
Peptide and protein nanoparticle conjugates: versatile platforms for biomedical applications

Abstract
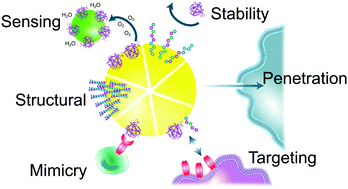
Peptide– and protein–nanoparticle conjugates have emerged as powerful tools for biomedical applications, enabling the treatment, diagnosis, and prevention of disease. In this review, we focus on the key roles played by peptides and proteins in improving, controlling, and defining the performance of nanotechnologies. Within this framework, we provide a comprehensive overview of the key sequences and structures utilised to provide biological and physical stability to nano-constructs, direct particles to their target and influence their cellular and tissue distribution, induce and control biological responses, and form polypeptide self-assembled nanoparticles. In doing so, we highlight the great advances made by the field, as well as the challenges still faced in achieving the clinical translation of peptide- and protein-functionalised nano-drug delivery vehicles, imaging species, and active therapeutics.
- This article is part of the themed collections: Peptide and protein nanotechnology and Probes for in vitro and in vivo fluorescence imaging
 Please wait while we load your content...
Please wait while we load your content...

