wt% HCl and water eutectic mix, up to 500+ °C.44 Several metals and alloys have TMs lower than 100 °C; others have TMs of 1000+ °C. The major advantage of metallic PCMs is their high thermal conductivity,45 but they have low storage density. The high mass of metals must also be considered for any practical applications such as their use in building materials, and makes them unsuitable for transportation of heat energy.23 Low temperature metallic PCMs such as gallium (TM = 29.8 °C) have been used to cool computer chips and USB memory drives.46,47 It is anticipated in the future that encapsulation techniques for high temperature PCMs will be developed which will make them available for storage of high temperature heat.29
Low thermal conductivity of commonly used PCMs is the biggest technological problem facing PCMs now.48 This leads to poor life stability of PCM containers and heat exchanger tubing, and also decrease the number of effective cycles they can undergo without any deterioration in their properties.49
Improvement of PCM performance by encapsulation
The practical use of PCMs is hindered by their limitations. For instance, the solid–liquid transition must be suitably contained to prevent leakage. No known PCM fulfils all of their ideal criteria (outlined by Abhat and listed above).10 There are two approaches to modify pure PCMs in order to improve their stability and performance. The first one is to make form-stable PCMs, which is a network on micro or macroscale where PCMs are trapped. These networks are open to the local environment and cannot prevent material exchange, which is particularly important for crystallohydrate PCMs to avoid water loss. This type of PCM control has been covered in a comprehensive review by Kenisarin et al.50The second is the encapsulation of PCMs into micro and nanocapsules possessing ‘smart shell’ properties: controlled thermal energy release, prevention of material exchange with environment, protection against degradation during heat uptake/release cycles, increased PCM surface area and heat conductivity and the possibility to use the capsules in powder or paste form as additives to convenient materials (concrete, foam, paint, etc.) to attain thermal energy storage/release properties. The aim of our review is to highlight the advances in this second approach – to involve readers in the interesting and rapidly expanding area of nanoencapsulation of energy-enriched materials.
2. Overview of encapsulation approaches
Capsules can be macro- (>1000 μm), micro- (1–1000 μm) or nanosized (1–1000 nm). Smaller capsules greatly increase the surface-area-to-volume-ratio of the material, which improves heat transfer. For example, it has been predicted that encapsulating PCMs in capsules of 1 mm in size would increase the surface area by 300 m2 m−3 when compared with the bulk PCM.1 Reducing their diameter to the nanometre range would vastly enhance this effect. Other advantages of encapsulating PCMs include prevention of both leakage and reactions with the external environment, corrosion protection for container materials, control over volume change upon melting and improved thermal cycling stability.51,52 All these properties are crucial to PCM usage in practical applications, so encapsulation can almost be thought of as a “one size fits all” solution. The shell is usually made up of a polymer, as they give a good balance between strength and flexibility. Inorganic shells can also be used, which have higher thermal conductivity (silica is a good example of an inorganic shell for PCMs due to its enhanced heat transfer) but are more brittle. It is also possible to form a composite polymer/inorganic shell combining advantages of each.To create capsules containing active energy materials, an emulsion of the desired droplet size must first be formed, followed by the formation of shell at the emulsion droplet interface. However, the future level of the encapsulation development requires not only the fabrication of the emulsion systems, but also their functionalization in order to realise multifunctional properties.53
Emulsions
An emulsion is a liquid dispersed in another liquid in which it is not soluble or miscible. This is achieved with surface active agents, widely known as surfactants. Due to their amphiphilic nature, they spontaneously form an initial shell around the dispersed liquid to create droplets known as micelles. Emulsions can be oil-in-water (O/W) or water-in-oil (W/O), depending on the PCM to be encapsulated. Usually, the liquid of least volume is dispersed within the other liquid. W/O emulsions (used for crystallohydrate encapsulation) require careful selection of the continuous oil phase and surfactants to give the highest solubilisation capacity for the dispersed phase.54,55 The shell material can either polymerise around the droplets, or can be premade and deposited.It is possible to reduce emulsion droplet size by an external energy input. Common energy inputs include homogenisation and sonication. An emulsion with nanosized droplets formed with a high energy input is known as a miniemulsion (or nanoemulsion). This is in contrast with microemulsions, which also have nanometre sized droplets, but form spontaneously. Miniemulsions are kinetically stable while microemulsions are thermodynamically stable.56 Microemulsions require a larger amount of surfactant than miniemulsions, usually at least 20 wt% of surfactants in the oil phase, whereas miniemulsions require 3–10 wt%.57 Regular emulsions and miniemulsions are thermodynamically unstable due to the spontaneous minimisation of interfacial area between the two immiscible phases.58 Microemulsions, in contrast, display thermodynamic stability as the large amount of surfactants overcome the interfacial energy.52 A comparison of the basic features of the emulsions types is displayed in Table 2.
| Characteristics | Emulsion | Miniemulsion | Microemulsion |
|---|---|---|---|
| Thermodynamic stability | No | No | Yes |
| Stability lifetime | Seconds to months | Hours to months | Infinite |
| Droplet size range | 1–10 μm | 20–200 nm | 10–100 nm |
| Polydispersity | Low | Very low | Very low |
| Typical particle size | 1+ μm | 100–300 nm | 30–100 nm |
Regular homogenisation (such as Ultraturrax) does not provide the required amount of energy to form a miniemulsion, as much energy is lost as heat due to friction.58 Regular homogenisation is therefore only effective at producing microcapsules >1 μm in size.
The use of ultrasound for a wide variety of applications is a recent development in the field of materials chemistry. Due to the reverse piezoelectric effect, electrical energy can be converted to mechanical energy using an ultrasonic transducer.60 Ultrasound is an advanced physico-chemical process, and has been used for applications including the removal of contaminants from water,61 driving reactions,62 materials synthesis,63 cleaning,64 creating new surfaces65 and breaking up aggregates of particles.65 Ultrasound is defined as sound at frequencies above 16 kHz, which is generally inaudible to adult humans, and creates a huge amount of energy produced through a process known as acoustic cavitation. Although not fully understood, the phenomenon is caused by microscopic bubbles forming and rapidly collapsing, producing localised temperatures above 5000 K and pressures of several thousand bars.66,67 This process is schematically shown in Fig. 3.
 | ||
| Fig. 3 Growth and collapse of bubbles, arising from acoustic cavitation. Once the bubble reaches an unstable size it collapses, giving enormous local temperatures and pressures. | ||
There are three major effects from acoustic cavitation: (i) primary sonochemistry – gas phase chemistry occurring inside the bubbles, (ii) secondary sonochemistry – solution phase chemistry occurring outside the bubbles and (iii) physical effects associated with bubble collapse – a shockwave causing strong turbulent effects such as interparticle collisions.67
Miniemulsions are an excellent precursor to the formation of nanocapsules, and sonication is highly efficient at reducing droplet size.68 All sonochemical applications require the optimisation of reaction conditions, such as the total time of sonicating and amplitude. Selecting a suitable probe will prevent splashing or foaming of the liquid, effects which lead to a reduction in the power delivered to the solution. A study was undertaken by Asakura et al.69 to determine how sonochemical efficiency was affected by the amount of liquid in the reactor. They found the optimum liquid height in the reactor is approximately 15 times the height of the wavelength, with the ideal frequency being 200–600 kHz. Lower frequencies mean too few bubbles for cavitation are formed, whereas higher frequencies result in many bubble collisions,70 leading to a reduction in internal bubble temperature and therefore energy.71 Ultrasonic probes are currently not suitable for industrial use, but have the potential to be scaled up in future, as long as several factors of practicality and safety are taken into account.
Additionally, sonication can initiate polymerisation reactions.72 Teo et al. polymerised various methacrylate monomers using pulsed sonication to drive the reaction.62 Capsule formation could theoretically be designed so that sonication facilitates both miniemulsion and shell formation. As a downside, if high molecular weight polymers are desired, sonication may not be suitable as initiator. Longer polymer chains can be degraded by energy from acoustic cavitation.65
Formation of the stable capsule shell
Capsules have found many uses in applications such as in food technology,73,74 dyes,75 catalysis,76,77 corrosion inhibition and self-healing78,79 and drug delivery.80–84 Their main purpose is to provide protection for the core material and control material and energy exchange between core and external environment. Encapsulation is found throughout nature, for example egg shells and cell membranes. Synthesised capsules can be used to encapsulate many species, including drugs, enzymes,85 PCMs and DNA.86,87 They can also be used as a reaction medium, for example Kang et al. showed the rate of a Diels–Alder reaction could be rapidly sped up due to the vastly increased concentration of reactants inside the capsule.88 With technological advances over the last half century, preparing capsules with diameters from 1000 μm down to around 40 nm has become possible. Searching the literature using Web of Science, the first mention of microcapsules is from 1964 where Chang reported the encapsulation of enzymes.89 Nanocapsules are referenced from 1976 onwards, with an early example being Couvreur et al., who demonstrated how polyacrylamide capsules 200 nm in diameter could encapsulate fluorescein.90Capsule size can play a role in functionality. For instance, in drug delivery nanosized drug-loaded capsules have advantages over regular drugs because certain membranes in the body only allow diffusion of molecules less than 100 nm,91 and specific locations in the body can be targeted by specific particles.92–94 The interest in extremely small capsules is essentially an attempt to increase the surface-area-to-volume ratio (SA/V) of the active material. Increasing SA/V is the driving force for all nanotechnology and is inspired by natural structures such as the alveoli in the lungs. Along with increased surface area, nanocapsules provide increased structural stability compared to microcapsules, which may break whilst being pumped round a heating system.52,95,96
There have been many techniques reported for creating micro- and nanocapsules. These include spray-drying,97,98 miniemulsion polymerisation,99,100 precipitation of pre-formed polymers,101 layer-by-layer assembly (LbL),78,85 or other more advanced polymerisation reactions such as radical addition–fragmentation chain-transfer (RAFT)102 and the creation of dendrimers.103 Each of these techniques has their own advantages and disadvantages. While the deposition of pre-formed materials is a simple process, polymerisation reactions are generally more adaptable.59
However, future development of energy capsules should not only concentrate on the fabrication of a single shell around emulsion drops, but also shell functionalization to impart multifunctional properties.53 One of the prospective approaches to attain additional functionality to the emulsion particles is the use of the layer-by-layer shell assembly on their surface. This technique permits the step-wise adsorption of various components (polyelectrolytes, nanoparticles, proteins, enzymes, etc.) and allows the formation of multilayer shells with nanometre precision.104 Capsules with LbL assembled polyelectrolyte shell are used for encapsulation and release of drugs, DNA, dendrimers and enzymes.105,106
This technique is very simple and based on the iterative adsorption of oppositely charged molecules or nanoparticles on a flat surface or template particle (Fig. 4). A charged surface is immersed into a solution of an oppositely charged polyelectrolyte, followed by a washing step to remove excess polyelectrolyte. This procedure results in changes of the surface charge.
 | ||
| Fig. 4 Schematic representation of electrostatically driven layer-by-layer deposition of polyelectrolytes. Reproduced from ref. 104 with permission from the American Association for the Advancement of Science, copyright 1997. | ||
In the next step, the substrate is dipped in a solution of a second, oppositely charged polyelectrolyte. This second polyelectrolyte adsorbs onto the first layer reversing the surface charge. This process can be repeated as many times as desired, yielding multilayered films. In most cases, the technique employs electrostatic forces between oppositely charged polymers and surfaces.107 However, other mechanisms can be employed: hydrogen bonding,108 covalent bonding,109 base-pair interactions,110 guest–host interactions,111 hydrophobic interactions112 or biological recognition.113 The use of the LbL technique to prepare capsule shells offers many attractive possibilities. The method allows control over the composition and thickness of the multilayers in the capsule shell. The reduction of the LbL capsule size from microns to nanometers leads to their higher mechanic stability under deformation forces.114
Several groups employed LbL technology to encapsulate stable oil-in-water emulsions with a high monodispersity.115,116 A usual preparation method for LbL coated emulsion carriers involves several steps (Fig. 5).117 To stabilize the dispersed oil phase (dodecane) of the initial emulsion, it was doped with a small amount of the cationic surfactant dioctadecyldimethylammonium bromide (DODAB). The colloidal stability of initial emulsion was achieved due to concentrated monolayer of strongly positively charged DODAB (z-potential was about +90 mV) at the surface of each droplet.
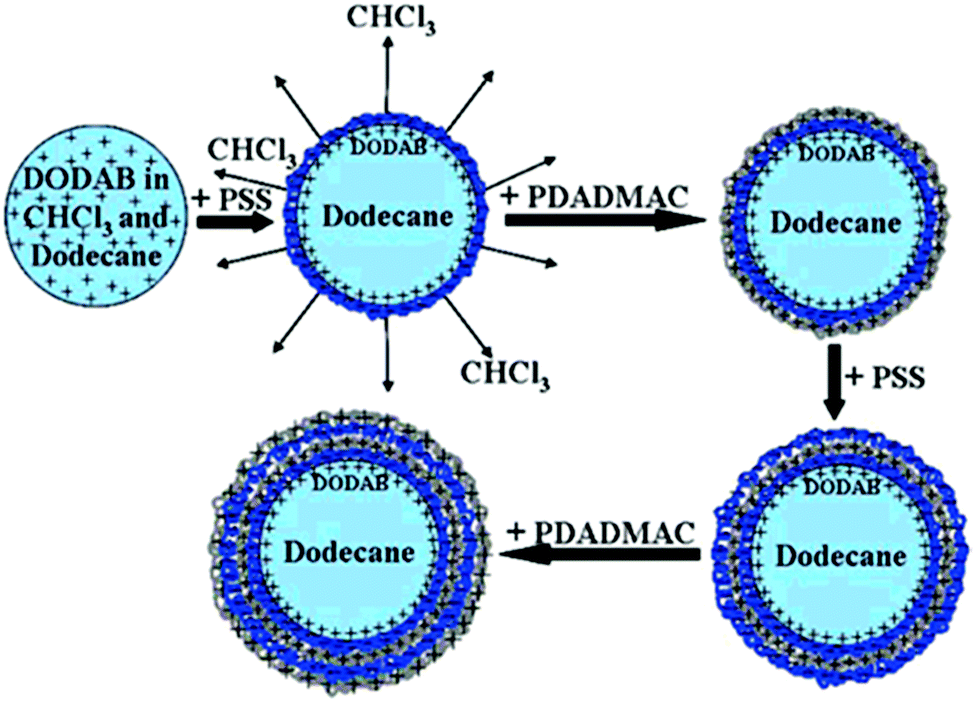 | ||
| Fig. 5 Schematic representation of several steps during LbL polyelectrolyte emulsion encapsulation. Reproduced from ref. 117 with permission from the American Chemical Society, copyright 2008. | ||
Then, the subsequent LbL deposition was performed from concentrated aqueous salt-free solutions of polyelectrolytes:poly(styrene sulfonate), PSS, and poly(diallyldimethyl ammonium chloride), PDADMAC. The further repetition of the alternating adsorption steps leads to the formation of oil-containing capsules with desired shell thickness depending on the particular final demand.
The main drawback of the polymer shell for PCM encapsulation is its low thermal conductivity. The LbL approach allows the addition of inorganic nanoparticles as one or several layers of the multilayer shell. A small amount of particles can lead to an enormous increase in thermal conductivity.118,119
The ultimate level of the capsule shell formation from nanoparticles is called Pickering emulsion (or colloidosomes). Colloidosomes are capsules with shell of nanoparticles localized at the oil–water interface. An economically attractive aspect is the simplicity of the fabrication procedure of such particle stabilized capsules. In principle, only the components (particles, oil, and water) need to be mixed and the application of high shear forces generates capsules with adjustable size. In comparison to surfactant based capsule production, no subsequent purification is required if precise process parameters are met, as all solids will self-assemble on the available oil–water interface. The application of the LbL assembly approach for colloidsomes will close the interstitial pores of the nanoparticulated shell thus preventing material exchange between capsule core and local environment.
The affinity of weak polyelectrolyte coated oxide particles to the oil–water interface can be controlled by the degree of dissociation and the thickness of the weak polyelectrolyte layer.120 To demonstrate this, weak polybase poly(allylamine hydrochloride) has been selected for the surface modification of oppositely charged alumina and silica colloids. Highly stable emulsions can be obtained when the degree of dissociation of the weak polyelectrolyte is below 80%. Cryo-SEM visualization showed that the regularity of the densely packed particles correlates with the degree of dissociation (Fig. 6).
 | ||
| Fig. 6 Cryo SEM images of dodecane droplets stabilized with silica–poly(allylamine hydrochloride) particles. Corresponding pH values of emulsions are (a) 8.5, (b) 9.1, and (c) 9.8. Scale bar equals 500 nm. Reproduced from ref. 120 with permission from the American Chemical Society, copyright 2011. | ||
Silica–poly(allylamine hydrochloride) particles arrange themselves in a monolayer which partially consists of some aggregates below pH 9.2. Above this pH value, flocculation of particles takes place; consequentially, the droplet shell consists almost entirely of particle aggregates.
Comparing to LbL deposition and colloidosome formation, polymerisation approaches for shell formation are more simple and robust, however they lack the possibility to make a PCM capsule shell possessing several functionalities like increased thermal conductivity, elasticity, targeted delivery and affinity to the surfaces of the heat transporting components (e.g., metals) of the macroscale heat capacitors. Additionally, doping of the LbL shell with nanoparticles significantly increases its stiffness and resistance to deformation.121
However, micro and nanocapsule polymerisation using an emulsion template is at a more advanced stage of research than LbL for the fabrication of PCM-loaded capsules. It is also feasible for industrial scale-up.122 Traditional emulsion polymerisation is a type of free radical polymerisation, initiated by radicals which enter monomer droplets.123 By producing a miniemulsion, monomer droplets become much smaller (around 100 nm compared to 1–10 μm). This gives a far greater surface area, and therefore a better chance of radical initiation. The miniemulsion therefore provides an enormous number of parallel reactions taking place inside 1018–1020 nanodroplets.124 Other important types of polymerisation for encapsulation are interfacial polymerisation, in situ polymerisation, polycondensation and polyaddition.48 Methods for polymer shell formation, not directly related to polymerisation, are solvent evaporation and coacervation.35
Many polymer materials can be used to fabricate capsules employing simple polymerisation step.125 These include differing glass transition and melting temperatures, and they may be hydrophilic, hydrophobic or amphiphilic. Materials for encapsulation should be compatible with the PCM, have a higher TM than the PCM and be stable to volume/pressure changes caused by melt–freeze cycles.
The selection of shell materials and structure of the composite PCM capsules is also very important for PCM stability and performance. As an example of lifetime requirements, PCMs incorporated into a building wall for air conditioning requires a life of at least 20 years. Assuming one melting/freezing cycle per day, the material must be stable for around 7300 cycles. An interesting potential solution to extend stability is the use of self-healing capsules. There are several examples in the literature of self-healing capsules which contain shell monomers in the core (e.g. diisocyanate in the core of a polyurethane capsules126). Additionally, self-healing effect can be achieved by combining polymerisation and LbL assembly methods for formation of PCM capsule shell. However, the combination of PCMs and self-healing materials has not yet been explored. The development of multi-compartmental capsules127 may also be of interest in order to use a combination of both organic and inorganic PCMs in one capsule for multitemperature heat storage, along with other beneficial materials such as corrosion inhibitors. Capsules containing multiple active materials are fabricated using multiple emulsions as a template. Zarzar et al. recently produced a simple one-pot method to fabricate complex three and four phase emulsions, demonstrating controllable and reconfigurable morphologies.128
3. Encapsulation of organic PCMs
As mentioned above, containment is crucial in giving PCMs the desirable properties for a wide-scale use. Micro- or nano-capsules can have different shapes such as spherical, tubular or an irregular one. An early example of an encapsulated PCM was a simulation by Theunissen and Buchlin,129 who determined a PCM storage system would require a volume 5 times less than that of a rock bed, in order to store an equal amount of energy. The encapsulation process for that study simply consisted of a large tank. Modern advances in emulsion and polymerisation chemistry allow for the fabrication of PCM capsules at the nanoscale, improving thermal characteristics.Polymer capsules loaded with organic PCMs
The most successful techniques for the fabrication of PCM-loaded capsules have been reactions of miniemulsion, in situ and interfacial polymerisations. During in situ and interfacial polymerisation, a polymer shell is formed at the oil/water interface, rather than within emulsion droplets. In situ polymerisation refers to systems in which the monomer is present only in one emulsion phase, whereas for interfacial polymerisations monomers are present in both phases. The morphology, physico-chemical and thermal properties of encapsulated organic PCMs are dependent on various synthetic parameters: stirring rate, content of emulsifier, content and type of initiator, core/shell weight ratio, shell/initiator weight ratio, polymerization temperature, polymerization time, etc. It is hard to predict the most appropriate combination of synthetic parameters for particular PCM-shell systems, requiring much experimentation to produce the most favourable properties. Typical monofunctional polymer shells for encapsulation of organic PCMs are polystyrene, polymethyl methacrylate (PMMA), urea–formaldehyde, melamine-formaldehyde and polyurethane.Felix et al. have fabricated docosane loaded capsules using polyurethane as shell material, controlling size by varying homogenisation speed.9,130 The capsules were stable over at least 100 heat uptake/release cycles. An interesting effect was that the smaller capsules displayed higher latent heat compared with larger capsules. This shows the benefits of reduced capsule size, resulting in improved thermal performance. Boosting heat transfer to the core material contributes to increased latent heat. Another effect found with decreasing capsule size has been the distinct appearance of multiple crystalline phases during freezing (Fig. 7). Spatial confinement clearly has a significant effect on PCM behaviour.130
 | ||
| Fig. 7 X-ray diffractograms of (a) bulk n-docosane, (b) polyurethane shell (hollow microcapsules), n-docosane loaded into (c) 10 m microcapsules, (d) 4 μm microcapsules and (e) 2 μm microcapsules. Reproduced from ref. 130 with permission from the John Wiley and Sons, copyright 2015. | ||
The diffractogram of polyurethane shell (Fig. 7b) shows two broad peaks, indicating low crystallinity of the shell itself. The encapsulated docosane demonstrates differing crystallinity when capsule size is altered. There are two amorphous peaks, previously seen in the polyurethane shell in the range of 15°–16° and between 16°–25°. The characteristic peaks, which are associated with the triclinic phase of docosane, appear at 19.29°, 19.70°, 22.31°, 23.28°, 23.79° and 24.79°. Face-centered orthorhombic phase appears as a sharp peak at 21.42°; the hexagonal packed phase appears at 21.80° as a small peak. Capsules of 2 μm size (Fig. 7e) display the previously mentioned peaks, which are noticeably more defined. Here, encapsulated docosane also has additional peaks associated with the triclinic phase at 20.07°, 25.37° and 15.95° which are observed in the bulk docosane, but not within the larger capsules of 10 μm (Fig. 7c) and 4 μm (Fig. 7d). This is in agreement with the higher melting and crystallization enthalpies (ΔHm = 79 J g−1 and ΔHc = 88 J g−1) found for the smaller capsules which is related to higher crystallinity. Capsules with 10 μm and 4 μm do not have the additional peaks from the triclinic phase and showed smaller melting and crystallization enthalpies (ΔHm = 47 J g−1 and ΔHc = 48 J g−1 for capsules with 10 μm and ΔHm = 53 J g−1 and ΔHc = 53 J g−1 for capsules with 4 μm).130
Two reports have noted monomer effects on encapsulation using toluene diisocyanate along with amines as crosslinkers for polyurea shell formation.131,132 Amines used have been ethylene diamine, diethylene triamine and Jeffamine (amine-terminated polyoxypropylene). Longer chain amines formed capsules of larger diameter, along with better coverage of the core material. The authors state this is due to the hydrophilic amines requiring migration into the oil phase to react with the diisocyanate. Monomer selection is therefore an important factor to consider when fabricating PCM loaded capsules. Alkan et al. used emulsion polymerization to encapsulate eicosane in polymethyl methacrylate.133 It has been also found that type of shell and core materials affect the capsule diameter, encapsulation ratio and heat storage capacity.
A polymer shell can also be used for encapsulation of paraffin mixtures and eutectics.134 Four alkane mixtures (heptadecane–tetracosane, octadecane–nonadecane, nonadecane–tetracosane and icosane–tetracosane) have been encapsulated into polymethyl methacrylate shell via emulsion polymerisation. DSC thermograms of encapsulated paraffin eutectics had transition temperatures similar to that of the bulk eutectics. The eutectic composition of the mixture is therefore preserved during the encapsulation process.
LbL assembly technique has been applied to build up ultrathin nanostructured shells for PCM capsules.135 The oppositely charged strong polyelectrolytes poly(diallyldimethylammonium chloride) and poly(4-styrenesulfonic acid) sodium salt have been employed to fabricate three multilayer shells with a thickness of ∼10 nm on emulsified octadecane droplets. Using bovine serum albumin as the surfactant, polyelectrolyte encapsulated octadecane spheres with a size of ∼500 nm were obtained with good shell integrity, high octadecane content (91.3 wt%), and thermal stability of 5 cycles.
Nanocapsules with polymer shell are structurally more stable than microcapsules as shown by Sukhorukov et al.114 Many authors have employed miniemulsion method for fabrication of organic PCM nanocapsules. Zhang et at., Fan et al., Konuklu et al. have employed miniemulsion polymerisation for docosane encapsulation into 100–150 nm melamine formaldehyde nanocapsules.136–138 The same method but with PMMA shell has been used by Chen et al. and Sari et al. for paraffins encapsulation into 150 nm capsules.139,140 Yang et al. synthesised poly(methyl methacrylate), poly(ethyl methacrylate) and polystyrene capsules containing tetradecane using in situ polymerisation, and found the acrylate capsules performed far better with regards to heat storage.141 Zhang et al. have also made poly(methyl methacrylate) and poly(ethyl methacrylate)capsules, but used sonication in order to perform a miniemulsion polymerisation.95 This resulted in nanocapsules of 100–150 nm in size and encapsulated octadecane with a high efficiency of 89–95 wt%. Both researchers noted that supercooling of the paraffin was reduced once encapsulated. Ultrasound has been applied by Fang et al. to reduce the size of n-dotriacontane/polystyrene capsules below 200 nm.142 The duration of the ultrasonic treatment does not have linear influence on the capsule size. First, the capsule size is reduced below 160 nm but after continued sonication, average size increases to 200 nm. This effect is caused by aggregation of PCM droplets due to the overall heating of the reaction mixture at prolonged sonication time.
Composite polystyrene/PMMA shell was successfully employed for synthesis of highly stable PCM nanocapsules with octadecane as PCM core.143Fig. 8 shows TEM images of the n-octadecane/polystyrene/PMMA nanocapsules. It can be seen that most of the nanocapsules have regular spherical shape. The shell/core morphology is clearly observed. Dark sections of the images show n-octadecane was located inside the shell, proving successful encapsulation. The nanocapsules are stable after 360 heating/cooling cycles and have phase change enthalpies of 107.9 J g−1 (melting) at 29.5 °C and 104.9 J g−1 (crystallization) at 24.6 °C.
 | ||
| Fig. 8 TEM images of the n-octadecane/polystyrene/(PMMA) capsules at (a) 100k magnification and (b) 30k magnification. Reproduced from ref. 143 with permission from the Elsevier, copyright 2014. | ||
Other methods such as interfacial polymerization and sol–gel precipitation have been used to a lesser extent.144,145 Wang et al. nanoencapsulated an eicosanoic–stearic acid eutectic in a PMMA shell by UV initiated emulsion polymerisation, finding particle size and the size distribution both decreased with agitation speed, use of cross-linking agent and reduction of monomer and initiator concentration. The nanocapsules of ∼46 nm diameter had good thermal stability and displayed decreased supercooling compared to the bulk eutectic.146
Organic PCM-loaded capsules with inorganic shell
The main drawback of organic shells used for encapsulation of organic PCMs is their very low thermal conductivity which, in addition to the low thermal conductivity of organic PCMs, results in very slow heat exchange with environment during heat uptake/release cycles, supercooling and overheating. To improve thermal conductivity, several attempts have been made to encapsulate organic PCMs into inorganic shells, mostly made of inert silica, alumina or clay materials.Yin et al. have made a hybrid SiO2/polystyrene/poly(divinyl benzene) shell using a Pickering emulsion template, resulting in capsules of approximately 100 μm diameter.125 Modified SiO2 nanoparticles were used as stabiliser and, due to the presence of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups on their surface, become embedded in the shell by covalent bonding. Other researchers have reported the formation of a full SiO2 shell by hydrolysing tetraethyl orthosilicate (TEOS) to create an encapsulation precursor. This gave capsules of 8–15 μm and a high encapsulation efficiency up to 87.5 wt%.147 Silica nanocapsules containing octadecane and n-dodecanol core were synthesized by using TEOS as an inorganic source through a sol–gel process.148 The authors noted the capsule thermal conductivity was 0.621 W m−1 K−1 compared with 0.151 W m−1 K−1 for bulk octadecane. They also showed the silica shell has a conductivity of 1.296 W m−1 K−1, compared with polymers which are around 0.20 W m−1 K−1.149 Fu et al. used polystyrene–silica shell for nanoencapsulation of n-tetradecane.150 The resulting capsules have 150 nm size and 83.38 J g−1 latent heat. Addition of silica to the capsule shell by just 1 wt% increased its thermal conductivity by 8.4%. Similar composite nanocapsules can be obtained substituting polystyrene with polymethylmetacrylate.151
C groups on their surface, become embedded in the shell by covalent bonding. Other researchers have reported the formation of a full SiO2 shell by hydrolysing tetraethyl orthosilicate (TEOS) to create an encapsulation precursor. This gave capsules of 8–15 μm and a high encapsulation efficiency up to 87.5 wt%.147 Silica nanocapsules containing octadecane and n-dodecanol core were synthesized by using TEOS as an inorganic source through a sol–gel process.148 The authors noted the capsule thermal conductivity was 0.621 W m−1 K−1 compared with 0.151 W m−1 K−1 for bulk octadecane. They also showed the silica shell has a conductivity of 1.296 W m−1 K−1, compared with polymers which are around 0.20 W m−1 K−1.149 Fu et al. used polystyrene–silica shell for nanoencapsulation of n-tetradecane.150 The resulting capsules have 150 nm size and 83.38 J g−1 latent heat. Addition of silica to the capsule shell by just 1 wt% increased its thermal conductivity by 8.4%. Similar composite nanocapsules can be obtained substituting polystyrene with polymethylmetacrylate.151
Changing silica content and hydrolysis rate, the morphologies of the PCM-loaded capsules can be regulated from thin-shelled nanocapsules with bowl-like, hemispherical or spherical geometries to thin-shelled spherical nanocapsules and even mesoporous particles (Fig. 9).152 At low CTAB concentration (15 mM), bowl like or spherical nanocapsules with well defined core–shell structure are obtained, and the shell thickness is relatively low. However, at higher CTAB concentration (30 or 35 mM), spherical nanocapsules with thicker shells are acquired. The PCM content in mesoporous silica nanocapsules is 30 wt% less than in hollow ones. Due to the confinement of n-octadecane in nanosized organosilica shells, the homogeneous nucleation is suppressed, resulting in a notable shift of crystallizing points. Encapsulated n-octadecane crystallises via shell induced heterogeneous nucleation and thicker organosilica shell induces the heterogeneous nucleation better. After 500 melting/solidifying cycles, the nanocapsules maintained their phase transition properties perfectly, indicating high thermal reliability.153 In addition, the hydrophobicity of the organosilica shell materials can be tuned by changing the volume ratios of silane precursors. The theoretical modelling showed that the mobility of the encapsulated n-octadecane is restrained within constrained SiO2 shell.154
 | ||
| Fig. 9 (a, b, e, f) SEM and (c, d, g, h) TEM images of the silica nanocapsules loaded with n-octadecane prepared at different CTAB concentrations. Reproduced from ref. 152 with permission from the Elsevier, copyright 2016. | ||
Besides improved thermal conductivity, inorganic shell materials can also provide flame-retardant properties to the encapsulated organic PCM. Demirbag et al. have reported a study on the thermal stability and flame-retardant properties of PCMs encapsulated into clay nanoparticles doped gelatine/sodium alginate shell.155 The capsules have been fabricated by the technique of complex coacervation using gelatine and sodium alginate as the shell and n-eicosane as PCM core. Nanocomposite structure of the capsule shell demonstrates improved flame retardant properties of cotton fabrics when treated with PCM-loaded capsules. Similar flame-retardant effect has been achieved for gelatine/gum arabic and gelatine/sodium alginate shell doped with alumina nanoparticles.156
Nanostructured shells from C-based materials
Although inorganic shells demonstrate thermal conductivity enhancement, their long-term stability is brought into question due to their brittleness. Despite good thermal and chemical stability, they may fracture due to stress formed by PCM volume change during melting. Hence, the next generation of shell materials for encapsulation of organic PCMs should combine both high thermal conductivity inherent to the inorganic shell and elasticity of the polymer shell. This can be done using nanostructured carbon-based materials as main shell component.An excellent example of a multifunctional shell for paraffin encapsulation was demonstrated by Zheng et al.157,158 The capsules are comprised of encapsulated paraffins and graphene oxide/carbon nanotube hybrids (GO–CNT) as core and shell material, respectively. Multiform carbon nanotubes stabilize GO capsule shell to resist volume-change induced rupture during heat uptake/release and enhance the thermal conductance of encapsulated paraffins.
The GO–CNT hybrids along with CNT clots (Fig. 10) were formed under pulsed tip-sonication and then directly applied in the following ultrasound-induced emulsification with melted docosane. Detailed observations at the sections of ultramicrotomed docosane/GO–CNT have unclosed the multiform CNTs in capsules: completely horizontal adherence on GO layers (Fig. 10a and d), partial adherence and partial inwards extension (Fig. 10b and e), and clot attached to the inner wall of capsules (Fig. 10c and f). The partial coverage is common among the CNTs longer than the persistence length, they are modeled as semi-flexible chain with configuration of partially adhering on GO layer and partially penetrating into the interior (Fig. 10e).
 | ||
| Fig. 10 Structural and morphological characterization of docosane/GO–CNT microcapsules. TEM images of ultrathin sections from ultramicrotomy showing (a) the CNTs are completely adhered on GO layers, (b) partially adhered and partially inwards extended, and (c) even led to clots embedded in docosane, respectively. (d), (e) and (f) Show schematic models of the three configurations of CNTs within docosane/GO–CNT microcapsules. The grey lamellar and yellow wires represent the GO sheet and CNTs, respectively. TEM images of GO–CNT hybrids in bright field imaging mode (g) and high-angle annular dark-field imaging mode (h) are shown. Molecular dynamics simulation of the GO–CNT assembly show side view (i top) and tilted top view (i bottom) of a snapshot during the relaxation process. The wire-like nanotube and silk-like GO can be distinguished. Besides, a CNT clot (j) is adhered by GO sheets. Scale bar: (a–c) 500 nm; (g and h) 50 nm; (i) 5 nm; (j) 200 nm. Reproduced from ref. 157 with permission from the American Chemistry Society, copyright 2016. | ||
The original shape of docosane/GO–CNT solid powder was retained at temperatures higher above the melting point of docosane; whereas the unprotected docosane deformed into liquid quickly. There is little change in docosane/GO–CNT thermal properties after 100 thermal cycles without notable supercooling. The average latent heat of encapsulated docosane remained around 240.8 J g−1, leading to an encapsulation ratio of paraffin as high as 96.7 wt% by comparing with the enthalpy of bulk state.
Through changing the cooling rate of original emulsions, it was possible to control the inner structure of the organic PCM capsules.158 The special geometry of PCMs is often hidden beneath the capsule shell and it influences whole system performance on molecular level. Hollow and solid structures of the PCM core determine the thermal properties of energy capsules with nanocarbon shell. The pronounced C–H⋯π interaction of hollow PCM core might be responsible for more stable encapsulation and greater heat diffusivity of melted PCMs, as compared with solid PCM core with weak PCM-shell interaction. Graphene nanosheets can be used to increase thermal conductivity in nanoencapsulated PCMs, however this leads to an undesirable rise in viscosity.159 The dramatic viscosity growth, up to over 100-fold at the highest loading, deteriorates significantly the intensity of natural convection, which has been identified as the dominant mode of heat transfer during melting of PCM core. The loss in natural convection was found to overweigh the enhancement in heat conduction, thus resulting in decelerated melting.
Eicosane-loaded capsules with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of ethyl cellulose (EC):methyl cellulose (MC) as shell material have been prepared through a novel liquid to solid encapsulation process.160 The obtained water suspension of microspheres was cooled below the melting point of eicosane, thus solidifying the core material. Solidification is accompanied by a decrease in volume, thus accounting for the observed dents in the capsules (Fig. 11).
:1 ratio of ethyl cellulose (EC):methyl cellulose (MC) as shell material have been prepared through a novel liquid to solid encapsulation process.160 The obtained water suspension of microspheres was cooled below the melting point of eicosane, thus solidifying the core material. Solidification is accompanied by a decrease in volume, thus accounting for the observed dents in the capsules (Fig. 11).
 | ||
| Fig. 11 (a) SEM, (b and c) TEM, and (d) optical microscopic images of the eicosane/EC/MC capsules prepared with a 9 wt% polymer content. Reproduced from ref. 160 with permission from the American Chemical Society, copyright 2011. | ||
The indentation of the wall of the capsules correlates well with the maximum interaction between eicosane molecules and the ethoxy moieties of the EC polymer. DSC measurements have demonstrated the increase of the absolute enthalpy value during the crystallization of the eicosane-loaded EC/MC capsules as compared to the pure eicosane, implying that the encapsulated eicosane molecules release more energy upon solidification. With the presence of 9 wt% of the EC/MC polymer, the capsules demonstrated a 24% increase in heat absorption and a 29% increase in heat release. It is likely that the interaction between the EC/MC shell and the encapsulated eicosane causes a different phase transition path for the encapsulated eicosane than for free eicosane. The new path contains multiple broad overlapped transition steps, releases more energy than the two-step crystallization of the pure eicosane and gives crystals with a different XRD pattern. An encapsulation process that allows for a good specific interaction between the polymer shell and the PCM is therefore crucial.
There are several other examples of using nanocellulose-based materials as PCM shells in form of Pickering emulsions. Capron et al. have produced Pickering emulsions of hexadecane in water stabilized with bacterial cellulose nanocrystals.161 High stability has been observed without any droplet size change after centrifugation. Svagan et al. have reported chemically cross-linked nanocellulose-based capsules with a hexadecane core.162 Although no thermal properties were recorded, the capsules had excellent elastic properties which are ideal to withstand volume changes. Li et al. have encapsulated RT25HC paraffin (Rubitherm Technologies GmbH) in nanocellulose, employing a Pickering emulsion method combined with ultrasonication.163 Nanocellulose diffuses into the paraffin core and stabilizes water/paraffin interface, forming a shell around paraffin droplets. Excess of nanocellulose forms a three-dimensional network with embedded energy capsules thus preventing their coalescence. The PCM composite has showed a solid content of paraffin of 72 wt%.
4. Encapsulation of inorganic PCMs
Salt hydrates are difficult to encapsulate due to their hydrophilicity, tendency to alter water content and surface polarity.164,165 Further added to these issues is their general chemical instability, which is well documented.10 Encapsulation can promote improved stability due to effects such as confinement of stoichiometry and improved heat transfer to decrease supercooling. It also acts as a barrier to prevent water loss from the crystallohydrate core, maintaining its thermal characteristics (Fig. 12). The enhanced energy storage properties of inorganic PCMs compared with organic ones make further research into their encapsulation highly beneficial.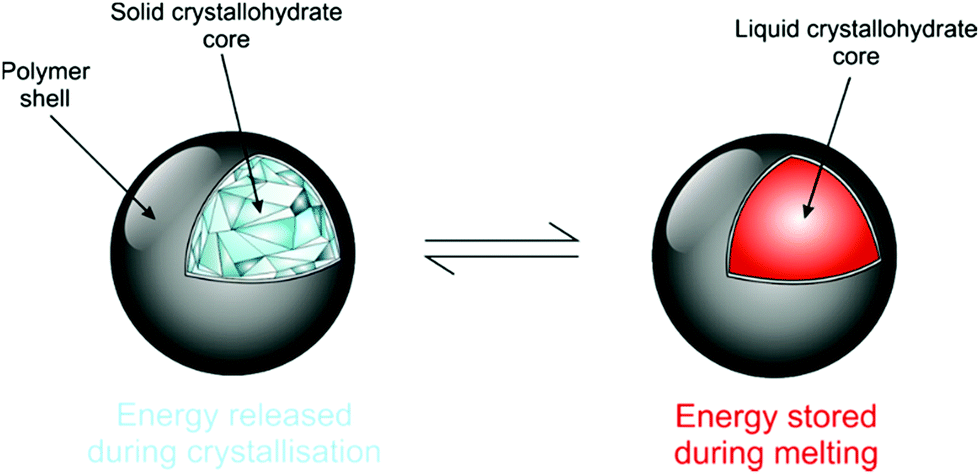 | ||
| Fig. 12 Cartoon showing ideal capsule behaviour for the salt hydrate core. Reproduced from ref. 173 with permission from the Royal Society of Chemistry, copyright 2017. | ||
Several chemical encapsulation methods have been reported in a review by Milian et al.166 Inverse Pickering emulsion, interfacial polymerisation and solvent evaporation–precipitation are the most common chemical methods described for the encapsulation of inorganic PCMs although there are relatively few examples to date.
In 2001, Gröhn et al. encapsulated chloroauric acid trihydrate within a dendrimer system, although this was not for energy storage purposes.103 To the best of the authors’ knowledge, the first attempted core–shell capsules containing salt hydrates specifically for energy storage were developed by Sarier and co-authors.167 They used a mix of PEG1000, hexadecane and sodium carbonate decahydrate (Na2CO3·10H2O) as core material encapsulated in an urea–formaldehyde shell. They noted the phase change behaviour resembled that of hexadecane and Na2CO3·10H2O did not contribute. The same authors also developed a method of trapping PCM micelles inside a polyurethane foam.168 One of the PCM combinations they used was octadecane and Na2CO3·10H2O. However, they found Na2CO3·10H2O acted merely as a blowing agent due to its water content, and did not contribute to the latent heat. As these researchers have been using O/W emulsions rather than W/O, the resulting capsules contained very little salt hydrate.
Salaün et al. have encapsulated Na2PO4·12H2O in a polyurea/polyurethane shell.169 They employed the solvent evaporation technique, dissolving cellulose acetate butyrate in a volatile solvent (chloroform). As the volatile solvent evaporates, polymerisation of cellulose acetate butyrate occurs. They used methylene bis(phenyl 1 isocyanate) as the crosslinker to form the polyurea shell. They have not demonstrated thermal energy storage characteristics or accurate size measurements (only stating capsules were >1 μm), although they did show the capsules contained a large amount of salt, up to 79 wt% of loading. In further research, they investigated the influence of solvent on capsule properties. They found that changing the solvents for the dispersed and continuous phases had a profound influence on the characteristics of capsules. Using chloroform as dispersed phase solvent instead of acetone facilitated the full coverage of the core with shell material. Using toluene as continuous phase instead of carbon tetrachloride reduced coacervation rate and produced better defined capsules. DSC results revealed that most of the formed capsules had incomplete crystallisation processes, meaning that by the 2nd thermal cycle, no latent heat storage was possible. The most successful capsules used toluene and chloroform as solvents with a latent heat of melting of 140.4 J g−1. The latent heat of crystallisation was only 48.9 J g−1, suggesting that long-term stability is not possible.
Similar work has been done by Liu et al., on the encapsulation of sodium thiosulfate pentahydrate into microcapsules with silica shell.170 The authors demonstrated successful encapsulation of inorganic PCM into silica shell by sol–gel method with high encapsulation rate (94.65 wt%). However, the stability of the capsules during the heat uptake/release cycles was not demonstrated.
Graham et al. have demonstrated a simple method to nanoencapsulate magnesium nitrate hexahydrate, employing an in situ miniemulsion polymerisation with ethyl-2-cyanoacrylate as monomer.171 Using sonication to prepare miniemulsions improved the synthesis by reducing the amount of surfactant required as stabiliser.
Before melting, pure Mg(NO3)2·6H2O (TM = 89 °C) is a crystalline solid (Fig. 13). After melting, it recrystallizes to the solid shown in Fig. 13c. This solid is surrounded by water, showing a volume change occurs during phase transition. The recrystallized solid forms a compact block, which prevents free diffusion of water vapour. In contrast to the bulk material, nanoencapsulated salt hydrate (Fig. 13b and d) shows no volume increase or change in appearance before and after heating to 100 °C. This indicates its chemical and structural stability at the transition temperature. The absence of leakage shows the salt hydrate is fully protected by encapsulation from the outside environment, helping prevent changes in salt hydrate composition in the nanocapsule core. These observations are consistent with their DSC results, which showed the Mg(NO3)2·6H2O was stable over at least 100 cycles, remaining fully hydrated due to the addition of extra water to the salt hydrate core, a principle previously demonstrated in macroscale systems.172
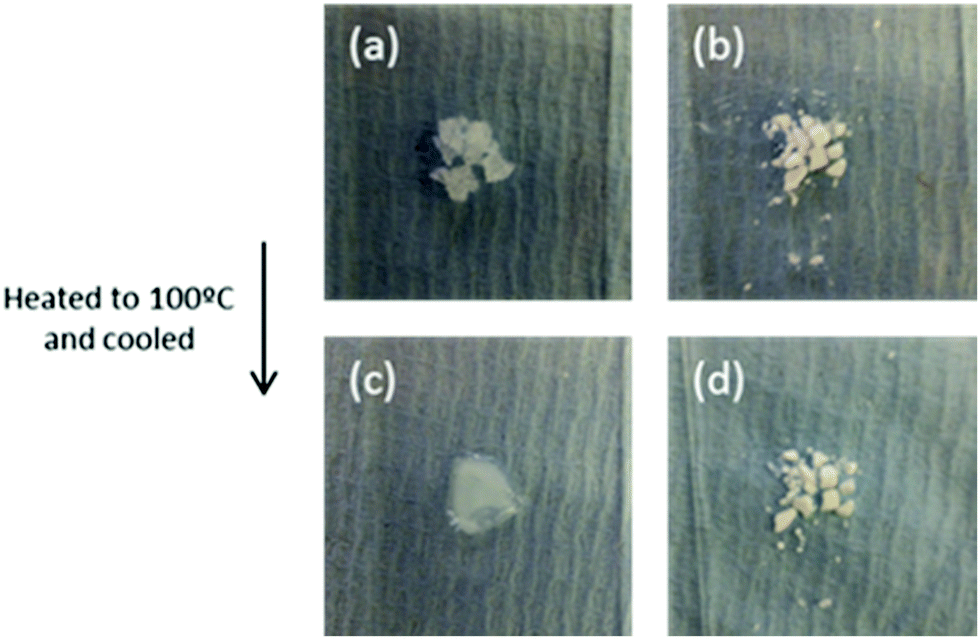 | ||
| Fig. 13 Bulk Mg(NO3)2·6H2O (a and c) and nanoencapsulated salt hydrate (b and d) before heating to 100 °C (top), and after letting them cool back to room temperature (bottom). Reproduced from ref. 171 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Their follow-up paper describes encapsulation of two crystallohydrates (Mg(NO3)2·6H2O and Na2SO4·10H2O) and their eutectic mixture.173 DSC results demonstrated high thermal stability of nanoencapsulated single and mixed crystallohydrates, which remained unchanged after 100 thermal cycles (Fig. 14). Encapsulation of the crystallohydrate mixtures prevents the loss of water during prolonged thermal cycling, resisting changes to the TM and latent heat, and also reduces supercooling (Fig. 14C and D). After encapsulation, the 1:2 Mg(NO3)2·6H2O:Na2SO4·10H2O eutectic ratio (Fig. 14D) was maintained, with one well defined phase transition peak at TM = 15.4 °C and TF = −1.1 °C and reduced supercooling of ΔT = 16.5 °C. The transition is stable over >100 heat uptake/release cycles and has latent heat capacity of 126.8 J g−1, which results in 67 wt% encapsulation efficiency. The effect of nanoencapsulation on salt hydrate mixtures is similar to its effect on single crystallohydrates, producing thermally stable energy capsules. The authors demonstrated that additive mixtures of nanocapsules containing single crystallohydrates have good potential for the design of multi-temperature heat storage systems containing energy capsules with different PCM cores sensitive to multiple transition temperatures.
 | ||
| Fig. 14 DSC data for (A) 1:1 wt% Mg(NO3)2·6H2O:Na2SO4·10H2O bulk mixture, (B) 1:2 wt% Mg(NO3)2·6H2O:Na2SO4·10H2O bulk mixture, (C) encapsulated 1:1 wt% Mg(NO3)2·6H2O:Na2SO4·10H2O and (D) encapsulated 1:2 wt% Mg(NO3)2·6H2O:Na2SO4·10H2O. Reproduced from ref. 173 with permission from the Royal Society of Chemistry, copyright 2017. | ||
Huang and collaborators fabricated capsules with Na2PO4·12H2O as core material and methyl methacrylate as monomer along with ethyl acrylate as crosslinker.164 The shell has been made by a suspension polymerisation combined with solvent evaporation. They found that, upon encapsulation, the PCM was partially dehydrated to form Na2PO4·7H2O which resulted in an increase in melting temperature from 36 °C to 51 °C. They also made a follow up article, where they compared their PMMA capsules to urea formaldehyde ones.174 The PMMA capsules demonstrated much better thermal characteristics, while urea formaldehyde capsules have very broad melting temperature peak at 41 °C, which is unsuitable for practical applications when a narrow temperature range is required. Importantly, upon encapsulation the thermal conductivity increased from 1.01 W m−1 K−1 for pure Na2PO4·12H2O to 1.426 W m−1 K−1 for the encapsulated Na2PO4·7H2O.166
Platte et al. have encapsulated different mixtures of sodium sulphate, sodium phosphate and sodium carbonate which were hydrated by dissolving in water.165 They used a surface-thiol Michael addition polymerisation using ORMOCER polymers as shell material, which are biodegradable inorganic–organic hybrid polymers developed by Fraunhofer ISC, Germany. ORMOCERs are impermeable to water, which is of great benefit to encapsulated salt hydrates to maintain the desired hydration state. Formed capsules are around 40 μm in diameter. Supercooling was still a problem, showing the capsules did not sufficiently improve heat transfer. Schoth et al. have developed a surfactant free method to encapsulate sodium sulphate decahydrate.175 They utilised the Pickering emulsion technique to create the initial emulsion, resulting in polyurethane nanocapsules with an average size of 750–1000 nm. It was also shown that Na2SO4·10H2O could be encapsulated up to 20 wt% (its solubility limit in water). Hassabo et al. prepared PCM capsules with a poly(ethoxysiloxane) shell. The capsules contained crystallohydrates and a Pickering emulsion template of silica nanoparticles was used.176 Silica dispersions mixed with inorganic salts in various ratios were dissolved in toluene, and pH was adjusted to pH = 1 using HCl. Encapsulation had minimal effect on the TM, however latent heat was low.
An interesting approach for salt hydrate encapsulation is the use of nanobowls, consisting of SiO2 matrix impregnated with Na2SO4·10H2O.177 The formation of the unusual bowl shape is due to non-synchronous rotation of droplets caused by viscosity differences between the liquid and solid phases. The SiO2 matrix improved heat conductivity of the PCM, as well as reducing phase separation. The sample also had high latent heat of 180.7 J g−1 which was relatively unchanged after 60 cycles. However, supercooling was only marginally reduced.
Strong interface interactions between core and shell materials can influence on the heat uptake/release of the capsules, similar to that observed for encapsulation of organic PCMs.159 Another factor is the nature of shell material, which presents different heat conduction and natural convection contributing to heat transfer as much as melting and glass-temperature.178
5. Cutting edge systems for thermal energy storage based on encapsulation
In this section of our review, we concentrate on three fields that have potential to be greatly accelerated by incorporating nanoencapsulated PCMs including heat-transfer fluid and PCM slurry, energy harvesting and conversion, and 3D printed functional composite. A few examples of microcapsule use for thermo-regulated textiles179,180 and building envelope materials181 have been reported but they are beyond the topic of this review. The toxicity issue of nanomaterials to the environment182 and human health is not yet fully understood,183 so these effects must be studied with regards to PCM nanocapsules before their widespread use.Heat-transfer fluid and PCM slurry
A heat-transfer fluid (HTF) is often utilized to transfer thermal energy from a hot source to a cold one, to maintain the stability of device performance and industrial safety.184 Of importance to determine the heat-transfer efficiency is the heat capacity of the fluid.184,185 PCMs exhibit a high latent thermal energy capacity near the melting point. Thus, properly selected PCMs can be dispersed in the liquids and enhance their heat capacity at the specific temperature range,186 leading to an enhanced heat-transfer capability of the fluid.187,188 The combination of HTF and dispersed PCMs is called a PCM slurry. Han et al.189 theoretically demonstrated the impact of volume fraction (Φ) and latent heat of dispersed PCMs to the effective specific heat (Ceff) of the HTF, as outlined in the equation:| Ceff = C0 + ΦHPCM/ΔT |
Advanced microchannel heat sinks (Fig. 15) have become popular in recent years due to overheating generated by the miniaturization and high integration of electronic components or systems.193 As a novel working medium, the nanoencapsulated PCM slurry enhances both the thermal capacity and conductivity during phase transition process.194 A typical example is silica nanoencapsulated indium PCMs applied into poly-a-olefin (PAO) thermal fluid for high temperature (150–180 °C) microchannel heat exchange.192 The silica shells not only minimize the leakage of liquid indium but also prevent agglomeration of nanocapsules at high temperature. Experiments with the microchannel heat exchanger indicated that the optimized heat transfer coefficient of nanoencapsulated PCM slurry could reach 47000 W m−2 K at a flow rate of 3.5 ml s−1. The magnitude of heat transfer coefficient represents a twofold improvement over that of single phase PAO. Seyf et al.195 have systematically investigated the influence of mass concentration and melting range of nanoencapsulated PCM, as well as Reynolds number on the thermal-hydraulic performance of a microchannel heat sink. They found that a combination of nanoencapsulated PCM and PAO fluid can enhance the thermal performance of the device in the aspects of lowering entropy generation and decreasing temperature uniformity. However, extra attention should be paid to eliminate the pressure drop as a consequence of increased mass concentration. Like many other applications involving PCMs, high thermal energy storage capacity and heat transfer efficiency of the nanoencapsulated PCM slurry are only possible near the phase transition range (PTR). Wang et al.186 found that the heat capacity of a nanoencapsulated PCM slurry below the melting point would likely decrease with increased PCM mass fraction due to the lower sensible heat capacity of PCM than the base fluid, which limits the use of single PCMs for this application. A solution to this problem may be to employ multiple PCMs with different TMs in a cascaded approach, arranged in order of decreasing TM with regards to the direction of HTF flow during charging. This has been shown to increase thermal efficiency, due to maintenance of a constant temperature difference between the PCMs and HTF.196 Qu et al. recently confirmed the enhancing role of nanoencapsulated PCM by computing the thermo-physical properties of slurries.197 In addition to the mass concentrations and Reynolds number, they also found higher heat transfer rates could be obtained by reducing the thermal boundary layer thickness at stagnation zone of slot jet impingement mode.
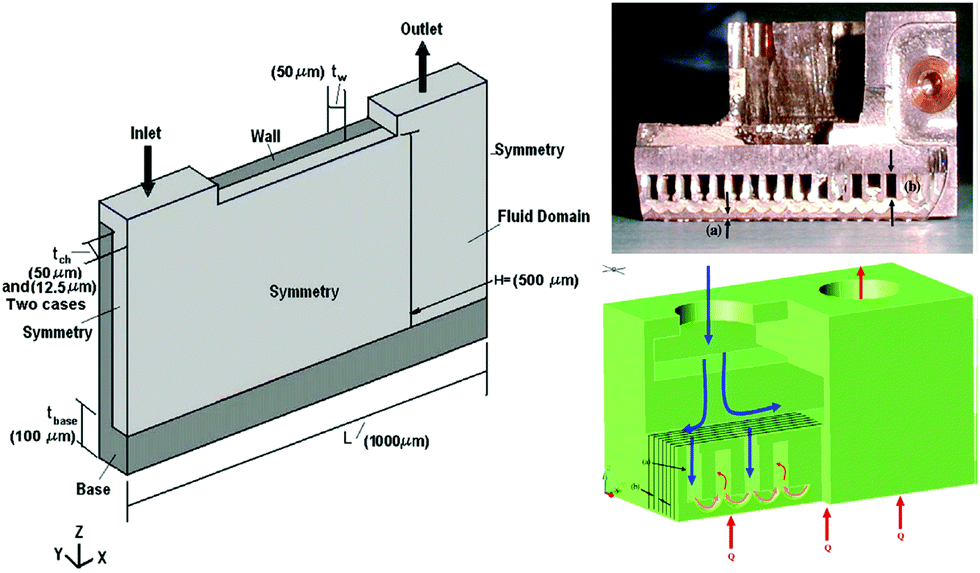 | ||
| Fig. 15 Design concept and cross-sectional view of microchannel heat exchanger. Reproduced from ref. 192 with permission from the Elsevier, copyright 2013. | ||
Energy harvesting and conversion
Solar-thermal and electro-thermal conversion, where either solar irradiation or electrical energy is harvested and converted to heat for beneficial usage, has been widely used in daily applications. The primary evaluation criterion is the output temperature at a certain input power, encouraging research to increase the energy conversion efficiency of the device further. Due to their large LHS capacity over the PTR, ideal PCMs will facilitate a decrease in size of cooling systems. This concept is emerging for using micro/nanoencapsulated PCMs as additives/dopants to thermal conversion systems.159 However, the main challenge is to increase the conversion efficiency not only at the PTR but also at temperatures lower or higher than the PTR. The PCMs store a higher accumulative energy (latent heat + sensible heat) above PTR than that within PTR, while exhibiting a much lower specific heat capacity. In other words, the liquid PCMs above the PTR can release the heat back to the heat-generating systems in the absence of phase transition. Especially at low volume/mass fraction, the synchronous temperature increase of incorporated PCMs and heat-generating structures is widespread. The heat generated by the incorporated PCMs, therefore, offsets the convective heat dissipation occurring at the outer surface,198 especially for sponge structures with a porous network and large surface area constantly enabling rapid exchange of heat with the environment but meanwhile suffer from severe convective heat dissipation.Based on the above considerations, Zheng et al. fabricated GO–CNT hybridized capsules containing long-chain alkane PCMs.158 The highly thermal conductive shell facilitates quick heat exchange between the confined alkanes and environment. Fig. 16a shows that the subcooling circle and delayed structural change of GO–CNT encapsulated PCMs were avoided, indicating a timely and sufficient structural change of alkane in response to temperature change. This enables the encapsulated PCM to homogeneously dope, resulting in the “built-in” structure (Fig. 16b). The composites with dopant at 25 wt% maintain similar temperature-dependent electrical resistivity curves with the pristine GO joule heater. Notably, the composite maintains an around 5–10% enhancement in output temperature, either within PTR or the temperature lower or higher than the PTR (Fig. 16c).
 | ||
| Fig. 16 (a) DSC curves of bulk docosane and docosane encapsulated by GO, CNT and GO–CNT. (b) High-resolution SEM shows the capsules are embedded at basal plane of GO sheets, resulting in a “built-in” structure. (c) As a function of voltage, the balanced surface temperatures are demonstrated for composites containing 0, 5, 25 and 50 wt% docosane/GO–CNT capsules. Reproduced from ref. 158 with permission from the American Chemical Society, copyright 2017. | ||
3D printed functional composites
Additive fabrication technologies, better known as 3D printing, have witnessed an incredible development in recent years, owing to the versatile and low-cost method for rapid casting and prototyping. In this technology, the computational architecture is realised by solidifying liquid or pre-melted material via a straightforward layer-by-layer fabrication process. Many nanoscale functional materials have been introduced as nanofiller into printable resins and/or included in the pre-blending of materials,199 resulting in 3D printed composites exhibiting unique characteristics and capabilities, especially for controlled thermal properties. For example, 3D printing of composites with a relatively low amount of thermal conductive nanofillers, such as carbon nanotubes, graphene or metal nanoparticles, allows one to build objects with high thermal conductivity. The enhanced thermal conductivity accelerates the utilization of 3D printed composites in the heat sink and cooling system for heat management applications. Similar concepts can be applied to produce thermal insulation materials which have a variety of applications, such as reactionware for the chemistry.200Commercial composite filaments for 3D printing systems are becoming widely available, produced by companies such as 3DXTech, Grafoid, Graphene 3D Lab, Haydale, etc. Relatively few technical problems are yet to be solved, such as temperature variations within a polymer matrix, aggregation at printing nozzle, inhomogeneity of resin, etc. Researchers have been shifting to print the functional nanoscale material itself without being used as the additive. We expect this emerging technique will facilitate the integration of nanoencapsulated PCM for advanced conceptual devices. Using the Digital Light Processing technique, Krajnc et al.201 printed a 3D hierarchical structure from a high internal phase emulsion (HIPE). The W/O emulsion contains 80 wt% droplet phase and photocurable continuous phase. Layer-by-layer assembly allows immobilization of droplets into free-standing complex 3D devices with excellent feature resolution. More recently, Magdassi et al.202 printed a monolithic porous structure from a 50–70 wt% O/W miniemulsion (Fig. 17).
 | ||
| Fig. 17 Images of printed emulsion with 60 wt% droplets phase through Digital Light Processing with an average diameter of 1.5 ± 0.03 mm (A) image of the device (B) SEM image of a cross-section (C) SEM image of the upper surface of the structure. Reproduced from ref. 202 with permission from the Royal Society of Chemistry, copyright 2015. | ||
Unlike the HIPE with high viscosity, the resin is composed of a UV polymerisable O/W dispersed phase which can feasibly be used in other printing techniques based on photo-polymerisation, for example photocurable inkjet printing.203 This method features simultaneous compartmentalization and device processing, allowing encapsulated materials to be directly made for applications. Besides light-based printing methods, direct ink writing of viscoelastic materials under ambient conditions offers a broad spectrum of printable materials for energy storage. Direct printing of nanoencapsulated PCMs is possible if the ink's viscosity, surface tension, shear yield stress, shear elastic and loss moduli can be properly tailored.
At the end, research on the applications of nanoencapsulated PCMs is still at an early stage, although some of researchers have approached the waste heat recovery, thermal management of electronic devices, energy conversion, intelligent building, “smart” air conditioning and thermal regulating fabric, etc. The main hurdle is to widen the choice of core and shell material through the optimal preparative process. Meanwhile, close-to-100% encapsulation efficiency, prolonged cyclability, tailorable thermal conductivity, more uniform particle size distribution and safety regards are the top criteria for the evaluation and commercialisation of nanoencapsulated PCM.
6. Conclusions & perspectives
Nanoencapsulation is one of the most promising solutions to increase the efficiency of PCMs, both organic and inorganic. It promotes high specific surface area, prevents exchange of encapsulated material with the environment, controls heat exchange across the capsule shell and initiates congruent melting/crystallisation due to the small core size. Energy nanocapsules can find new application fields in thermal energy storage, such as cascaded multi-temperature energy systems, additives to thermal paints or other building materials, etc.However, current level of development of PCM encapsulation is mostly represented by macro and microencapsulation. The capsules consist of a single-layer polymer or oxide shell with only one functionality – prevention of material exchange with the environment. Future research should focus on the design of multifunctional composite PCM nanocapsules with enhanced thermal conductivity. Moreover, the effect of the nanoconfinement can change the crystallinity of PCMs in solid phase, which, in turn, can increase (or decrease) the melting enthalpy (heat uptake/release) in the confined nanovolume. This aspect of the nanoencapsulated PCMs is very poorly explored but has great potential to study nanosized effects in common salts or alkanes. We believe that there is an optimal size of the capsules, which can provide high efficiency of the storage of thermal energy. The nanoconfinement effect can overcome the losses of the latent heat energy caused be encapsulation. PCM-loaded macro and microcapsules do not have 100% encapsulation yield and, in spite of high stability, the heat capacity is usually lower than pure PCMs.
The widespread use of PCMs as energy storage materials can have vital consequences to aid humanity's drive for clean and renewable energy. Their various applications have highly advantageous effects, such as reduction in energy demand, reduced waste heat and improved efficiency for concentrated solar power plants. New methodology for capsule production needs to be developed further, such as complex emulsions, layer-by-layer assembly, microfluidics and industrial-scale sonication. These high-throughput manufacturing methods will lead to simple and wide-scale fabrication of PCM nanocapsules, reducing costs and increasing viability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from ERC Consolidator grant ENERCAPSULE No. 647969 and ERC PoC grant ENERPAINT No. 767173.Notes and references
- B. Xu, P. Li and C. Chan, Appl. Energy, 2015, 160, 286–307 CrossRef
.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- G. Walther, E. Post, P. Convey, A. Menzel, C. Parmesan, T. J. C. Beebee, J. Fromentin, O. Hoegh-Guldberg and F. Bairlein, Nature, 2002, 416, 389–395 CrossRef CAS PubMed
- W. Thuiller, C. Albert, M. B. Araújo, P. M. Berry, M. Cabeza, A. Guisan, T. Hickler, G. F. Midgley, J. Paterson, F. M. Schurr, M. T. Sykes and N. E. Zimmermann, Perspect. Plant Ecol. Evol. Syst., 2008, 9, 137–152 CrossRef
- M. Asif and T. Muneer, Renewable Sustainable Energy Rev., 2007, 11, 1388–1413 CrossRef
- V. Siva Reddy, S. C. Kaushik, K. R. Ranjan and S. K. Tyagi, Renewable Sustainable Energy Rev., 2013, 27, 258–273 CrossRef
- J. Tsao, N. Lewis and G. Crabtree, US Dep. Energy, 2006, 1–24 Search PubMed
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed
- P. Felix De Castro, A. Ahmed and D. G. Shchukin, Chem. – Eur. J., 2016, 22, 4389–4394 CrossRef PubMed
- A. Abhat, Sol. Energy, 1983, 30, 313–332 CrossRef CAS
- P. Pardo, A. Deydier, Z. Anxionnaz-Minvielle, S. Rougé, M. Cabassud and P. Cognet, Renewable Sustainable Energy Rev., 2014, 32, 591–610 CrossRef CAS
- Cr. Prieto, P. Cooper, A. I. Fernandez and L. F. Cabez, Renewable Sustainable Energy Rev., 2016, 60, 909–929 CrossRef CAS
- M. Kenisarin and K. Makhamov, Renewable Sustainable Energy Rev., 2016, 55, 371–398 CrossRef
- N. Soares, J. J. Costa, A. R. Gaspar and P. Santos, Energy Build., 2013, 60, 82–103 CrossRef
- H. Akeiber, P. Nejat, M. Z. A. Majid, M. A. Wahid, F. Jomehzadeh, I. Zeinali Famileh, J. K. Kalautit, B. R. Hughes and S. A. Zaki, Renewable Sustainable Energy Rev., 2016, 60, 1470–1497 CrossRef
- S. H. Khateeb, S. Amiruddin, M. Farid, J. R. Selman and S. Al-Hallaj, J. Power Sources, 2005, 142, 345–353 CrossRef CAS
- N. O. Moraga, J. P. Xaman and R. H. Araya, Appl. Therm. Eng., 2016, 108, 1041–1054 CrossRef CAS
- G. Jiang, J. Huang, Y. Fu, M. Cao and M. Liu, Appl. Therm. Eng., 2016, 108, 1119–1125 CrossRef CAS
- Z. Ling, Z. Zhang, G. Shi, X. Fang, L. Wang, X. Gao, Y. Fang, T. Xu, S. Wang and X. Liu, Renewable Sustainable Energy Rev., 2014, 60, 427–438 CrossRef
- M. C. Browne, B. Norton and S. J. McCormack, Renewable Sustainable Energy Rev., 2015, 47, 762–782 CrossRef CAS
- C. J. Smith, P. M. Forster and R. Crook, Appl. Energy, 2014, 126, 21–28 CrossRef
- T. Y. Kim, J. J. Lee and J. Rhee, Aerosp. Sci. Technol., 2013, 27, 10–16 CrossRef
- X. Liu and Y. Lou, Fibres Text. East. Eur., 2015, 23, 63–67 CAS
- N. Sarier and E. Onder, Thermochim. Acta, 2012, 540, 7–60 CrossRef CAS
- S. Mondal, Appl. Therm. Eng., 2008, 28, 1536–1550 CrossRef CAS
- N. Maruoka, T. Mizuochi, H. Purwanto and T. Akiyama, ISIJ Int., 2004, 44, 257–262 CrossRef CAS
- K. Nagano, K. Ogawa, T. Mochida, K. Hayashi and H. Ogoshi, Appl. Therm. Eng., 2004, 24, 221–232 CrossRef CAS
- M. T. Johansson and M. Söderström, Energy Effic., 2014, 7, 203–215 CrossRef
- T. Nomura, N. Okinaka and T. Akiyama, ISIJ Int., 2010, 50, 1229–1239 CrossRef CAS
- Y. Zheng, J. L. Barton, K. Tuzla, J. C. Chen, S. Neti, A. Oztekin and W. Z. Misiolek, Sol. Energy, 2015, 115, 180–194 CrossRef CAS
- P. Gimenez-Gavarrell and S. Fereres, Renewable Energy, 2017, 107, 497–507 CrossRef CAS
- J. Li, W. Lu, Z. Luo and Y. Zeng, Sol. Energy Mater. Sol. Cells, 2017, 171, 106–117 CrossRef CAS
- M. Karthikeyan and T. Ramachandran, Mater. Res. Innovations, 2014, 18, 541–554 CrossRef CAS
- C. Liu, Z. Rao, J. Zhao, Y. Huo and Y. Li, Nano Energy, 2015, 13, 814–826 CrossRef CAS
- S. S. Chandel and T. Agarwal, Renewable Sustainable Energy Rev., 2017, 31, 581–596 CrossRef
- B. Zalba, J. M. Marin, L. F. Cabeza and H. Mehling, Appl. Therm. Eng., 2003, 23, 251–283 CrossRef CAS
- S. A. Mohamed, F. A. Al-Sulaiman, N. I. Ibrahim, M. H. Zahir, A. Al-Ahmed, R. Saidur, B. S. Yılbaş and A. Z. Sahin, Renewable Sustainable Energy Rev., 2017, 70, 1072–1089 CrossRef CAS
- R. Naumann and H. H. Emons, J. Therm. Anal. Calorim., 1989, 35, 1009–1031 CrossRef CAS
- P. W. Stott, A. C. Williams and B. W. Barry, J. Controlled Release, 1998, 50, 297–308 CrossRef CAS PubMed
- P. D. Myers and D. Y. Goswami, Appl. Therm. Eng., 2016, 109, 889–900 CrossRef
- M. Kenisarin and K. Mahkamov, Sol. Energy Mater. Sol. Cells, 2016, 145, 255–286 CrossRef CAS
- L. F. Cabeza, J. Illa, J. Roca, F. Badia, H. Mehling, S. Hiebler and F. Ziegler, Mater. Corros., 2001, 52, 140–146 CrossRef CAS
- F. C. Porisini, Sol. Energy, 1988, 41, 193–197 CrossRef CAS
- E. Oró, A. de Gracia, A. Castell, M. M. Farid and L. F. Cabeza, Appl. Energy, 2012, 99, 513–533 CrossRef
- H. S. Ge, H. Y. Li, S. F. Mei and J. Liu, Renewable Sustainable Energy Rev., 2013, 21, 331–346 CrossRef CAS
- K. Ma and J. Liu, Frontiers of Energy Power Engineering in China, 2007, 1, 384–402 CrossRef
- H. Ge and J. Liu, Front. Energy, 2012, 6, 207–209 CrossRef
- J. Pereira da Cunha and P. Eames, Appl. Energy, 2016, 177, 227–238 CrossRef CAS
- A. Shukla, D. Buddhi and R. L. Sawhney, Renewable Energy, 2008, 33, 2606–2614 CrossRef CAS
- M. Kenisarin and K. Kenisarina, Renewable Sustainable Energy Rev., 2012, 16, 1999–2040 CrossRef CAS
- P. Schossig, H. M. Henning, S. Gschwander and T. Haussmann, Sol. Energy Mater. Sol. Cells, 2005, 89, 297–306 CrossRef CAS
- P. B. Salunkhe and P. S. Shembekar, Renewable Sustainable Energy Rev., 2012, 16, 5603–5616 CrossRef CAS
- D. G. Shchukin and E. Shchukina, Curr. Opin. Pharmacol., 2014, 18, 42–46 CrossRef CAS PubMed
- B. K. Paul and R. K. Mitra, J. Colloid Interface Sci., 2005, 288, 261–279 CrossRef CAS PubMed
- K. Kundu and B. K. Paul, Colloids Surf., A, 2013, 433, 154–165 CrossRef CAS
- D. J. McClements, Soft Matter, 2012, 8, 1719–1729 RSC
- L. C. Peng, C. H. Liu, C. C. Kwan and K. F. Huang, Colloids Surf., A, 2010, 370, 136–142 CrossRef CAS
- N. Anton, J. P. Benoit and P. Saulnier, J. Controlled Release, 2008, 128, 185–199 CrossRef CAS PubMed
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS
- S. De La Rochebrochard, J. Suptil, J. F. Blais and E. Naffrechoux, Ultrason. Sonochem., 2012, 19, 280–285 CrossRef CAS PubMed
- R. Xiao, Z. Wei, D. Chen and L. K. Weavers, Environ. Sci. Technol., 2014, 48, 9675–9683 CrossRef CAS PubMed
- B. Teo, S. Prescott, M. Ashokkumar and F. Grieser, Ultrason. Sonochem., 2008, 15, 89–94 CrossRef CAS PubMed
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039–1059 CrossRef CAS PubMed
- T. J. Mason, Ultrason. Sonochem., 2000, 7, 145–149 CrossRef CAS PubMed
- K. S. Suslick and G. J. Price, Annu. Rev. Mater. Sci., 1999, 29, 295–326 CrossRef CAS
- K. S. Suslick and D. J. Flannigan, Annu. Rev. Phys. Chem., 2008, 59, 659–683 CrossRef CAS PubMed
- H. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555–2567 RSC
- C. Solans, P. Izquierdo, J. Nolla, N. Azemar and M. J. Garcia-Celma, Curr. Opin. Colloid Interface Sci., 2005, 10, 102–110 CrossRef CAS
- Y. Asakura, T. Nishida, T. Matsuoka and S. Koda, Ultrason. Sonochem., 2008, 15, 244–250 CrossRef CAS PubMed
- P. Sathishkumar, R. V. Mangalaraja and S. Anandan, Renewable Sustainable Energy Rev., 2016, 55, 426–454 CrossRef CAS
- A. Henglein and M. Gutierrez, J. Phys. Chem., 1990, 94, 5169–5172 CrossRef CAS
- G. J. Price, Ultrason. Sonochem., 2003, 10, 277–283 CrossRef CAS PubMed
- B. F. Gibbs, S. Kermasha, I. Alli and C. Mulligan, Int. J. Food Sci. Nutr., 1999, 50, 213–224 CrossRef CAS PubMed
- A. Madene, M. Jacquot, J. Scher and S. Desobry, Int. J. Food Sci. Technol., 2006, 41, 1–21 CrossRef CAS
- L. Cheng, W. He, H. Gong, C. Wang, Q. Chen, Z. Cheng and Z. Liu, Adv. Funct. Mater., 2013, 23, 5893–5902 CrossRef CAS
- P. M. Arnal, M. Comotti and F. Schüth, Angew. Chem., Int. Ed., 2006, 45, 8224–8227 CrossRef CAS PubMed
- A. C. Bijlard, A. Hansen, I. Lieberwirth, K. Landfester and A. Taden, Adv. Mater., 2016, 28, 6372–6377 CrossRef CAS PubMed
- D. G. Shchukin, M. Zheludkevich, K. Yasakau, S. Lamaka, M. G. S. Ferreira and H. Möhwald, Adv. Mater., 2006, 18, 1672–1678 CrossRef CAS
- D. G. Shchukin and H. Möhwald, Adv. Funct. Mater., 2007, 17, 1451–1458 CrossRef CAS
- A. P. Kafka, B. J. McLeod, T. Rades and A. McDowell, J. Controlled Release, 2011, 149, 307–313 CrossRef CAS PubMed
- D. Chitkara and N. Kumar, Pharm. Res., 2013, 30, 2396–2409 CrossRef CAS PubMed
- S. Vrignaud, N. Anton, C. Passirani, J. P. Benoit and P. Saulnier, Drug Dev. Ind. Pharm., 2013, 39, 1706–1711 CrossRef CAS PubMed
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS PubMed
- C. E. Mora-Huertas, H. Fessi and A. Elaissari, Int. J. Pharm., 2010, 385, 113–142 CrossRef CAS PubMed
- F. Caruso, D. Trau, H. Moehwald and R. Renneberg, Langmuir, 2000, 16, 1485–1488 CrossRef CAS
- H. Cohen, R. J. Levy, J. Gao, I. Fishbein, V. Kousaev, S. Sosnowski, S. Slomkowski and G. Golomb, Gene Ther., 2000, 7, 1896–1905 CrossRef CAS PubMed
- H. Lomas, I. Canton, S. MacNeil, J. Du, S. P. Armes, A. J. Ryan, A. L. Lewis and G. Battaglia, Adv. Mater., 2007, 19, 4238–4243 CrossRef CAS
- J. Kang and J. Rebek, Nature, 1997, 385, 50–52 CrossRef CAS PubMed
- T. M. S. Chang, Science, 1964, 146, 524–525 CAS
- P. Couvreur, P. Speiser, P. Tulkens, M. Roland and A. Trouet, FEBS Lett., 1977, 84, 323–326 CrossRef CAS PubMed
- J. Ballenger and R. Post, Am. J. Psychiatry, 1980, 137, 782–790 CrossRef CAS PubMed
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed
- S. M. S. M. Moghimi, A. C. A. C. Hunter and J. C. J. C. Murray, Pharmacol. Rev., 2001, 53, 283–318 CAS
- J. Rytkönen, R. Miettinen, M. Kaasalainen, V. P. Lehto, J. Salonen and A. Närvänen, J. Nanomater., 2012, 2012, 896562 Search PubMed
- G. H. Zhang, S. A. F. Bon and C. Y. Zhao, Sol. Energy, 2012, 86, 1149–1154 CrossRef CAS
- A. Jamekhorshid, S. M. Sadrameli and M. Farid, Renewable Sustainable Energy Rev., 2014, 31, 531–542 CrossRef CAS
- A. Soottitantawat, H. Yoshii, T. Furuta, M. Ohkawara and P. Linko, J. Food Sci., 2003, 68, 2256–2262 CrossRef CAS
- A. Gharsallaoui, G. Roudaut, O. Chambin, A. Voilley and R. Saurel, Food Res. Int., 2007, 40, 1107–1121 CrossRef CAS
- Y. W. Luo and X. D. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2145–2154 CrossRef CAS
- F. Tiarks, K. Landfester and M. Antonietti, Langmuir, 2001, 17, 908–918 CrossRef CAS
- U. Paiphansiri, P. Tangboriboonrat and K. Landfester, Macromol. Biosci., 2006, 6, 33–40 CrossRef CAS PubMed
- R. H. Utama, M. H. Stenzel and P. B. Zetterlund, Macromolecules, 2013, 46, 2118–2127 CrossRef CAS
- F. Gröhn, B. J. Bauer and E. J. Amis, Macromolecules, 2001, 34, 6701–6707 CrossRef
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS
- G. B. Sukhorukov and H. Möhwald, Trends Biotechnol., 2008, 25, 93–98 CrossRef PubMed
- D. G. Shchukin, E. Ustinovich, G. B. Sukhorukov, H. Möhwald and D. V. Sviridov, Adv. Mater., 2005, 17, 468–472 CrossRef CAS
- X. Zhang, H. Chen and H. Zhang, Chem. Commun., 2007, 1395–1405 RSC
- W. B. Stockton and M. F. Rubner, Macromolecules, 1997, 30, 2717–2725 CrossRef CAS
- M. M. Fang, D. M. Kaschak, A. C. Sutorik and T. E. Mallouk, J. Am. Chem. Soc., 1997, 119, 12184–12191 CrossRef CAS
- A. P. R. Johnston, E. S. Read and F. Caruso, Nano Lett., 2005, 5, 953–956 CrossRef CAS PubMed
- Z. P. Wang, Z. Q. Feng and C. Y. Gao, Chem. Mater., 2008, 20, 4194–4199 CrossRef CAS
- T. Lojou and P. Bianco, Langmuir, 2004, 20, 748–755 CrossRef PubMed
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, Chem. Commun., 1995, 2313–2314 RSC
- G. Sukhorukov, A. Fery and H. Möhwald, Prog. Polym. Sci., 2005, 30, 885–897 CrossRef CAS
- S. Moya, G. B. Sukhorukov, M. Auch, E. Donath and H. Mohwald, J. Colloid Interface Sci., 1999, 216, 297–302 CrossRef CAS PubMed
- E. M. Shchukina and D. G. Shchukin, Adv. Drug Delivery Rev., 2011, 63, 837–846 CrossRef CAS PubMed
- D. O. Grigoriev, T. Bukreeva, H. Möhwald and D. G. Shchukin, Langmuir, 2008, 24, 999–1004 CrossRef CAS PubMed
- T. Oya, T. Nomura, M. Tsubota, N. Okinaka and T. Akiyama, Appl. Therm. Eng., 2013, 61, 825–828 CrossRef CAS
- G. Q. Qi, J. Yang, R. Y. Bao, Z. Y. Liu, W. Yang, B. H. Xie and M. B. Yang, Carbon, 2015, 88, 196–205 CrossRef CAS
- M. F. Haase, D. Grigoriev, H. Möhwald, B. Tiersch and D. G. Shchukin, Langmuir, 2011, 27, 74–82 CrossRef CAS PubMed
- M. Bedard, A. Munoz-Javier, R. Mueller, P. Del Pino, A. Feri, W. J. Parak, A. Skirtach and G. B. Sukurukov, Soft Matter, 2009, 5, 148–155 RSC
- J. M. Asua, Prog. Polym. Sci., 2014, 39, 1797–1826 CrossRef CAS
- J. M. Asua, Prog. Polym. Sci., 2002, 27, 1283–1346 CrossRef CAS
- K. Landfester, J. Eisenblätter and R. Rothe, J. Coat. Technol. Res., 2004, 1, 65–68 CrossRef CAS
- D. Z. Yin, L. Ma, J. J. Liu and Q. Y. Zhang, Energy, 2014, 64, 575–581 CrossRef CAS
- J. Yang, M. W. Keller, J. S. Moore, S. R. White and N. R. Sottos, Macromolecules, 2008, 41, 9650–9655 CrossRef CAS
- X. Huang and B. Voit, Polym. Chem., 2013, 435–443 RSC
- L. D. Zarzar, V. Sresht, E. M. Sletten, J. A. Kaliw, D. Blankschtein and T. M. Swager, Nature, 2015, 518, 520–524 CrossRef CAS PubMed
- P.-H. Theunissen and J.-M. Buchlin, Sol. Energy, 1983, 31, 271–277 CrossRef CAS
- P. Felix De Castro and D. G. Shchukin, Chem. – Eur. J., 2015, 21, 11174–11179 CrossRef CAS PubMed
- H. Zhang and X. Wang, Sol. Energy Mater. Sol. Cells, 2009, 93, 1366–1376 CrossRef CAS
- S. Barlak, O. N. Sara, A. Karaipekli and S. Yapici, Nanoscale Microscale Thermophys. Eng., 2016, 20, 85–96 CrossRef CAS
- C. Alkan, A. Sari and A. Karaipekli, Energy Convers. Manage., 2011, 52, 687–692 CrossRef CAS
- C. Alkan, A. Sari and C. Biglin, Appl. Energy, 2014, 136, 217–227 CrossRef
- Q. Yi, G. B. Sukhorukov, J. Ma, X. Yang and Z. Gu, Int. J. Polym. Sci., 2015, 2015, 756237 Search PubMed
- X. X. Zhang, Y. F. Fan, X. M. Tao and K. L. Yick, Mater. Chem. Phys., 2004, 88, 300–307 CrossRef CAS
- Y. F. Fan, X. X. Zhang, X. C. Wang, J. Li and Q. B. Zhu, Thermochim. Acta, 2004, 413, 1–6 CrossRef CAS
- Y. Konuklu, M. Unal and H. O. Paksoy, Sol. Energy Mater. Sol. Cells, 2014, 120, 536–542 CrossRef CAS
- Z. H. Chen, X. R. Zeng and G. Zhang, Appl. Energy, 2012, 91, 7–12 CrossRef CAS
- A. Sari, C. Alkan and A. Karaipekli, Appl. Energy, 2010, 87, 1529–1534 CrossRef CAS
- R. Yang, H. Xu and Y. Zhang, Sol. Energy Mater. Sol. Cells, 2003, 80, 405–416 CrossRef CAS
- Y. Fang, X. Liu, X. Liang, H. Liu, X. Gao and Z. Zhang, Appl. Energy, 2014, 132, 551–556 CrossRef CAS
- K. Tumirah, M. Z. Hussein, Z. Zulkarnain and R. Rafeadah, Energy, 2014, 66, 881–890 CrossRef CAS
- M. Fuensanta, U. Paibhansiri, M. D. Romero-Sanches, C. Guillem, A. M. Lopez-Buendia and K. Landfester, Thermochim. Acta, 2013, 565, 95–101 CrossRef CAS
- S. T. Latibari, M. Mehrali, M. Mehrali, T. Meurah, T. M. I. Malia and H. S. C. Metselaar, Energy, 2013, 61, 664–672 CrossRef
- Y. Wang, Y. Zhang, T. Xia, W. Zhao and W. Yang, Sol. Energy Mater. Sol. Cells, 2014, 120, 481–490 CrossRef CAS
- G. Fang, Z. Chen and H. Li, Chem. Eng. J., 2010, 163, 154–159 CrossRef CAS
- H. Zhang, X. Wang and D. Wu, J. Colloid Interface Sci., 2010, 343, 246–255 CrossRef CAS PubMed
- L. X. Geng, S. F. Wang, T. Y. Wang and R. L. Luo, Energy Fuels, 2016, 30, 6153–6160 CrossRef CAS
- W. Fu, X. Liang, H. Xie, S. Wang, X. Gao, Z. Zhang and Y. Fang, Energy Build., 2017, 136, 26–32 CrossRef
- J. Shi, X. Wu, X. Fu and R. Sun, Thermochim. Acta, 2015, 617, 90–94 CrossRef CAS
- Y. Zhu, S. Liang, H. Wang, K. Zhang, X. Jia, C. Tian, Y. Zhou and J. Wang, Energy Convers. Manage., 2016, 119, 151–162 CrossRef CAS
- Y. Zhu, S. Liang, K. Chen, X. Gao, P. Chang, C. Tian, J. Wang and Y. Huang, Energy Convers. Manage., 2015, 105, 908–917 CrossRef CAS
- Z. Rao, S. Wang and F. Peng, Appl. Energy, 2012, 100, 303–308 CrossRef CAS
- S. Demirbag and S. A. Aksoy, Fibers Polym., 2016, 17, 408–417 CrossRef CAS
- S. Demirbag and S. A. Aksoy, J. Text. Eng., 2013, 20, 27–35 CrossRef CAS
- Z. Zheng, J. Jin, G.-K. Xu, J. Zou, U. Wais, A. Beckett, T. Heil, S. Higgins, L. Guan, Y. Wang and D. Shchukin, ACS Nano, 2016, 10, 4695–4703 CrossRef CAS PubMed
- Z. Zheng, Z. Chang, G. K. Xu, F. McBride, A. Ho, Z. Zhuola, M. Michailidis, R. Raval, R. Akhtar and D. G. Shchukin, ACS Nano, 2017, 11, 721–729 CrossRef CAS PubMed
- L. W. Fan, Z. Q. Zhu, M. J. Liu, C. L. Xu, Y. Zeng, H. Lu and Z. T. Yu, J. Heat Transfer, 2016, 138, 122402 CrossRef
- S. Phadungphatthanakoon, S. Pumpradub and S. Wanichwecharungruang, ACS Appl. Mater. Interfaces, 2011, 3, 3691–3696 CAS
- I. Kalashnikova, H. Bizot, B. Kathala and I. Capron, Langmuir, 2011, 27, 7471–7479 CrossRef CAS PubMed
- A. J. Svagan, A. Musyanovich, M. Cappl, M. Bernhardt, G. Glasser, C. Wohnhaas, L. A. Berglund, J. Risbo and K. Landfester, Biomacromolecules, 2014, 15, 1852–1859 CrossRef CAS PubMed
- Y. Li, S. Yu, P. Chen, R. Rojas, A. Hajan and L. Berglund, Nano Energy, 2017, 34, 541–548 CrossRef CAS
- J. Huang, T. Y. Wang, P. P. Zhu and J. B. Xiao, Thermochim. Acta, 2013, 557, 1–6 CrossRef CAS
- D. Platte, U. Helbig, R. Houbertz and G. Sextl, Macromol. Mater. Eng., 2013, 298, 67–77 CrossRef CAS
- Y. E. Milian, A. Gutierrez, M. Gragada and S. Ushak, Renewable Sustainable Energy Rev., 2017, 73, 983–999 CrossRef CAS
- N. Sarier and E. Onder, Thermochim. Acta, 2007, 452, 149–160 CrossRef CAS
- N. Sarier and E. Onder, Thermochim. Acta, 2007, 454, 90–98 CrossRef CAS
- F. Salaün, E. Devaux, S. Bourbigot and P. Rumeau, Carbohydr. Polym., 2008, 73, 231–240 CrossRef
- C. Liu, C. Wang, Y. Li and Z. Rao, RSC Adv., 2017, 7, 7238–7249 RSC
- M. Graham, E. Shchukina, P. Felix De Castro and D. G. Shchukin, J. Mater. Chem. A, 2016, 4, 16906–16912 CAS
- A. A. El-Sebaii, S. A-Heniti, F. Al-Agel, A. A. Al-ghamidi and F. Al-Marzouki, Energy Convers. Manage., 2001, 52, 1771–1777 CrossRef
- M. Graham, J. Cola-Clemente, E. Shchukina and D. G. Shchukin, J. Mater. Chem., 2017, 5, 13683–13691 RSC
- T. Wang, J. Huang, P. Zhu and J. Xiao, Colloid Polym. Sci., 2013, 291, 2463–2468 CAS
- A. Schoth, K. Landfester and R. Muñoz-Espí, Langmuir, 2015, 31, 3784–3788 CrossRef CAS PubMed
- A. G. Hassabo, A. L. Mohamed, H. Wang, C. Popescu and M. Moller, Inorg. Chem.: Indian J., 2015, 10, 59–65 CAS
- J. Zhang, S. S. Wang, S. D. Zhang, Q. H. Tao, L. Pan, Z. Y. Wang, Z. P. Zhang, Y. Lei, S. K. Yang and H. P. Zhao, J. Phys. Chem. C, 2011, 115, 20061–20066 CAS
- G. Zhang, J. Li, Y. Chen, H. Xiang, B. Ma, Z. Xu and X. Ma, Sol. Energy Mater. Sol. Cells, 2014, 128, 131–137 CrossRef CAS
- M. Karthikeyan, T. Ramachandran and O. L. S. Sundaram, J. Ind. Text., 2014, 44, 130–146 CrossRef
- M. Karthikeyan, T. Ramachandran and O. L. Shanmugasundaram, J. Text. Inst., 2014, 105, 1279–1286 CrossRef CAS
- A. Sarı, C. Alkan, A. Biçer, A. Altuntaş and C. Bilgin, Energy Convers. Manage., 2014, 86, 614–621 CrossRef
- P. C. Ray, H. T. Yu and P. P. Fu, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2009, 27, 1–35 CrossRef CAS PubMed
- S. Sharifi, S. Behzadi, S. Laurent, M. L. Forrest, P. Stroeve and M. Mahmoudi, Chem. Soc. Rev., 2012, 41, 2323–2343 RSC
- M. Goel, S. K. Roy and S. Sengupta, Int. J. Heat Mass Transfer, 1994, 37, 593–604 CrossRef CAS
- J. M. Khodadadi and S. F. Hosseinizadeh, Int. Commun. Heat Mass Transfer, 2007, 34, 534–543 CrossRef CAS
- Y. Wang, Z. Q. Chen and X. Ling, Appl. Therm. Eng., 2016, 98, 835–840 CrossRef CAS
- J. P. Dumas, M. Krichi, M. Strub and Y. Zeraouli, Int. J. Heat Mass Transfer, 1994, 37, 737–746 CrossRef CAS
- X. Hu and Y. Zhang, Int. J. Heat Mass Transfer, 2002, 45, 3163–3172 CrossRef
- Z. H. Han, F. Y. Cao and B. Yang, Appl. Phys. Lett., 2008, 92, 243104 CrossRef
- C. Z. Liu, Z. H. Rao, J. T. Zhao, Y. T. Huo and Y. M. Li, Nano Energy, 2015, 13, 814–826 CrossRef CAS
- M. M. U. Rehman, Z. G. Qu, R. P. Fu and H. T. Xu, Int. J. Heat Mass Transfer, 2017, 109, 312–325 CrossRef
- W. Wu, H. Bostanci, L. C. Chow, Y. Hong, C. M. Wang, M. Su and J. P. Kizito, Int. J. Heat Mass Transfer, 2013, 58, 348–355 CrossRef CAS
- M. Liu, F. Bruno and W. Saman, Sol. Energy, 2011, 85, 3017–3027 CrossRef CAS
- Z. Youssef, A. Delahaye, L. Huang, F. Trinquet, L. Fournaison, C. Pollerberg and C. Doetsch, Energy Convers. Manage., 2013, 65, 120–132 CrossRef CAS
- H. R. Seyf, Z. Zhou, H. B. Ma and Y. Zhang, Int. J. Heat Mass Transfer, 2013, 56, 561–573 CrossRef CAS
- H. Michels and R. Pitz-Paal, Sol. Energy, 2007, 81, 829–837 CrossRef CAS
- M. M. U. Rehman, Z. G. Qu and R. P. Fu, J. Therm. Sci., 2016, 25, 431–439 CrossRef
- J. J. Bae, S. C. Lim, G. H. Han, Y. W. Jo, D. L. Doung, E. S. Kim, S. J. Chae, T. Q. Huy, N. V. Luan and Y. H. Lee, Adv. Funct. Mater., 2012, 22, 4819–4826 CrossRef CAS
- U. Kalsoom, P. N. Nesterenko and B. Paull, RSC Adv., 2016, 6, 60355–60371 RSC
- M. D. Symes, P. J. Kitson, J. Yan, C. J. Richmond, G. J. T. Cooper, R. W. Bowman, T. Vilbrandt and L. Cronin, Nat. Chem., 2012, 4, 349–354 CrossRef CAS PubMed
- M. Sušec, S. C. Ligon, J. Stampfl, R. Liska and P. Krajnc, Macromol. Rapid Commun., 2013, 34, 938–943 CrossRef PubMed
- I. Cooperstein, M. Layani and S. Magdassi, J. Mater. Chem. C, 2015, 3, 2040–2044 RSC
- R. L. Truby and J. A. Lewis, Nature, 2016, 540, 371–378 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2018 |