
The evolution of artificial light actuators in living systems: from planar to nanostructured interfaces
Francesca
Di Maria
 *a,
Francesco
Lodola
b,
Elena
Zucchetti
bc,
Fabio
Benfenati
d and
Guglielmo
Lanzani
*bc
*a,
Francesco
Lodola
b,
Elena
Zucchetti
bc,
Fabio
Benfenati
d and
Guglielmo
Lanzani
*bc
aCNR-ISOF, Consiglio Nazionale delle Ricerche, Bologna, Italy. E-mail: francesca.dimaria@isof.cnr.it
bCenter for Nano Science and Technology, Istituto Italiano di Tecnologia, Milano, Italy. E-mail: Guglielmo.Lanzani@iit.it
cDepartment of Physics, Politecnico di Milano, Milano, Italy
dCenter for Synaptic Neuroscience and Technology, Istituto Italiano di Tecnologia, Genova, Italy
First published on 17th April 2018
Abstract
Artificially enhancing light sensitivity in living cells allows control of neuronal paths or vital functions avoiding the wiring associated with the use of stimulation electrodes. Many possible strategies can be adopted for reaching this goal, including the direct photoexcitation of biological matter, the genetic modification of cells or the use of opto-bio interfaces. In this review we describe different light actuators based on both inorganic and organic semiconductors, from planar abiotic/biotic interfaces to nanoparticles, that allow transduction of a light signal into a signal which in turn affects the biological activity of the hosting system. In particular, we will focus on the application of thiophene-based materials which, thanks to their unique chemical–physical properties, geometrical adaptability, great biocompatibility and stability, have allowed the development of a new generation of fully organic light actuators for in vivo applications.
 Francesca Di Maria | Francesca Di Maria received her BSc in Chemistry from the University of Catania and her PhD in Chemical Sciences from the University of Bologna in 2016. Her current research interests are focused on the synthesis and characterization of thiophene-based materials and their organization into supramolecular nanostructures for application in optoelectronics, photonics and biological systems. She has published more than 30 research papers in peer-reviewed journals. Currently, she is working at CNR-ISOF in Bologna as a Post-doctoral researcher under the guidance of Dr G. Barbarella. |
 Francesco Lodola | Francesco Lodola graduated in Biotechnology in 2007 and received his PhD in Physiology and Neuroscience under the supervision of Prof. Tanzi and Prof. Moccia from the University of Pavia in 2012. Later he joined Prof. Priori's group at ICS Maugeri in Pavia focusing on the study of arrhythmogenic mechanisms in cardiac disorders and on the development of targeted therapies to reduce mortality in affected individuals. Since 2016, Francesco has been working at the Center for Nano Science and Technology, IIT@PoliMI in Milan directed by Prof. Lanzani. His research concerns the use of optically active polymer-based interfaces for light-induced control of living systems. |
 Elena Zucchetti | Elena Zucchetti obtained her master's degree in Engineering Physics from Politecnico di Milano in 2014, with a study on bio-polymer interfaces for optical stimulation of living cell cultures. She is a PhD student at the Center for Nano Science and Technology at Italian Institute of Technology in Milan. Her current PhD thesis focuses on fabrication and characterization of organic polymer nanoparticles and their interaction with biological systems, from both physiological and photophysical perspective, with the final aim of realizing photoactive devices for light-control of biological activity. |
 Fabio Benfenati | Fabio Benfenati is the director of the Center for Synaptic Neuroscience and Technology, The Italian Institute of Technology, professor of Neurophysiology at the University of Genova and adjunct professor at The Rockefeller University, New York, NY. He is the author of over 300 research papers with an h-index of 72 and over 15 |
 Guglielmo Lanzani | Guglielmo Lanzani graduated in Physics at the University of Milan (I) in 1987 and received PhD in Chemical Physics from the University of Genova in 1992. He was a visiting scientist at the University of Utah in 1989 and 1990. Since 2011 he has been “Full Professor” in Physics at Politecnico di Milano (I). Since 2010 he has been Coordinator of the Center for Nano Science and Technology of the Istituto Italiano di Tecnologia. His research concerns the science and technology of carbon based materials and nanostructures for application in energy and bio-photonics. He has given about 180 invited talks and published about 290 papers in international journals. |
1. Introduction
The overarching goal of the research activity described here is to induce light sensitivity in living tissue in order to (i) rescue lost functions (e.g. vision); (ii) interrogate single cells in cell/neuronal networks by light driven remote stimulation (non-genetic approach to opto cell stimulation); (iii) achieve light control of physiological functions such as movement in impaired limbs; (iv) realize phototactic guidance of animaloid soft robots or cyborgs. The approach is based on the realization of suitable light actuators, i.e. interfaces that transduce light into a biological (bioelectrical, biochemical or biomechanical) signal. Typical target cells are neurons, due to their prominent role in signal processing and control, or muscle cells for addressing light-controlled movement, but other non-excitable cells can be considered as model systems for understanding fundamental biological processes.The use of light as a stimulation tool has emerged in the last decade as a valid alternative to electrical stimulation, since it allows targeting single cells and even discrete regions of a single cell in a temporally and spatially precise manner offering, at the same time, the opportunity to exploit various excitation geometries.1 Moreover, light allows decoupling recording from stimulation and avoids physical contact between the stimulation source and the target cell, thus escaping problems related to contact impedance and, more generally, to biocompatibility issues.2
Since the vast majority of animal cells do not display any specific response to light, several strategies have been proposed in order to elicit light-sensitivity. In the following, starting from a general background on light–matter interactions we review a number of ongoing endeavors.
1.1 Light–matter interaction
Sunlight is the primary source of energy for all life on Earth, even though cells are almost transparent to visible light. To cope with this apparent contradiction, Nature has developed specific strategies to transduce light into biochemical energy or information.3–5 Vision in animal is a remarkable example of how evolution has led to a complex apparatus with advanced photonic performance and sophisticated signal processing.6–8 In the retina, light transmitted by the eye lens goes through several neuronal layers of the inner retina with virtually zero losses before reaching the photoreceptors that efficiently absorb light and transduce it into an electrical/biochemical signal to the bipolar cells. Through a complex path, the signal reaches the optic nerve and the visual cortex in the brain. The active chromophore, namely retinal, is located in a pocket within a protein that belongs to the opsin family.9 These proteins are present in most creatures endowed with vision and in many photosynthetic organisms as well.10–13 Retinal is a polyene chromophore with a carbon-conjugated backbone. When it absorbs light, it undergoes isomerization from cis to trans starting the phototransduction cascade.14–16Vision is a straightforward consequence of light sensitivity in living organisms, but it is not the unique outcome. In humans, sunlight regulates the mood, creates vitamin D, can kill bacteria and parasites, and the UV portion of the spectrum may cause cancer in skin cells.5,17 The endogenous absorption, that is responsible for these effects, is usually assigned to chromophores in the cell mitochondria.18 This is however far too small to allow an immediate response to a stimulus that could be exploited for the control of the abiotic/biotic interface. Accordingly, the artificial enhancement of light sensitivity in living organisms could enlarge the potential opening up new opportunities in photo-medicine and photo-biology.
“Optogenetics” is a possible way to answer this challenge.19 It refers to the use of genetic modification of cells to express light-sensitive actuators (ion channels/pumps or molecular switches).20,21 Optogenetics allows controlling in a very selective and specific way transmembrane as well as intracellular events, but has the drawback of requiring the introduction of exogenous genetic material using viral vectors.22,23 In addition to the safety issues related to the choice of a suitable viral vector (diffusion, inflammation, gene incorporation in the genome), another pitfall is due to the expression of heterologous proteins from very distant species that can trigger an appropriate, but undesirable, immune response in the host organism.22 Avoiding these problems would open up a much broader field of applications. For this reason, geneless opto-stimulation of living cells and tissues is a growing field of research at the border between photonics, materials science and biology. The use of infrared radiation (IR) has been investigated for many years. IR is absorbed by water, and this in turn causes a local rise of temperature that can affect biochemical parameters.24–27 This approach has the great advantage of simplicity, since it does not require any modifications of the cell specimen. However, it has a number of drawbacks stemming from the very weak absorption cross-section of water in the explored range.1 Considering the available radiation sources, the practical wavelength range is between 1 and 3 μm. Very often non-linear multiphoton transitions with tiny cross sections are used to get absorption. However, the high radiation intensity that is required may damage cell organelles by direct absorption.28,29 Furthermore, the excitation volume is not well controlled and is rather unspecific towards diverse biological targets. Overall, while IR can be useful to demonstrate temperature-induced effects, it seems hard to develop a technology based on it. An alternative approach considers light actuators of different shape and composition to be interfaced with living matter in order to exploit a variety of photoinduced physical phenomena for signal transduction. This approach, that avoids genetic manipulation, may in principle provide enough selectivity and be less invasive and non-toxic. In this review, we describe materials and devices adopted for geneless cell opto-stimulation, focusing on those based on organic semiconductors.
1.2 Materials used for light transduction
Metals, inorganic and organic semiconductors, or just molecular chromophores have been used as phototransducers in different shapes, from planar interfaces to micro-structured surfaces and nano- or micro-particles.30–42 These systems have very different electronic structures and elementary photo-excitations. Metals are generally used for their ability to absorb light and generate heat. This is particularly efficient in small dots, due to the very large oscillator strength of the surface plasmons.43 Photo-excited carriers in metals are short lived, as they quickly decay back to the Fermi distribution by intra-band phonon scattering, with typical time constants of 1–10 ps.44–47 This provides an efficient channel of energy dissipation into heat and local temperature that can rise following photoexcitation. It is the large absorption cross section of the plasmon modes that makes these systems appealing for thermal processes, because it allows the use of lower excitation intensities and better localization of the effect with respect to direct water excitation using IR.Inorganic semiconductors initially store light energy above a certain energy gap in electron and hole pairs, with lifetime in the 1–10 ns time domain.48–50 Recombination across the energy gap may occur radiatively, by photon emission, or non-radiatively by dissipation into phonons.51 This leads to temperature increase in the lattice with typical lifetimes in the micro to millisecond time regime, depending on the dissipation path.52,53 Trap states, due to a variety of defects and impurities, may heavily affect the dynamics and sometimes govern the whole process. Doping alters the transport properties54 and interestingly affects the interface setting, causing depletion regions and local fields often exploited in devices,55 including the coupling to cells.56–59 In the presence of strong Coulomb correlation or spatial confinement, electron–hole bound states known as excitons may be formed, with remarkable changes in the below gap electronic structure and optical spectrum.43,60 Due to their rich phenomenology, inorganic semiconductors can photo-transduce light into a biological signal by the thermal effect, capacitive coupling, faradic currents or photo-catalysis. In addition, it is well known that the quantum confinement regime allows tuning of the nanoparticle properties, practically providing a tool for optoelectronics function design and engineering. Quantum dots61 and nanorods62 have been investigated with the purpose of cell photostimulation since these structures display interesting physical properties, such as an intrinsic dipole moment and piezoelectric polarization, that contribute to the building of an internal field. However in biology, the use of inorganic semiconductors is limited due to the potential toxicity for cell cultures.63
Organic semiconductors are fundamentally different from inorganic ones. The molecular backbone is made up of strong carbon–carbon covalent bonds, but they are soft materials with a high degree of mechanical conformability due to weak supramolecular interactions.64 Since they are based on carbon, akin to biological molecules, organic semiconductors are often highly biocompatible and prone to seamless interfacing with living tissues.65 They have a large optical cross-section associated with π electron delocalization along the carbon-conjugated backbone.66,67 However, absorption of light rarely results in the generation of separated charge carriers. On the contrary, the primary photoexcitations are neutral, molecular-like states.68 In the biological environment, which is essentially water, organic semiconductors are subject to ion penetration at the molecular level and undergo p-doping due to the effect of hydrated oxygen that behaves as a strong electron acceptor.69 Such a process occurs at a faster rate under light.70 Primary neutral photoexcitations require extra-energy to split into oppositely charged carriers. Once generated, carriers have mobility orders of magnitude smaller than those of inorganic semiconductors, and many transport processes are thus slowed down with respect to the inorganic counterpart. Due to slow motion and localization, charged states (polarons) can be extremely long lived, up to the ms time domain.66,71 This may favor the photocatalytic activity at the interface with an electrolyte.
Organic-based semiconducting materials have been less investigated in neural interfaces compared to inorganic ones, but they offer several advantages owing to their multiple functional properties, synthetic versatility and easy functionalization.72–74
Thiophene-based materials represent one of the most investigated families of organic semiconductors extensively applied in photovoltaics, photodetection and photonics.75–79
Absorption, emission and semiconducting properties of polythiophene derivatives, such as poly-3-hexylthiophene (P3HT), have been employed for a long time to detect biological molecules, from early studies on colorimetric and fluorimetric detection of DNA80 to recent polythiophene-based photodynamic therapy studies and investigations on their use as photosensitizers for cancer treatment.81,82 Even more recently, the interest in the mechanism of light–biomatter interaction has led to the investigation of polythiophene derivatives as phototransducers in living cells with the objective of controlling cell properties, such as the membrane potential, by light (Fig. 1).
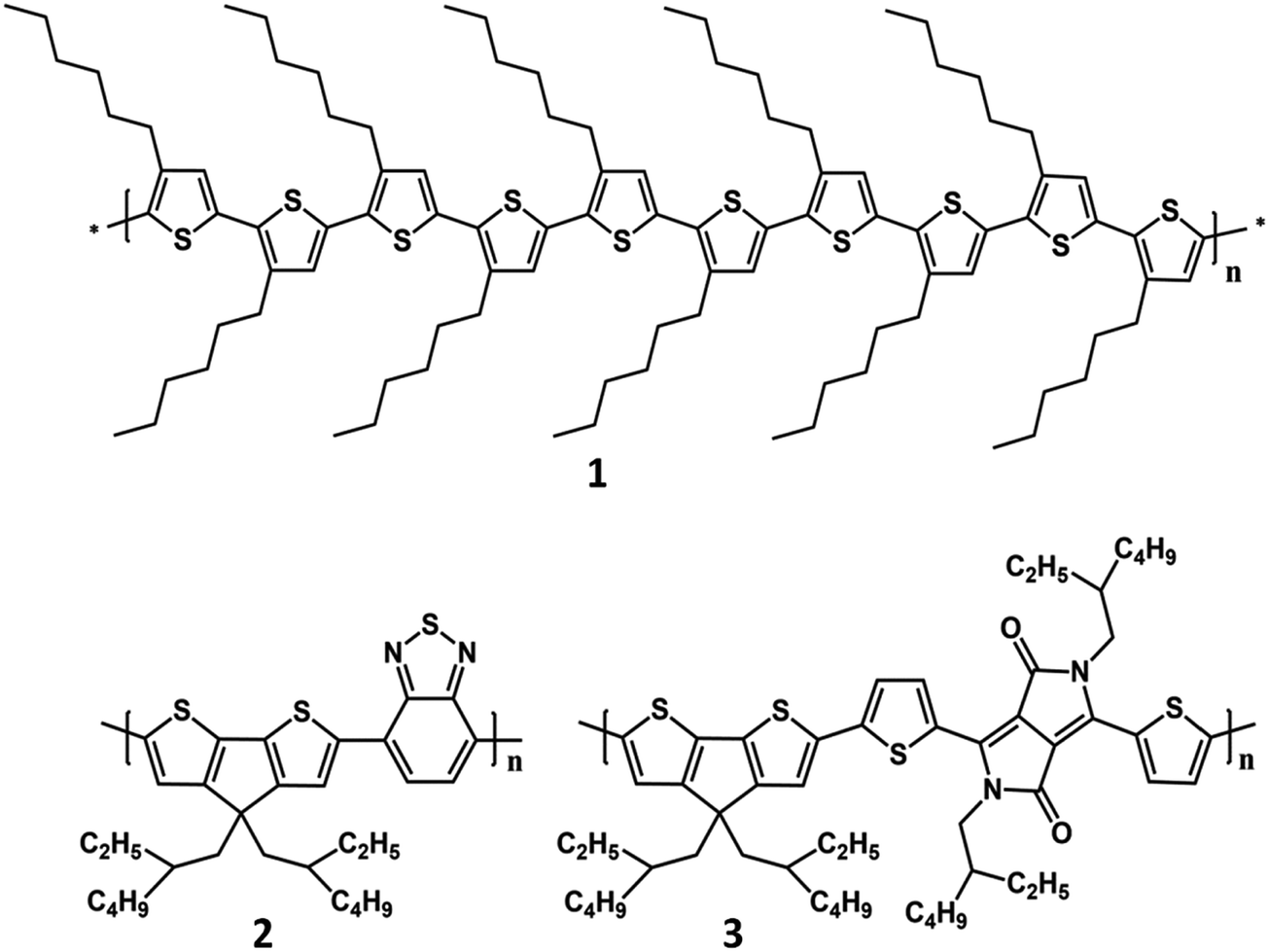 | ||
| Fig. 1 Molecular structures of polythiophene derivatives used as phototransducers in living cells: (1) head-to-tail poly(3-hexylthiophene) (P3HT); (2) poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta [2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)] (PCPDTBT); (3) poly(cyclopentadithiophene-alt-diketopyrrolopyrrole). | ||
1.3 Physical mechanisms of light transduction
Cells are surrounded by a membrane composed of a double layer of lipids. This wall separates the cell interior from the outside environment and largely consists of highly specialized lipids, the phospholipids. Its basic scope is to act as an insulator; however it is also selectively permeable to ions and organic molecules serving as a regulated diffusion barrier.83 The traffic of ions across cell membranes is controlled by two types of integral proteins: ion pumps and ion channels.84 The former actively push ions across the membrane and establish concentration gradients across the lipid bilayer, and the latter allow specific ions to pass through the channel pore along their electrochemical gradient.83 Altogether, they act as biological batteries and resistors, thus creating a voltage difference between the two sides of the cell membrane. The difference in electric potential between the cytoplasm and the exterior of a biological cell is known as membrane potential.84 In non-excitable cells it is held at a relatively stable value (typically ranging from −90 mV to −20 mV) defined as resting potential.85 The opening and closing of specific ion channels can induce departures from this resting condition. This phenomenon is called hyperpolarization if the interior becomes more negative or depolarization if, conversely, the interior becomes less negative.85 In excitable cells (i.e. neurons, muscle cells) a sufficiently strong depolarization can evoke a rapid (1–3 ms) reversal of the membrane potential.85Live cells can be seeded on top of suitable functional surfaces like metal electrodes, inorganic semiconductors or organic polymers. Among the organic polymers, thiophene-based materials, such as P3HT, show the highest biocompatibility, providing the chance for optical stimulation. Indeed, under working conditions, cells should preserve all their natural features, like shape, vitality, equilibrium parameters and, when excitable, their spontaneous activity (e.g. firing capability in neurons).65,86 Protein layers, such as those formed by poly-L-lysine, may help cell adhesion, but are not strictly required and usually do not affect the coupling mechanism.65 Simplifying a complex situation, the thin gap between the cell and the functional surface, the so-called cleft, can be described as a thin electrolyte layer separating the material surface and the cells.87
From the studies conducted so far, it is possible to identify more than one phenomenon implicated in the conversion of light into a physiological signal of the cell. These processes will be discussed in more detail later in the review; however, in general terms, phototransduction can be induced by (i) thermally mediated effects, which primarily alter the cell membrane parameters; (ii) a capacitive coupling between electronic charges accumulated at the surface of the active material; (iii) electrical current injection (Faradaic coupling); and (iv) chemical phenomena (photodynamics), in which photo-activated reactions at the surface lead to a modulation of local osmolarity and/or pH, in turn affecting membrane excitability.
1.4 Types of interfaces (e.g., planar 2D vs. 1D vs. 0D interfaces)
An interface should be as seamless as possible having the ability to transduce a signal that is selectively detected on one side and transferred to the other. Such coupling is the major mission of the interface, on which its performance is evaluated. However, the mechanical, biological and chemical affinities are of paramount importance for an interface, which should be fully harmless and biocompatible.Two-dimensional (2D) flat or planar interfaces are the easiest to fabricate, although in some cases they can feature many layers and have a surface patterning. The substrates for in vitro experiments are usually inert and rigid, like glass. However, in vivo flexible and conformable substrates are needed to have a stable and biocompatible interfacing with the living matter.88,89 In 2D interfaces, one can usually distinguish a bulk that receives the stimulus (e.g. light absorption) and a surface where the signal is transduced. While for inorganic interfaces the surface is well defined, in the organic counterpart this is not so. In the latter case, the interface is distributed and the molecules on the surface get in deep contact with those of the living matter. Moreover, ion penetration into the organic semiconductor takes place at distances that can be tens or even hundreds of nm.2,90 The deployment of 2D interfaces might be hard, as they are relatively large structures that should be in contact with the cell or tissue through a macroscopic interface. Nevertheless, two ways are possible, namely an “external” plaster-like application or an “internal” surgical implantation. In this contest, the use of lower dimensional interfaces (<2D) could offer several advantages. One is the possibility of being delivered in a drug-like fashion, thus simplifying the administration and allowing a pervasive contact with the bulk of the biological tissue. One-dimensional (1D) systems such as carbon nanotubes, inorganic semiconductor nanorods or wires and fibers have been considered for this purpose.34,62,91–96 Recently, optical neuromodulation was demonstrated using silicon nanowires with a diameter of a few hundred nm and consisting of core/shell sections with different electrical properties that mimic the structure of standard microelectronic devices. In simple terms, the nanowires are nano-photovoltaic cells operating in open circuit conditions.97 This exemplifies the advantage of using nanosystems: the ability to engineer the charge distribution and exploit an easy access to the surface.98 However, 1D systems have other issues; for example very narrow samples may penetrate the cell membrane and become harmful by inducing asbestos-like reactions, or be too bulky to have a close contact with cells.99 Zero-dimensional systems (0D), like quantum dots or nanoparticles, have the highest surface to volume ratio, allow for quantum engineering of their properties (like tuning of their optical properties) and can be easily administered.100–102 Size can vary from a few nm for metallic dots to hundreds of nm for polymer nanoparticles, with semiconductor nanocrystals lying in between. Even in this case, the control of their localization might be hard, but recent findings show that this is not crucial for their applications.103 The photoexcitations and consequent effects of such light actuators are discussed below.
2. Planar interfaces
2.1 General considerations
The easiest way for investigating the abiotic/biotic coupling is to grow cells on top of a material of choice, like a semiconductor or a device, and measure the transducing efficiency upon light stimulation. For this reason, a number of experimental data are dealing with cells cultured on top of planar interfaces.104–106 This method is certainly useful for the general understanding although, due to its limited spatial and temporal resolution and the need for bulky supporting electronics, it does not represent the most appealing approach for applications. Important exceptions are flexible/conformable devices that can be used as wearable patches (on the skin surfaces or implanted subcutaneously), or for wrapping internal organs.89,107–111 The planar interface is complex, due to the formation of multiple regions with different properties. The equilibration of the chemical potential (Fermi level) leads to charge redistribution at the interface generating a depletion layer (a charged region) inside the semiconductor and an ion concentration gradient in the electrolyte (described by the Helmholtz–Gouy–Chapman–Stern theory).112,113 In addition, the semiconductor is contaminated at the surface by adsorbed ion species that contribute to generate a local electric field. Organic semiconductors, differently from inorganic ones, allow a more substantial ion penetration and chemical interaction. Last but not least, a variety of proteins protrude from the cell membrane towards the substrate modulating the dielectric response.114,115 A full quantitative and universal model, that can capture the essence of the abiotic/biotic interface, is not available yet, although partial rationalizations of the complex multi-scale problem exist. In addition, the planar interface is a model system for the nanoparticle (NP)–surface/medium interaction that can help in understanding the working mechanism of NPs in a biological environment.2.2 Planar silicon interfaces
During the last decade, the interest in interfaces able to restore the function of the nervous system lost during injury or disease has constantly increased as witnessed by numerous developments in the current state-of–the-art of neural technologies.116–119 A neural interface communicates with the nervous system through implantable electrodes capable of acquiring bioelectric signals and transducing them into electric currents.120 The most popular techniques to monitor the activities of a large number of neurons and their network activity are microfabricated multielectrode arrays (MEAs) and active planar silicon field effect transistor arrays (FET).121,122 Different shapes, sizes and materials of the interface have been also explored to optimize the electrical and mechanical properties, biocompatibility, performance and long-term stability of neural electrodes.122–125Recently, non-invasive extracellular stimulation has been achieved by exploiting the ability of light to alter the conductivity of silicon.32,126–128 In this way, the targeted cell is excited with minimal physiological manipulation, permitting long-term modulation of activity patterns. Similar to transistor-based neuronal interfaces (FET), the photoconductive stimulation method has the advantage of being able to extracellularly excite any neuron in a network, regardless of its spatial position on the silicon substrate.126
Peter Fromherz and coworkers in their pioneering work studied the electrical interfacing of individual nerve cells and silicon microstructures.129,130 Without electrochemical processes, coupling of the semiconductor with neurons relies on a close contact of the cell membrane and oxidized silicon with high resistance of the junction and high conductance of the attached membrane. Extracellular capacitive stimulation in the geometry of cell adhesion was demonstrated under conditions where cell damage by Faradaic current and electroporation could be excluded.131 Furthermore, using an optical technique, Fromherz and coworkers could measure the gap between the cell membrane and the semiconductor surface, namely the cleft, finding experimental values between 40 nm and 100 nm.56,87,132–134 The electrical resistance of the cleft, assumed to be filled by the electrolyte, was also considered. Indeed, it turned out that the conductance of the cleft is a crucial parameter in the coupling equation (Fig. 2, panels A and B).
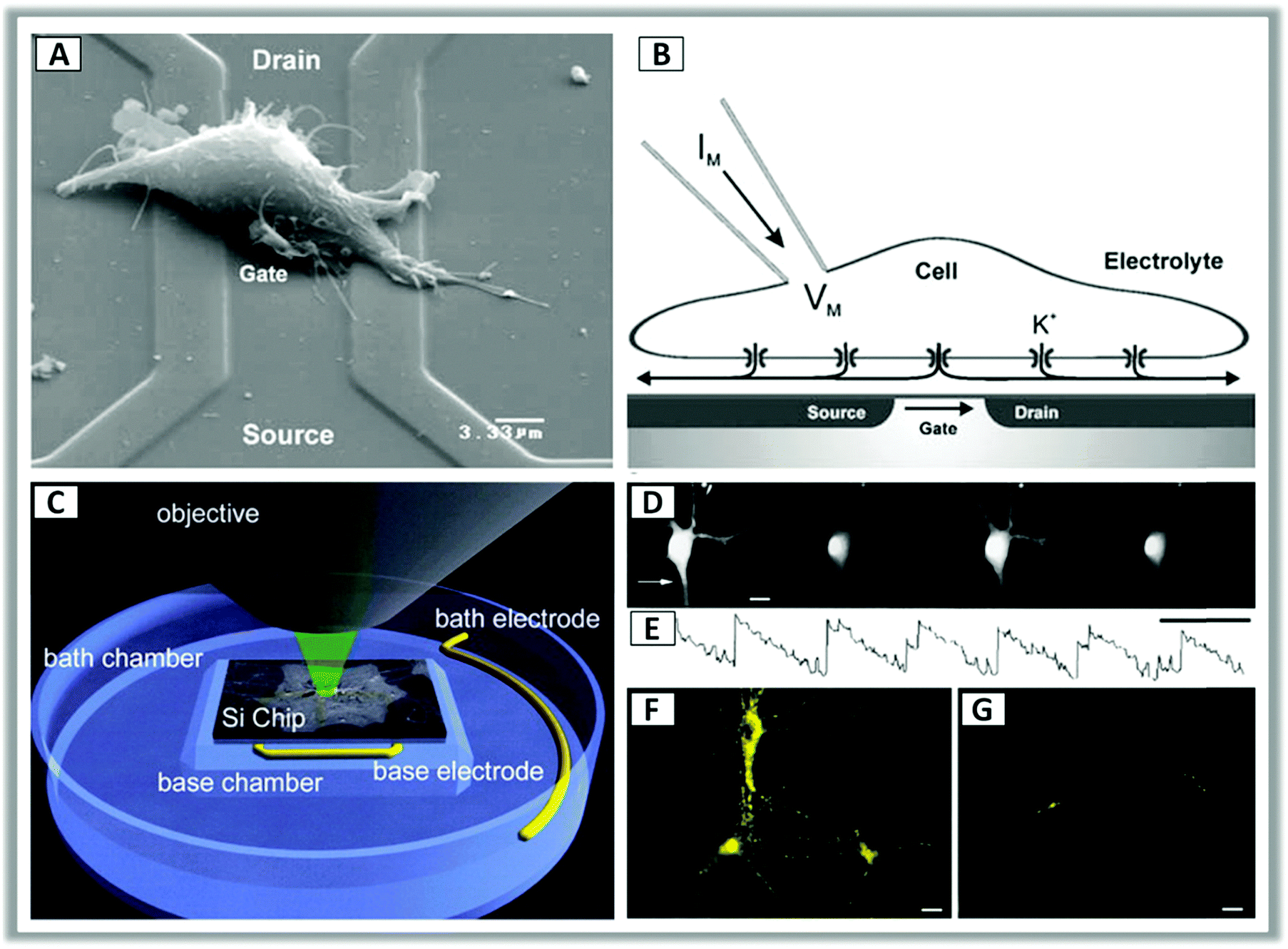 | ||
| Fig. 2 (A) Scanning electron microscope image of a HEK-293 cell lying on top of a silicon field-effect transistor with a metal-free gate of silicon dioxide. (B) Cross sectional sketch of a typical whole-cell patch clamp recording. Application of a voltage across the transistor determines a change in the ion concentration at the cell–transistor interface, leading to potassium (K+) channels opening and a change in the membrane potential. Reproduced from ref. 133 with permission from Springer Science + Business Media, copyright 2005. (C) Schematic configuration for photoconductive experiments of neurons grown on silicon wafers. (D and E) Representative video frames of intracellular calcium (Ca2+) transients in response to 1 Hz pulse stimulation under continuous illumination. (F and G) Synaptic activation was confirmed by labelling neurons with a presynaptic membrane probe that monitors exo-endocytosis (FM1-43) during the application of a 50 Hz stimulus train for 10 s under continuous illumination. Reproduced from ref. 128 with permission from Cell Press, copyright 2001. | ||
More recently light control of the silicon interface allowed to obtain sub-millisecond precision, 100 μm spatial resolution and stimulating rates exceeding 100 Hz128 (Fig. 2, panels C–G). In this configuration, p-doped silicon is usually reverse-biased with respect to the hybrid interface with the electrolyte, having a majority-carrier-depleted region at the surface. Photoexcitation leads to electron migration to the interface, driven by the local field associated with band bending. This in turn changes the voltage gradient in the ionic solution and affects the cell membrane.128
Colicos et al. demonstrated that rat hippocampal cells, cultured on an untreated silicon surface in a physiological solution, respond to constant illumination by modulating the bias voltage between the silicon and an external platinum counter electrode.128 In a similar way, Starovoytov et al., by using single crystal silicon, as a photoconducting substrate, showed that it is possible to achieve non-invasive depolarization of neurons.36 In this case, the stimulation of neurons occurred by modulating laser beam illumination and holding a fixed voltage between the p-type silicon and the counter electrode. It was also observed that the temporal and spatial resolution of silicon photoexcitation can be improved by patterning the silicon surface and thinning the chip.33,135 In addition to p-type silicon, other photoconductive substrates have also been employed, including single crystals and hydrogenated amorphous silicon (a-Si:H).36,136–139
Thus, silicon interfaces provide a powerful tool for examining in vitro neuronal network behavior, since they can be directly coupled to any standard electrophysiology setting. However, the mechanical mismatch between the inorganic material and the biological soft tissue limits its biocompatibility and use as neuroprostheses, since the stiffness of the implanted devices might cause tissue damage and promote an inflammatory reaction in the surrounding tissue.119,140 To overcome these shortcomings, soft, lightweight, and flexible organic materials mimicking the nature of the living tissue are the main focus of recent research to develop biocompatible neural interfaces for in vivo applications.140 The photostimulation mechanism first observed for planar silicon interfaces has inspired the early interpretation of the results obtained using organic semiconductors. Yet the two systems, organic versus inorganic semiconductor interfaces, are very different and hardly comparable.43
2.3 Planar polythiophene interfaces
Organic semiconductors are radically different from inorganic ones. Although the charge concentration achievable in organic semiconductors is smaller than that of silicon, their interface with tissue is a diffuse intermixed region where electrolyte molecules, protons and other ions penetrate and interact, leading to a unique behavior of organic semiconductors for interaction and communication with neural tissue. In some fields of application, organic semiconductors can compete and outperform inorganic ones, with the further advantage of a cheap and simple fabrication technology.141 Moreover, the high flexibility and conformability of the organic materials can provide a tool for applications such as active plaster or prosthesis.77,108,142 P3HT is a prototypic conjugated polymer that can be useful for this purpose, and for bio applications, as we stated above, it seems also to be a gifted one.143,144A P3HT film in contact with water or an electrolyte behaves as a p-doped semiconductor by virtue of the strong electron affinity and appropriate energy matching of the penetrated hydrated oxygen.69,145–148 At equilibrium, due to polarization of the water molecules at the surface, the orbital energy approaching the interface gets reduced, mimicking band bending.113 The polarization of water molecules is consistent with an effective positive charge in the electrolyte close to the polymer surface and a negative charge inside the polymer (depletion region).113 The phenomenon is only formally equivalent to a semiconductor metal interface, since polymers do not have extended band states and the involved charge carrier mobility is much smaller. The watery dipole layer at the polymer surface participates in the formation of the Stern layer, together with ions in the solution (Helmholtz layer).112 In addition, it was shown that the polymer surface changes its affinity for water becoming less hydrophobic (in H2O and under illumination the contact angle drops below 90°). However, the interface is dispersed on several nm due to ions, protons and water molecules penetration. Doping concentrations up to 1018 cm−3 in the soaked P3HT polymer layer have been observed in electrochemical cells by the Mott–Schottky plot technique.69,149 Due to doping, photoluminescence is almost completely quenched.113,150,151 The reason is an efficient energy transfer from the singlet excited state to the polaron state and a consequent internal conversion of the excited polaron state. This process prevails on electron transfer to acceptors, like oxygen, when doping is elevated and contributes to reduce the excited state lifetime from ns to ps.113 Upon photoexcitation, negative charges tend to migrate to the surface causing a local ionic readjustment in the electrolyte.113 The electron poor layer, that results from this process, has been indirectly detected in a variety of experiments that include local probing of photoluminescence in nanodots,113 changes in astrocytes’ electrophysiological properties152 and activation of the mammalian Transient Receptor Potential Channel Vanilloid 1 (TRPV1) channel.153 Accordingly, it is reasonable to hypothesize that following photoexcitation the local pH can be reduced near the surface.113
Simplifying a complex situation, the cleft, i.e. the thin gap between the cell and the polymer surface, can be described as a polymer–electrolyte interface.87 Accordingly, what we said above describes the abiotic/biotic interface. Three possible coupling mechanisms, between the cell and the polymer, that are capable of transducing light energy into a bio-chemical signal have been advanced.153–155
The first is thermal (Fig. 3, panel A). This effect was initially characterized in Human Embryonic Kidney 293 (HEK-293) cells.155–157 By using this simple model, it was demonstrated that light absorption deposits energy to increase the polymer temperature. The temperature gradient normal to a 100 μm spot of light at the polymer surface decays in space with a 100 μm space constant, running over the whole cell. With the light power that is typically used (between 1 and 50 mW mm−2), the increase in temperature observed after 200 ms illumination can reach 7 °C, with a buildup time constant of about 50 ms (Fig. 3, panel B).
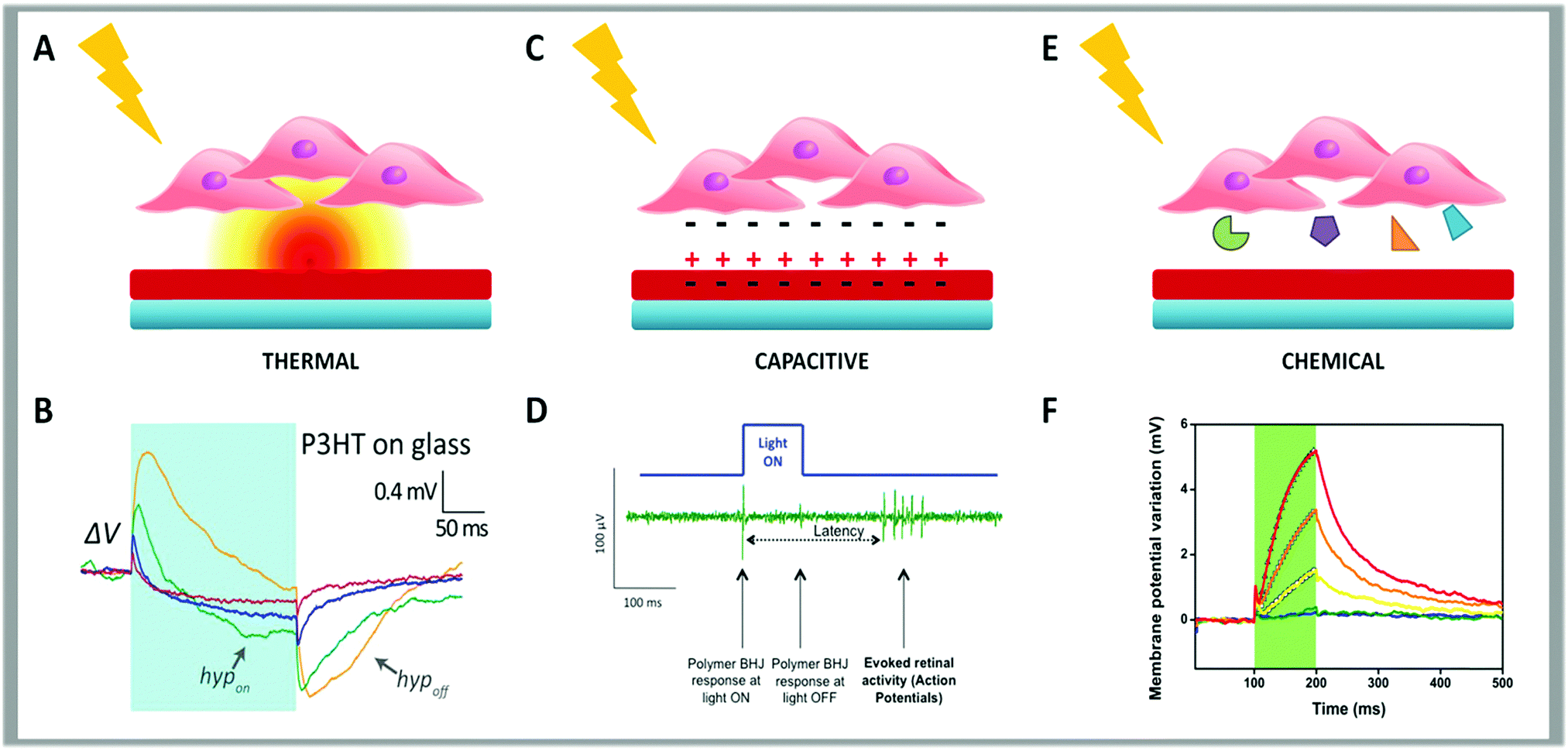 | ||
| Fig. 3 Schematic representation of the three possible coupling mechanisms between cells and conjugated polymers. The thermal effect is depicted in panel (A). (B) Membrane potential variation upon illumination with long light pulses (200 ms pulse, 57 mW mm−2) for HEK-293 cells seeded on a thin film of P3HT spin-coated on a glass coverslip. Reproduced from ref. 155 with permission from Nature Publishing Group, copyright 2015. In panel (C) is represented the capacitive effect. (D) Typical neuronal response elicited in the retina upon polymer photoexcitation using the experimental set-up for simultaneous photoexcitation of the bulk heterojunction layer and recording of the retinal activity using a MEA. Reproduced from ref. 154 with permission from Wiley-VCH, copyright 2014. (E) Schematic cartoon summarizing the chemical effect. (F) Membrane potential variation measured in HEK-293 cells stably expressing human TRPV1 seeded on P3HT when photostimulated with 100 ms visible light pulses, represented by green shaded areas, at increasing photoexcitation densities (from 26 to 343 mW mm−2). Reproduced from ref. 153 with permission from Nature Publishing Group, copyright 2017. | ||
Martino et al. evidenced that the cell response, measured as a change in membrane potential by whole-cell patch-clamp, takes place with two time scales.155 One is associated to a variation of the membrane potential towards more positive values (depolarization). Depolarization initially follows the temperature increase, reaches a maximum and then decays. Based on experimental evidence, this is assigned to a temperature-induced increase in membrane capacitance that is probably attributable to a phase transition in the phospholipid ordering in space that leads to a local thinning of the membrane, as recently supported by numerical modeling.158 A second response, which steadily builds up with temperature, causes a shift of the membrane potential to more negative values (hyperpolarization). This can be due to the readjustment of the ion distribution across the membrane caused by a change in the ion channels’ conductance that, in the case of HEK-293 cells, has been assigned to a change in the K+ conductance.159 This phenomenon can be due to the change in temperature or a consequence of the membrane phase transition via strain changes in the ion channels’ properties. The overlap of depolarization and hyperpolarization with different time scales gives rise to the characteristic shape in the potential–time plot measured.
For the sake of completeness, it is worth mentioning that the optical modulation of the membrane potential, related to the thermal effect, was also observed under similar conditions with other polymers (i.e. PCPDTBT, MEH-PPV) pointing to a universal behavior and suggesting a limit associated with the photocatalytic potentials. However, polyfluorene, a wide energy gap polymer, resulted in phototoxicity probably due to the energy level alignment with oxidants, that induces a very high rate of free radical formation.157
The thermal phenomenon described above has potential biological applications. In fact, it was recently exploited to obtain a reliable, robust and temporally precise control of the previously mentioned TRPV1, the best-studied member of the family of temperature-activated transient receptor potential ion channels (thermoTRPs). To this goal, HEK-293 cells were stably transfected with the human TRPV1 channel (HEK-293T cells).153 In this case the activation of the channel is due to the combination of two different locally confined effects, namely the release of thermal energy from the polymer surface and the variation of the local ionic concentration at the cell/polymer interface, that will be discussed later in the review. Moreover, polymer-mediated optical excitation has been exploited for controlling firing activity in primary neuronal cultures, according to the stimuli duration. In fact, it was demonstrated that, for short light pulses (5–20 ms), by using an organic photovoltaic blend, action potentials could be evoked with short latencies and a very high temporal and spatial resolution.160 The highly localized conjugated polymer response to light with a space constant of <50 μm produces a spatial resolution that is comparable to that obtained by previous approaches using microelectrode or photodiode arrays of fixed, discrete geometries. On the opposite, for light pulses exceeding 200 ms, hyperpolarization was the dominant response. As a consequence, both induced and spontaneous neuron firing could be silenced by light in the presence of the polymer interface.161 Also, the firing activity in explanted retinas and brain slices, as monitored by using MEA, was similarly affected showing a statistical inhibition of neural activity during illumination, followed by a rebound excitation on light switch-off.77
The second mechanism is electrical, due to a capacitive coupling (Fig. 3, panel C). When a polymeric layer is placed in contact with an electrode and soaked in water-based media, photoexcitation can induce ion readjustment. Thus, the surface charge displacement can cause cell depolarization or hyperpolarization.
Gautam et al. reported that by accurately controlling the polymer thickness of a photoactive bulk heterojunction (BHJ), i.e. an intimately mixed phase of donor and acceptor molecules able to favor charge separation and increase photocurrent generation, it is possible to obtain a vertical phase distribution, between a polythiophene based polymer (P3OT) and a naphthalene derivative (N2200), inducing different surface voltage polarity in aqueous media depending on the specific incident wavelength.162,163 Since this approach had interesting similarities with the function of retinal cone cell photoreceptors, it gave support to a successive study in which the same authors performed photoelectrical stimulation of blind chick retina (Fig. 3, panel D).154 Further investigations demonstrated a correlation between the polymer-mediated evoked neuronal signals in the explanted retina and the photovoltage profile with charge accumulation induced at the BHJ surface upon illumination.33,135,160,164 In addition to capacitive coupling, another electrical-induced sub-mechanism is due to current injection into the cell membrane. This process is well known in inorganic semiconductor interfaces, but it has not yet been identified in polymer–cell interfaces.
Finally, the third mechanism is photo-electro chemical (Fig. 3, panel E). Under illumination the polymer surface may support redox reactions. This is well documented in electrochemical cells where the polymer behaves as a photocathode for oxygen or hydrogen reduction. The formation of superoxide could interfere with biological paths and result in a photo-transducing action.165
Somewhat in between capacitive and electrochemical mechanism is the photo-induced local change in pH. Although difficult to prove at the nm scale, at least three distinct experiments suggest that the pH decreases under illumination: (i) the apparent shift in the resting potential observed in current–voltage (I–V) characteristics of astrocytes grown onto the polymer;152 (ii) the lower threshold for stimulation observed in TRPV1 HEK-293T cells (Fig. 3, panel D);153 and (iii) the photoluminescence quenching in core–shell dots conveniently placed on the polymer surface at the interface with the electrolyte.113 All these observations can be rationalized assuming that photoexcitation generates a local electron poor environment. The local change in pH and photocatalytic activity, together with the aforesaid photothermal and photoelectrical mechanisms, suggests a possible mechanism of in vivo photo transduction corroborating the hypothesis that all-organic devices may play, in the next future, a pivotal role in retinal prosthesis technology.77
Vaquero et al. studied four different conjugated polymers with different chemical and physical properties – two polythiophene derivatives with different optical bandgaps (P3HT and the lower band gap polymer PCPDTBT; Fig. 1), a polyphenylene vinylene derivative (MEH-PPV) and a polyfluorene derivative (PFO) – to optically excite the electrical activity of HEK-293 cells. The authors found that not all the tested materials are suitable for optical stimulation, highlighting that thiophene-based materials possess superior biocompatibility and stability compared to the other two polymers and suggesting their possible in vivo long-term compatibility. Both thiophene-based polymers were able to optically modulate the membrane potential according to the previously described thermal effect.157
Feyen et al. studied thin films of P3HT and PCPDTBT interfaced with primary hippocampal neurons, explanted retinas and brain slices to investigate their modulatory effects on the electrical activity of these excitable cells and tissues.161 Prolonged illumination (500 ms) of the polymeric interface, by using visible or infrared light depending on the polymer used, triggered hyperpolarization of the neuronal membrane that significantly reduced both spontaneous and evoked action potential firing. These results prove that conjugated polymer thin films are a tool for the photo-inhibition of neuronal activity, suggesting their potential for in vivo application in pathologies characterized by neural hyperactivity, such as epilepsy, or as retina prosthesis for the rescue of light sensitivity in retina degeneration.161
More recently, Benfenati et al. studied the in vivo application of planar polythiophene interfaces.77,167 They developed a fully organic multi-layered device composed of photoactive layers of P3HT deposited in a flexible and highly conformable silk substrate and implanted it in the subretinal space of dystrophic Royal College of Surgeons (RCS) rats, a widely recognized model of retinitis pigmentosa. The dimension, shape and multilayer structure of the prosthesis are shown in Fig. 4. Functional analysis showed that significant recovery of visual parameters persisted up to 6–10 months after implantation in RCS dystrophic rats. This indicates the full structural and functional preservation of the prosthesis and its efficient rescue of vision. This experimental approach demonstrates the possibility to work without implanted metal- or silicon-based electronics, bypassing the need for power supply or external cameras.
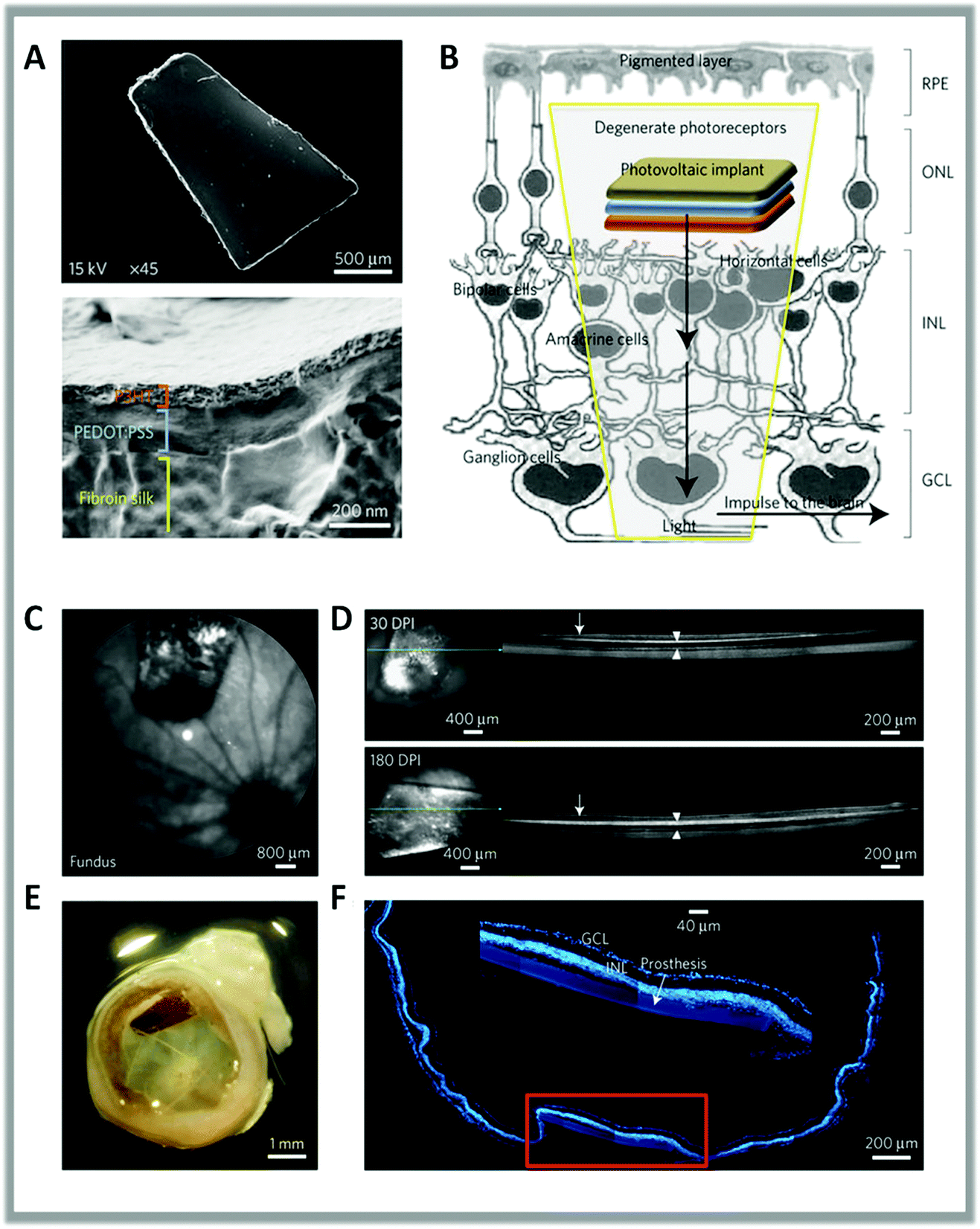 | ||
| Fig. 4 (A) Scanning electron microscopy images of the full prosthetic device and of its cross-section. (B) Scheme of the subretinal implant strategy. (C) Sample confocal scanning laser ophthalmoscopy image of the surgical prosthesis placement in the eye fundus of a dystrophic RCS rat. (D) OCT analysis showing the strict contact between the retina (arrows) and the implant (arrowheads) at 30 and 180 dots per inch (DPI). (E) Explanted eye stained with bisbenzimide and acquired by confocal microscopy (F) to identify retinal nuclear layers and the position of the device. The inset shows the integrity and location of the implant in the retina. Reproduced from ref. 77 with permission from Nature Publishing Group, copyright 2017. | ||
3. Nanostructured interfaces
3.1 General considerations
It is well known that materials’ properties strictly depend on the three-dimensional arrangement of their building blocks. Downscaling one, two or all three dimensions of a material to the nanoscale level allows the fabrication of 2D (e.g. thin films, membranes, graphene), 1D (e.g. nanorods, nanotubes, nanofibers) or 0D (e.g. hollow capsules, nanoparticles) architectures, whose structural and functional properties, owing to confinement effects and large surface area to volume ratio, are substantially different from those observed for bulk materials.168,169 The engineering of these highly organized architectures allows the manipulation of mechanical, electric, magnetic, optical and electronic properties providing new application opportunities via the selection of structures with appropriate dimensionality.100,170In the last decade, the use of nanomaterials for research, diagnosis and therapy of neurological diseases has received great attention from the scientific community, due to their ability to stimulate neurons by converting primary stimuli (light, magnetic field or ultrasound) into localized secondary stimuli (electric field or heat), with a high degree of spatiotemporal resolution and cell specificity.140
In particular, the use of nanoparticles (NPs) – i.e. materials with all three dimensions downsized to the nanoscale – is emerging as an innovative approach to create non-invasive and highly specific light-activated actuators for localized neural stimulation.2,171–173 Indeed, due to their size, comparable to those of receptors embedded in neuronal membranes, NPs may operate as camouflaged actuators capable of controlling and interacting with receptors or even acting as receptors themselves to repair/restore lost or damaged sensory functions. Consequently, their application in neural interfaces may overcome the limitations resulting from the current therapeutic techniques, such as invasive surgical implant or genetic modification, thus opening new perspectives for wireless neurostimulation approaches.42,174
NPs can be divided into two general classes: hard nanoparticles made up of inorganic materials (e.g. quantum dots, silica NPs, carbon NPs, metal nanoclusters), in which the presence of strong covalent bonds among the atoms results in the formation of nano-objects with a rigid shape and a constant size, and soft nanoparticles made up of organic materials (e.g. polymeric NPs, nanogels, micelles), in which the weak interactions among the self-assembled molecules lead to nanostructures with a dynamic character, susceptible to shape and size variations depending on the environment. The diversity in the chemical composition of NPs, together with variation in size, internal arrangement, charge distribution and surface functionalization, allows for a remarkable modulation of their chemical–physical properties, such as electron density, optical absorption, photoluminescence or magnetic properties as well as a radically different interaction with biological systems that determines their biocompatibility and stability.175,176
Thanks to this functional flexibility and adaptability, NPs are currently being investigated as next-generation neural interfaces.2,173 Herein we will highlight the recent investigations related to their use as light-sensitive actuators, aiming at inducing a change in neuronal activity upon illumination, to control physiological functions. In the second part, we will specifically focus on the preparation and in vitro/in vivo applications of organic semiconducting NPs, that possess a higher number of key enabling features than the inorganic counterpart such as biocompatibility, biodegradability and adaptability.177,178
3.2 Inorganic nanoparticles activated by optical stimulation
Neurons are able to respond to a variety of stimuli: electrical, thermal, physical and chemical. The methods traditionally employed to trigger neural activity are based on electrical stimulation, involving invasive and bulky implanted brain electrodes.116,179 Lately, as previously discussed, optogenetics and high-power infrared light stimulation have been explored as non-invasive alternatives to direct electrical stimulation.1,27,180 Moreover, it has been highlighted that NPs, upon opportune functionalization, are able to recognize and bound specific targets inside live cells and act as transducers for neural stimulation. NPs allow modulation of neural activity through the conversion of a diffused primary input into a highly spatiotemporally localized secondary neuronal output, avoiding the need for genetic manipulation and with the added benefit of requiring low power densities for stimulation, preventing tissue heating, one of the main causes of cell damage and death. In particular, optically active NPs can efficiently transform non-local optical signal into local neuronal stimulation by converting light into localized heat or electrical current (Fig. 5).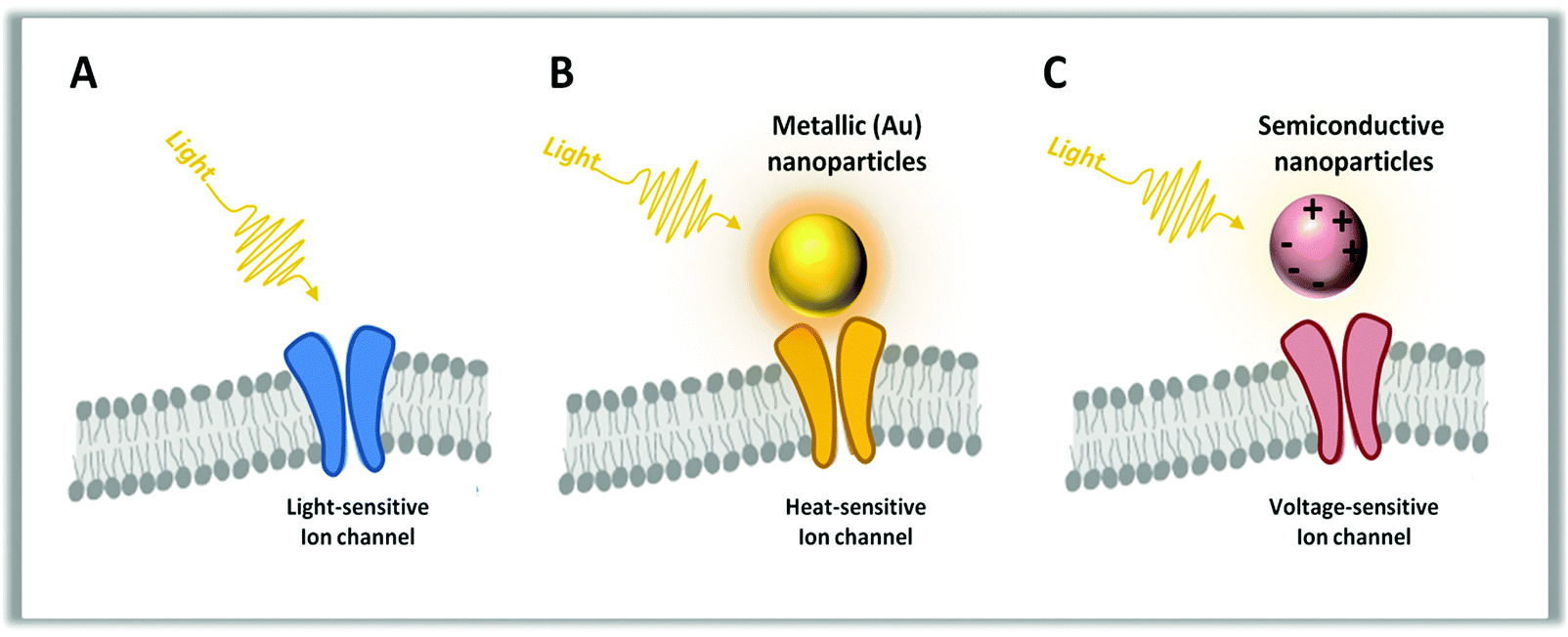 | ||
| Fig. 5 Different approaches for neural stimulation: optogenetic stimulation (A), photo-thermal stimulation with metallic nanoparticles (B) and photo-electric stimulation with semiconducting nanoparticles (C). | ||
Among the different types of inorganic NPs, gold nanomaterials have attracted a lot of interest for their ability to thermally activate neurons by absorbing visible or infrared light and converting it into localized heat that changes the neuronal membrane capacitance.40,181–183 The interaction of gold nanomaterials with light strongly depends on their size, shape, surface and aggregation state. In particular, by changing their shape it is possible to fine-tune their optical properties through alteration of the surface plasmon resonance,184,185 determining absorption maxima ranging from 500 nm observed in the case of spherical gold nanoparticles (AuNPs), to the near-infrared observed for gold nanorods, nanoshells, nanostars or nanocages.186 The optical absorption characteristics of AuNPs make these systems good candidates for the restoration of sight in patients with photoreceptor degenerative diseases, as they show plasma resonance absorption near 500 nm, a wavelength which is present in daylight ambient illumination. Recently, Bezanilla et al. demonstrated that 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm gold nanoparticles conjugated with cell-targeting biomolecules, such as the Ts1 neurotoxin, or with anti-TRPV1 or anti-P2X3 antibodies, can specifically bind neuronal membrane proteins of dorsal root ganglion neurons conferring light-responsiveness to these cells upon irradiation with visible wavelength (532 nm) via activation of voltage-gated Na+ channels. This type of functionalization allows NPs to directly link to the cell of interest, allowing one to employ a lower amount of NPs while achieving a high level of signal transduction (Fig. 6, panel A). Moreover, functionalized NPs show an increased resistance to washout compared to non-conjugated AuNPs, where the lack of specific functionalization causes rapid displacement from the target neurons by convective solution exchange and, consequently, loss of the optical excitability of the cells.187
nm gold nanoparticles conjugated with cell-targeting biomolecules, such as the Ts1 neurotoxin, or with anti-TRPV1 or anti-P2X3 antibodies, can specifically bind neuronal membrane proteins of dorsal root ganglion neurons conferring light-responsiveness to these cells upon irradiation with visible wavelength (532 nm) via activation of voltage-gated Na+ channels. This type of functionalization allows NPs to directly link to the cell of interest, allowing one to employ a lower amount of NPs while achieving a high level of signal transduction (Fig. 6, panel A). Moreover, functionalized NPs show an increased resistance to washout compared to non-conjugated AuNPs, where the lack of specific functionalization causes rapid displacement from the target neurons by convective solution exchange and, consequently, loss of the optical excitability of the cells.187
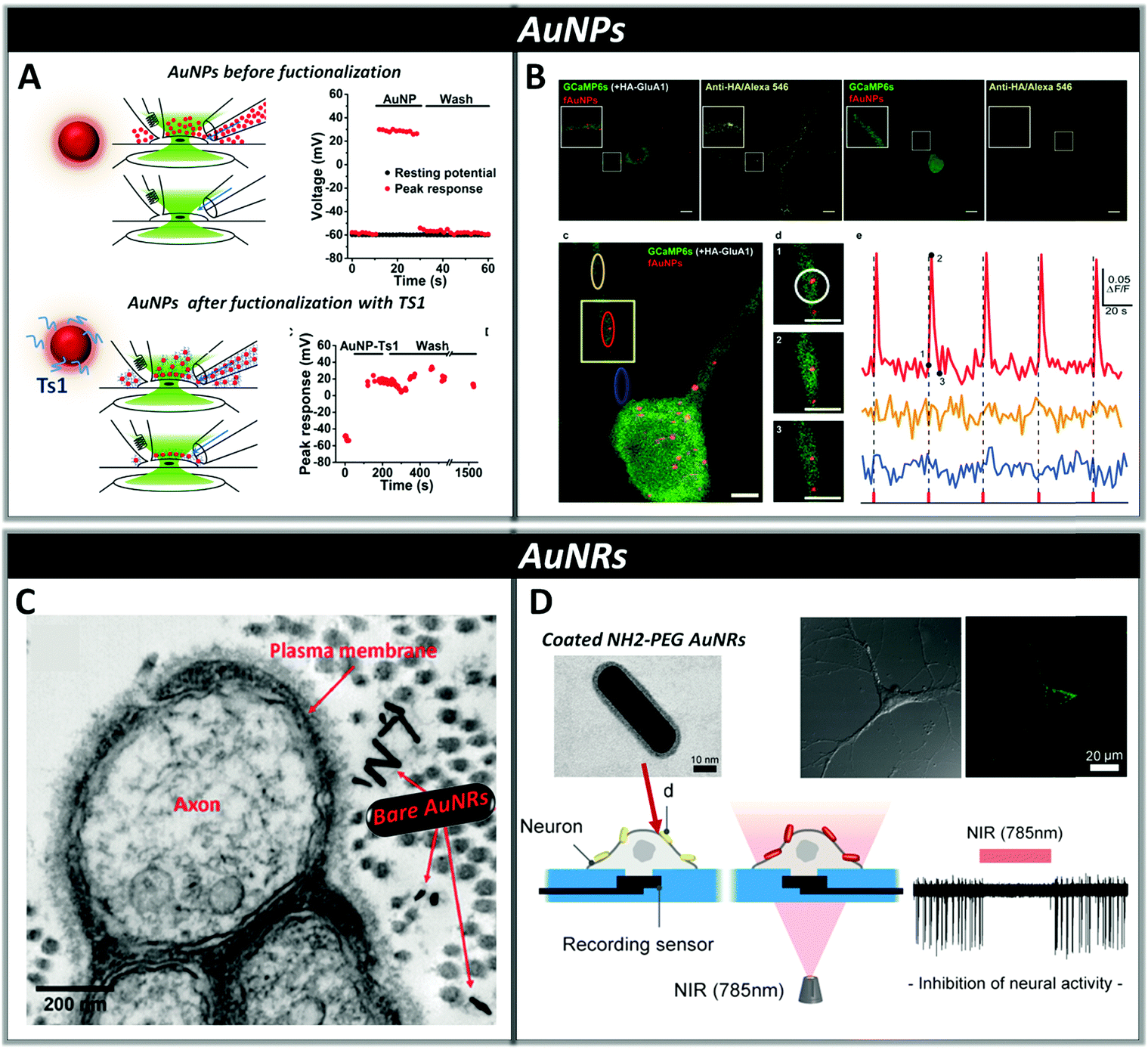 | ||
| Fig. 6 Panel A: Dorsal root ganglion neurons can be excited with visible light in the presence of bare or functionalized AuNPs (AuNP-Ts1). In both cases, NPs are added via perfusion through one side of a theta capillary and then washed with fresh buffer through the other side of the capillary (a and c). In the case of bare AuNPs, washing drastically reduces their concentration near the cell and the laser effect rapidly disappears within seconds (b), while in functionalized AuNPs-Ts1, in which Ts1 promoted stable labeling of the neuronal membrane with AuNPs, even after 20 min of washing, neurons remain optically excitable (d). Reproduced from ref. 187 with permission from Cell Press, copyright 2015. Panel B: Neurons expressing HA-GluA1 and GCaMP6s (a) or only GCaMP6s (b) were incubated for 90 min with functionalized AuNPs. In the absence of HA-GluA1, AuNPs were not able to bind neurons. Nanoparticle-Assisted Localized Optical Stimulation (NALOS) applied to neurons labeled with AuNPs (d) triggers the induction of local and repeated Ca2+ spikes (e). (Scale bars (a, b) 10 μm and (c, d) 5 μm.) Reproduced from ref. 188 with permission from Nature Publishing Group, copyright 2016. Panel C: Transmission electron microscopy (TEM) image of a cross-sectional view of the rat sciatic nerve; bare AuNRs are located near the surface of the plasma membrane after injection. Reproduced from ref. 189 with permission from Wiley-VCH, copyright 2014. Panel D: Representation of AuNRs functionalized with NH2-PEG localized on a neuronal membrane and the inhibition of neural activity through photo-thermal conversion of near-infrared energy. TEM image (a) of the NH2-PEG AuNR evidencing the presence of a brighter region associated with PEG coating. Confocal fluorescence microscopy (b) showing the localization of the NH2-PEG AuNR on the plasma membrane of neurons. Reproduced from ref. 193 with permission from American Chemical Society, copyright 2014. | ||
This result highlights the importance of chemical functionalization that offers a unique and powerful engineering platform to tailor chemical and electronic surface properties and drives target–ligand interactions.
Bio-functionalization of AuNPs with monoclonal antibodies has also recently allowed the photo-stimulation of subcellular regions in cultured hippocampal neurons by using laser assisted confocal microscopy, triggering action potentials through activation of local Ca2+ transients and Ca2+ signalling via CaMKII in dendritic domains (Fig. 6, panel B).188 The ability of gold nanomaterials to absorb in the near infrared region (700–900 nm) makes them advantageous for in vivo applications, since tissues in this range of wavelength display the highest transmissivity.
Eom et al. showed that gold nanorods (AuNRs) targeted to the neuronal cell membrane activate temperature-sensitive channels in the rat sciatic nerve in vivo upon irradiation with pulsed infrared light at a wavelength of 980 nm. This localized photo-thermal effect triggers action potentials by lowering the firing threshold stimulation level by approximately two orders of magnitude compared to direct infrared neural stimulation methods, thus allowing reduction of radiant exposure and consequently the risk of tissue damage (Fig. 6, panel C).189 Yong et al. used AuNRs coated with silica to increase their biocompatibility and chemical stability,190 for the stimulation of primary auditory neurons in vitro using pulsed-laser irradiation at 780 nm. By using temperature measurements, they demonstrated that the presence of AuNRs determines a change in the temperature of the local environment between 0.5 and 6 °C depending on the pulse duration, allowing a significant increase in electrical activity.40 Both bare AuNRs and silica-coated AuNRs were also cultured with NG108-15 cells and irradiated at 780nm showing that after internalization, they can promote neurite outgrowth in vitro and induce intracellular Ca2+ transients.191,192
Yoo et al. found that functionalizing AuNRs with positively charged amine-terminated polyethylene glycol increases their biocompatibility and favors their binding to the negatively charged membrane phospholipids of primary cultured hippocampal neurons via electrostatic interactions. This ensures minimal cellular uptake and inhibition of the spatiotemporal suppression of neural activity in networks after exposure to continuous NIR laser irradiation (Fig. 6, panel D).193 Moreover, they discovered that NIR-sensitive AuNRs, deposited on the electrode–neuron interface of a microelectrode, are capable of enabling simultaneous electrical excitation and optical inhibition of neural activity with high temporal precision.194
Another family of inorganic nanomaterials employed as mediators of optical neural stimulation is that of semiconductive quantum dots (QDs).164,195–200 The interest in QDs, generally composed by atoms from II–VI or III–V groups of the periodic table of elements, derives from their unique optoelectronic properties due to quantum confinement effects, i.e. spectrally sharp emission peaks, broad band absorption, high quantum efficiency, excellent photostability and the ability to generate photocurrents.201 Their interaction with excitation light creates a transient electric dipole moment, through the formation of electron–hole pairs,202 creating localized electric fields able to trigger action potentials in neurons.
QDs have mainly been applied in the form of thin films in close proximity of cells membranes due to their ability to perturb the electrochemical equilibrium between the inside and outside of the cells by acting as efficient opto-electrical transducers.203 Lugo et al. demonstrated that it is possible to activate voltage-gated ion channels with light in both prostate cancer (LnCap) cells grown on cadmium telluride (CdTe) QD films and cortical neurons grown on cadmium selenide (CdSe) QD films. The electric fields produced by the photo-generated dipoles of the QDs activate the ion channels of the cell membrane determining hyperpolarization or depolarization of the cell.196 Moreover, Pappas et al. established that by engineering a multi-layer interface between cells and a thin film of semiconducting NPs made of HgTe, it is possible to enhance the coupling between electrical properties of the NPs and the ion fluxes through the cell membrane.198
Direct introduction of QDs inside cells is currently investigated to overcome the problems associated with the use of thin films, i.e. planar interfaces, which generally suffer from low reproducibility of the neuron/film interface due to uncontrollable neuronal growth, and poor electronic coupling arising from relatively large separation, in the order of 1 μm, between the cell and the stimulating surface.204,205 On the other hand, QDs lacking specific ligands able to recognize cell membranes undergo rapid endocytosis within 5 min of exposure, as observed for mercaptoacetic acid-coated cadmium sulfide (CdS) and CdTe QDs with three different types of nerve cells, i.e. SK-N-SH and PC12 cells as well as primary rat cortical neurons.195 Consequently, the labeling of QDs with recognition groups can offer a viable strategy for long-term active interfacing with cells under physiological conditions. This approach was investigated by Winter et al., who described two pathways to attach semiconductor QDs to the SK-N-SH neuronal cell line by employing either antibody–antigen recognition or peptide recognition groups. Both methods allowed targeting receptors on the neuron surface, localizing semiconductor–biomolecule binding to the exterior of the cell, even if the use of peptide recognition groups provides a better control over the targeting and separation distance between the cell and QDs (Fig. 7, panel A).200 More recently, Pellegrino et al. demonstrated that, by selecting the surface charge of CdS–CdSe QDs and quantum rods (QRs), it is possible to specifically localize QDs on the neuronal membrane, resulting in electrophysiological responses at both single neuron and network levels. In particular, they found that only negatively charged NPs, obtained by functionalization with amino-PEG-carboxyl (COOH), interact in vitro with neuronal membranes of primary hippocampal neuronal cultures and cause neuronal spiking activity. Conversely, the same type of NPs functionalized with positive or neutral amino-PEG derivatives display only non-specific interactions with the cell membrane and no effect on bioelectric activity (Fig. 7, panel B).206
 | ||
| Fig. 7 Panel A: Labelling of neurons with QDs by targeting specific cell surface receptors with antibodies (a) or peptides (b). Peptide functionalization results in a smaller separation distance between QDs and cells (∼3 nm based on an octamer peptide recognition sequence used). Bright-field (c and e) and fluorescence (d and f) optical microscopy images of SK-N-SH human neuroblastoma cells labelled with CdS QDs functionalized with antibodies (c and d) and peptides (e and f). Reproduced from ref. 199 with permission from American Chemical Society, copyright 2007. Panel B: Type I (a) and type II (b) QRs functionalized with amino-PEG derivatives bearing carboxyl (COOH), methoxy (OCH3) or amino (NH2) end groups. Negatively charged QRs interact with the cell membrane of neurons (c and d), while positively charged QRs do not (e and f). The staining of the cell nuclei with DAPI is shown in blue. (Scale bars: 100 μm.) Reproduced from ref. 206 with permission from American Chemical Society, copyright 2007. | ||
QDs have also been employed as transducers to monitor neuronal spiking activity in a large population of individual neurons207 and to quantify temperature fluctuations in live cells by using fluorescence imaging techniques.208 Furthermore, Rowland et al. have shown that the quenching of QDs’ photoluminescence can be used to track the action potential profile of a firing neuron with ms time resolution, opening the possibility of using QDs in live cells, tissue and animal systems to achieve real time imaging of neuronal activity.209
As clearly evidenced by the reported examples, inorganic NPs are highly promising phototransducers for diagnostic and therapeutic applications; however, their in vivo application is hampered by a variety of potential risks.210 NP toxicity depends on physiochemical parameters such as particle size, shape, surface charge and chemical composition. For example, gold which is known to be inert and biocompatible in the bulk can become toxic in the form of nanoparticles.211 Soderstjerna et al. have shown that Au NPs can have significant neurotoxic effects especially on retinal neurons by causing an increase in oxidative cell stress and apoptosis.212 Regarding QDs, their toxicity most likely arises from their degradation and the leaching of the heavy metal ions.213–215 As a consequence, inorganic NPs need further investigation to better understand how they chronically interact with cells and animal models before clinical use. Toward this goal, the evaluation of the toxicity of some Au- and QD nanosystems in Hydra vulgaris, an efficient model system to assess in vivo the ecotoxicological impact of colloidal nanomaterials, revealed that there is no straightforward link between the composition of an inorganic nanomaterial and its toxicity.62,216–220
3.3 Organic nanoparticles activated by optical stimulation
The interest in organic semiconducting NPs for applications involving light stimulation originated from the fact that they show high brightness, good stability toward light and oxygen, large absorption cross sections and vast specific surface area. Moreover, their chemical–physical properties can be finely tuned by modifying the chemical structure of the starting material. Semiconductive NPs are made of optically and electronically active oligomers or polymers characterized by an extended π-conjugation along the backbone.221 Examples of these NPs include acenes, oligothiophenes, polythiophenes, polypyrrole, poly(p-phenylene vinylene) as well as polyacetylene and their derivatives.222 Most of these materials have been successfully implemented in opto-electronic devices, such as organic light-emitting diodes (OLEDs),223 organic photovoltaic devices (OPVs)224–227 and organic field-effect transistors (OFETs),228–230 achieving higher performances through a better control of the nanomorphology by means of the use of pre-aggregated nanostructures with the added value of low-cost and eco-friendly large-scale fabrication.231The lack of heavy metals has made organic NPs very appealing systems for in vivo biological applications.178,232 Indeed, over the last decade a steadily increasing number of publications regarding their use in photoacoustic imaging, photothermal therapy of tumors, optogenetics as well as phototheranostics has been reported.106,233–241
Nano-precipitation and mini-emulsification are considered the most versatile methods to prepare water-suspended NPs249,250 due to the wider variety of chemical moieties that can be employed compared to other methods. Moreover, the ability to modify in a controlled way the molecular structure of the starting materials through easy synthetic procedures allows for a very wide property–function tuning of frontier orbital energies and energy gap, optical properties and charge transport characteristics.251 However, in most cases the resulting polydispersity – i.e. the broadness of size distribution – is greater than that of NPs prepared by direct polymerization methods.
Fig. 8 (panels A and B) shows a schematic representation of nano-precipitation and mini-emulsification methods. In both cases, the starting material is first dissolved in an organic solvent and then injected into water, under magnetic stirring or ultrasound assistance. The rapid change in solvent polarity favours π–π stacking and hydrophobic interactions determining the formation of nanoparticles. However, while in mini-emulsification the organic solvent used to dissolve the starting material is immiscible with water and requires the presence of surfactants, such as sodium dodecyl sulfate, to avoid coalescence of the emulsified droplets, in nano-precipitation the organic solvent is miscible with water and the resulting NP dispersion is free of added surfactants.
 | ||
| Fig. 8 Scheme of reprecipitation (A) and mini-emulsion (B) methods for the synthesis of semiconductive polymer nanoparticles. (C) Examples of functionalization of the NP surface. | ||
Generally, the nano-precipitation method yields smaller nanoparticles compared to mini-emulsification and also allows the formation of NPs with dimensions down to few nanometers, corresponding to single chain polymer particles called polymer dots (pDOTs).251–253 The preparation method and experimental conditions – organic solvent (or mixture of solvents), starting material concentration, polydispersity and regioregularity of the polymer, surfactant concentration, stirring speed, etc. – strongly affect the size and the internal supramolecular organization of the material in the NPs, which are in turn intimately related to their optoelectronic, charge transfer and charge transport properties.222,230,248,254,255
The NP surface plays an important role in determining the stability, solubility, toxicity and reactivity of the particles. For this reason the possibility to perform selective chemical functionalization on the surface of organic NPs is an important possibility providing an effective way to control their in vitro and in vivo activity and interactions with biological systems.256
The functionalization of organic NPs with specific moieties can be conducted before (pre-assembly) or after (post-assembly) their formation (Fig. 8, panel C). In the pre-assembly approach, chemically reactive groups (amino, carboxylic, active esters) and/or specific ligands (peptides, proteins, sugars) are incorporated either by direct conjugation to the starting material through covalent bonding257–259 or by encapsulation within the polymer through electrostatic or hydrophobic interactions.260–262 In the post-assembly approach, functionalities can be attached specifically to the NP surface either by covalent bonding through reactions carried out in aqueous media, such as oxidation,251 esterification263,264 and click reactions,263 or by non-covalent bonding through electrostatic, hydrogen bonding or hydrophobic interactions.265 The main difference between post-assembly and pre-assembly functionalization of NPs is that in the former case the functional groups intervene during the self-assembly of the NPs and are randomly distributed both inside and outside the NPs, while in the latter case they are confined in the outer layer of the NPs without modifying their core.
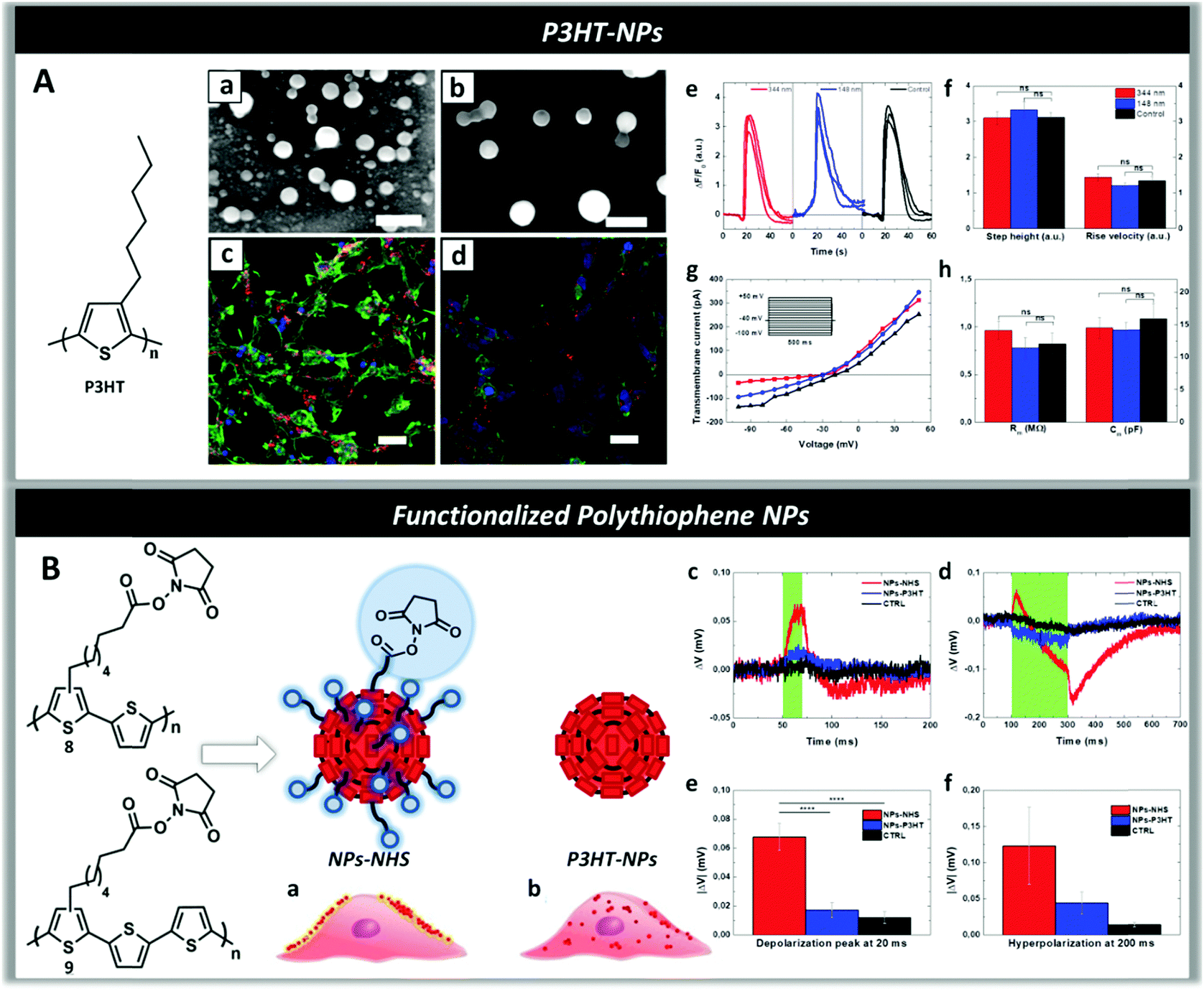 | ||
| Fig. 9 Panel A: Scanning electron microscope (SEM) images of P3HT-NPs of size 148 nm (a) and 344 nm (b). (Scale bars, 500 nm.) Fluorescence imaging of HEK-293 cells loaded with P3HT-NPs (c and d) and stained with DAPI (nuclei, blue) and phalloidin (actin filaments, green) showing the internalization of NPs by the cells. (Scale bars, 30 μm.) (e) ATP-evoked Ca2+ transients in HEK-293 cells treated with P3HT-NPs and relative untreated control. (f) Amplitudes and dynamics of Ca2+ signals after addition of ATP. (g) I–V characteristics of treated (blue and red lines, 148 nm- and 344 nm-sized NPs, respectively) and untreated (black line) cells, extracted by voltage-clamp measurements. The stimulation protocol is represented in the inset. (h) No changes are observed in the average values of membrane resistance and capacitance. Reproduced from ref. 269 with permission from Royal Society of Chemistry, copyright 2016. Panel B: Pictorial illustration shows that the presence of N-hydroxysuccinimidyl ester groups (NHS) on the surface of P3HT-NPs allows docking them on the cell membrane (a) differently from P3HT-NPs which are not being retained by the membrane and are rapidly internalized by the cells (b). Membrane potential variation traces for in vitro samples treated with NPs-NHS (in red), P3HT-NPs (in blue) and untreated cells (in black) and illuminated for 20 ms (c) and 200 ms (d). Average values of depolarization (e) and hyperpolarization (f) during 20 and 200 ms light stimulus respectively. Reproduced from ref. 259 with permission from Royal Society of Chemistry, copyright 2017. | ||
To direct the binding of P3HT-NPs toward the cell membrane in order to modulate the membrane polarization under illumination, novel polythiophenes functionalized with alkyl side chains terminating with N-hydroxysuccinimidyl ester groups (NHS) were synthesized and the corresponding NPs (NPs-NHS) were prepared through nano-precipitation (Fig. 9, panel B).2591H and DOSY NMR spectra enabled one to obtain information on the NP structure and distribution of NHS reactive groups on the surface of the NPs, as well as on their chemical stability at physiological pH. Reaction with chiral tryptophan at physiological pH was employed to test the reactivity of the NHS groups, while circular dichroism spectra proved the formation of a chiral shell around the NPs. Laser scanning confocal microscopy demonstrated that in contrast to P3HT-NPs, NPs-NHS were specifically docked to the plasma membrane in live HEK-293 cells, probably due to the reaction between the NHS groups of the NPs and the primary amines of membrane proteins270,271 and formation of amidic bonds docking the NPs to the cell membrane (Fig. 9, panel B(a)). Electrophysiology experiments showed that upon illumination with a radiation wavelength resonant with the P3HT absorption peak (550 nm) there was a statistically significant biphasic change in the membrane potential of the cells treated with NPs-NHS – depolarization (ΔV > 0) followed by hyperpolarization (ΔV < 0) – that was not observed in cells treated with NPs-P3HT (Fig. 9, panel B(c–f)), in agreement with the data obtained on HEK-293 cells deposited onto P3HT polymer films.86,160
Recently, Pu et al. reported that NPs based on properly bio-functionalized polythiophene act as photothermal nanomodulators capable of controlling thermosensitive ion channels in neurons. Push–pull polythiophene derivatives absorbing NIR light at 808 nm were synthesized (Fig. 10, panel C) and made water-soluble via co-nanoprecipitation with polystyrene-b-poly(acrylic acid) (PS-PAA) (Fig. 9, panel A). The presence of PS-PAA allowed bio-conjugation with anti-TRPV1 antibodies, via carbodiimide coupling between the carboxylic groups of the NPs and amine groups of anti-TRPV1 antibodies. This bio-functionalization protocol allows a precise targeting to the thermosensitive ion channels (Fig. 9, panel B) and a consequent localized heating. Upon transient NIR laser irradiation at 808 nm, these NPs act as wireless remote nano-modulators inducing an intracellular Ca2+ influx in neurons within milliseconds in a reversible manner (Fig. 10, panel D). Moreover, these NPs display good cytocompatibility also after laser irradiation and, despite the sub-optimal characteristics, a good photothermal conversion efficiency, faster heating capability and better photothermal stability compared to inorganic gold nanorods, confirming the great and growing potential of organic materials as phototransducers.272
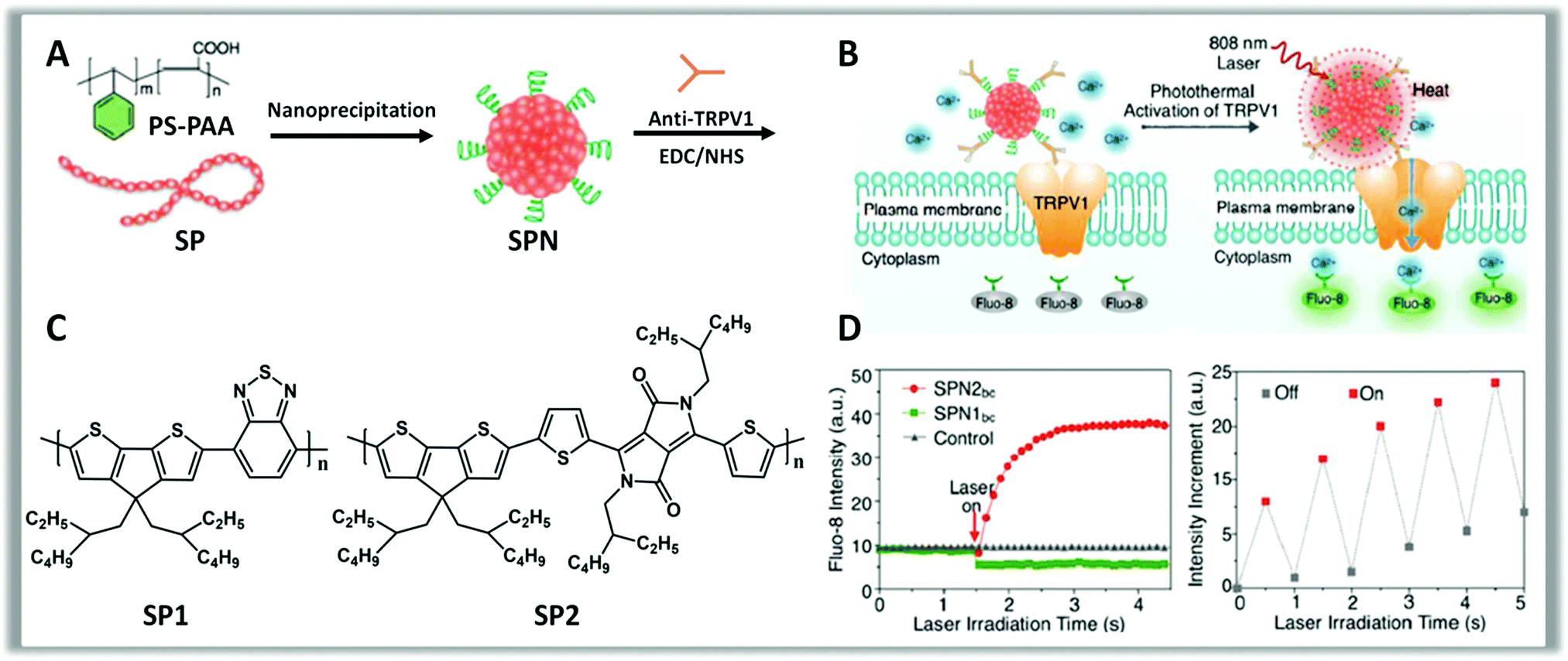 | ||
| Fig. 10 Schematics showing the preparation of SPNs (A) starting from the polymers SP1, SP2 (C). Conjugation of SPNs with anti-TRPV1 antibodies, via carbodiimide coupling, allows the controlled photothermal activation of Ca2+ channels in neurons (B). The intracellular concentration of Ca2+ is checked in real-time by using Fluo-8, a fluorescent intracellular Ca2+ indicator (D). Change in the fluorescence intensity of Fluo-8 upon switching on and off laser irradiation (808 nm) with an interval of 0.5 s, indicating the reversible activation and silencing of TRPV-1 Ca2+ channels by laser irradiation. Reproduced from ref. 272 with permission from American Chemical Society, copyright 2016. | ||
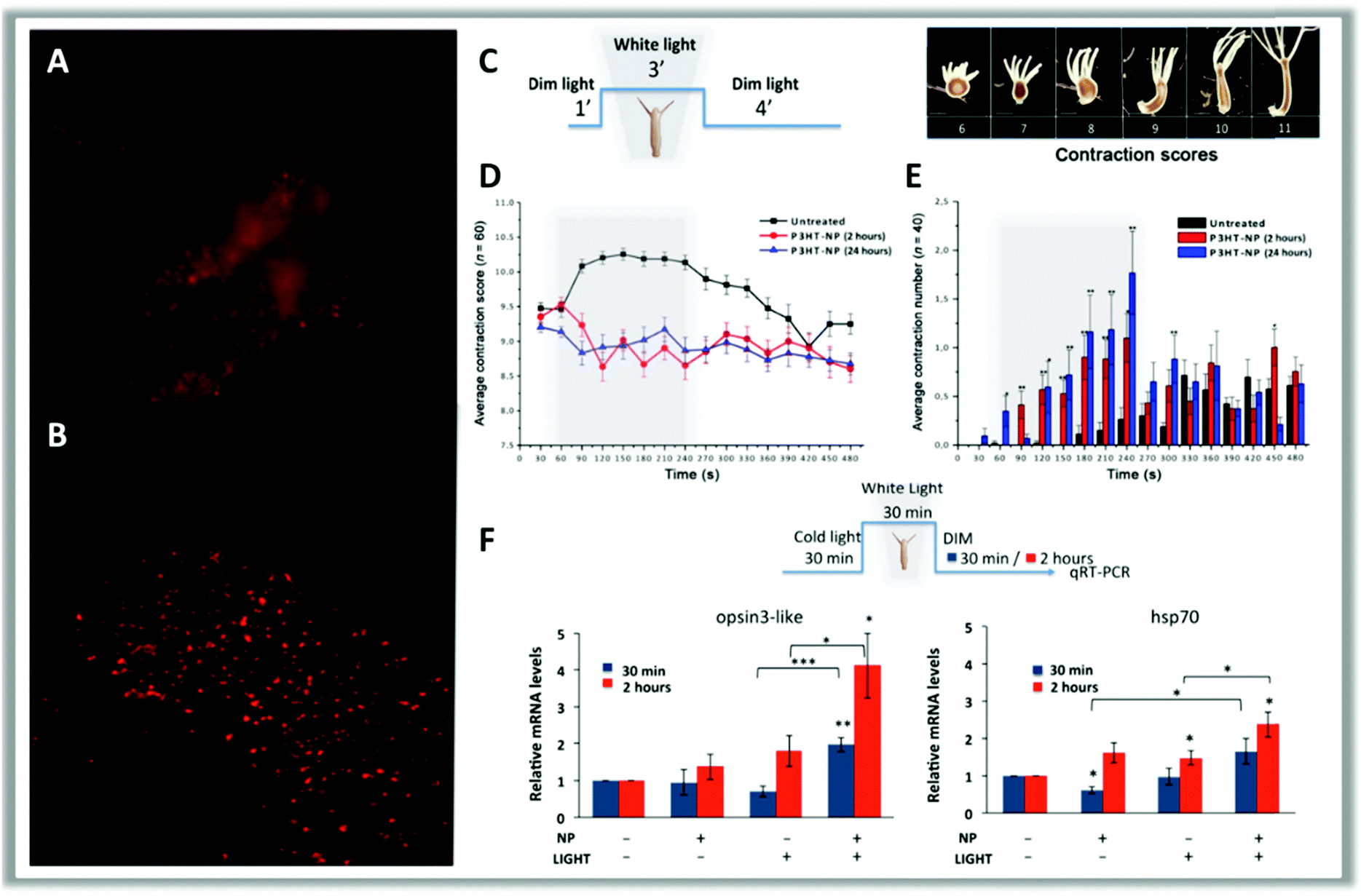 | ||
| Fig. 11 In vivo fluorescence imaging of a head (A) and a tentacle tip (B) of a polyp treated with P3HT-NPs. At the beginning, polyps treated with 0.25 μM P3HT-NPs show fluorescent staining of all tissues but, after a few hours, NPs are located inside the ectodermal cells and appear as fluorescent spots. The behavioral response of Hydra to the presence of P3HT-NPs was estimated through a contraction scoring system ranging from 6 (highly contracted) to 11 (elongated polyp) by using the illumination protocol reported in scheme (C) (scale bars, 500 μm). (D) Average contraction scores resulting from behavioral analysis. Grey boxes indicate the 3 min light illumination period; black curves show the contraction behavior of untreated polyps; red and blue curves show the contraction behavior of polyps treated with P3HT-NPs for 2 or 24 hours, respectively. (E) Average number of contraction events estimated on treated (2 and 24 hours, red and blue lines, respectively) and untreated (black) polyps. (F) Hydra exposure to P3HT-NPs and/or white light illumination elicits a number of molecular reactions, which may provide useful clues on the transduction pathways activated by each stimulus. Genes selected in this study belong to the light transductions pathway (opsin3-like) and heat response (hsp70). Opsin3-like gene expression shows great activation in response to both NPs and white light illumination, as compared to other experimental conditions. Reproduced from ref. 103 with permission from American Association for the Advancement of Science, copyright 2017. | ||
4. Conclusion and outlook
Photo-control of the activity of organisms may lead to function recovery (e.g. vision77), function loss (e.g. neuron silencing161) or function gain (e.g. enhanced light response103). Parallel to electro medicine, usually referred to as electroceutical when it concerns the control of the neuronal activity by electrical pulses,274 artificially enhanced light-sensitivity can be exploited in enlarging the scope of photo-medicine. Furthermore, light allows remote control, getting rid of the cumbersome wiring and cross talk problems. As an example, this could be exploited in guiding cyborgs (being with both organic and biomechatronic body parts), as recently demonstrated in a hybrid soft robotic ray.275 In this system, an artificial flexible scaffold is powered by an integrated network of muscle cells obtained from lab animals that are stimulated by light via optogenetics. Recent results on Hydra, a tiny creature similar to jellyfish, demonstrate that in spite of the lack of any specific localization of the polymer nanoparticles on target cells, light stimulation affects motion in a simple and reproducible way.103Light is the only external source of energy for our planet. As we mentioned at the beginning light does affect life on earth, in spite of the generally weak absorption of cells and tissues. Yet artificially enhancing this sensitivity seems an interesting challenge to enlarge such interaction in a controlled and wholesome way. Phototoxicity is indeed the limit of this approach. Nanotechnology offers opportunities for developing photo-medicine and photobiology by coupling mesoscale probes with photonics. Miniaturized probes can behave as artificial light actuators, as discussed here, or as biointegrated light sources, as demonstrated by the recently reported lasing in cells.276 The use of light for these purposes appears as a seamless extension of a natural feature that can provide control of the activity of organisms at the cellular level, and the investigation of new bio-compatible materials and devices for this application is the way to realize this vision.
Conflicts of interest
There are no conflicts to declare.References
- A. C. Thompson, P. R. Stoddart and E. D. Jansen, Curr. Mol. Imaging, 2014, 3, 162–177 CrossRef CAS PubMed.
- J. Rivnay, H. Wang, L. Fenno, K. Deisseroth and G. G. Malliaras, Sci. Adv., 2017, 3, e1601649 CrossRef PubMed.
- R. R. Birge, Annu. Rev. Biophys. Bioeng., 1981, 10, 315–354 CrossRef CAS PubMed.
- M. Chen, J. Chory and C. Fankhauser, Annu. Rev. Genet., 2004, 38, 87–117 CrossRef CAS PubMed.
- H. Chung, T. Dai, S. K. Sharma, Y.-Y. Huang, J. D. Carroll and M. R. Hamblin, Ann. Biomed. Eng., 2012, 40, 516–533 CrossRef PubMed.
- G. L. Fain, H. R. Matthews, M. C. Cornwall and Y. Koutalos, Physiol. Rev., 2001, 81, 117–151 CrossRef CAS PubMed.
- Y.-T. Hsu and R. S. Molday, Nature, 1993, 361, 76 CrossRef CAS PubMed.
- I. B. Leskov, V. A. Klenchin, J. W. Handy, G. G. Whitlock, V. I. Govardovskii, M. D. Bownds, T. D. Lamb, E. N. Pugh and V. Y. Arshavsky, Neuron, 2000, 27, 525–537 CrossRef CAS PubMed.
- I. Provencio, I. R. Rodriguez, G. Jiang, W. P. Hayes, E. F. Moreira and M. D. Rollag, J. Neurosci., 2000, 20, 600–605 CrossRef CAS PubMed.
- D. Arendt, Int. J. Dev. Biol., 2003, 47, 563–571 Search PubMed.
- A. R. Choi, L. Shi, L. S. Brown and K.-H. Jung, PLoS One, 2014, 9, e110643 Search PubMed.
- P. Hegemann, Annu. Rev. Plant Biol., 2008, 59, 167–189 CrossRef CAS PubMed.
- J. Purschwitz, S. Müller, C. Kastner and R. Fischer, Curr. Opin. Microbiol., 2006, 9, 566–571 CrossRef CAS PubMed.
- W. L. Hubbell, C. Altenbach, C. M. Hubbell and H. G. Khorana, Adv. Protein Chem., 2003, 63, 243–290 CrossRef CAS PubMed.
- T. Kobayashi, T. Saito and H. Ohtani, Nature, 2001, 414, 531–534 CrossRef CAS PubMed.
- Q. Wang, R. W. Schoenlein, L. A. Peteanu, R. A. Mathies and C. V. Shank, Science, 1994, 266, 422–424 CAS.
- D. E. Brash, J. A. Rudolph, J. A. Simon, A. Lin, G. J. McKenna, H. P. Baden, A. J. Halperin and J. Ponten, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10124–10128 CrossRef CAS.
- T. Karu, J. Photochem. Photobiol., B, 1999, 49, 1–17 CrossRef CAS.
- M. T. Alt, E. Fiedler, L. Rudmann, J. S. Ordonez, P. Ruther and T. Stieglitz, Proc. IEEE, 2017, 105, 101–138 CrossRef.
- K. Deisseroth, Nat. Neurosci., 2015, 18, 1213–1225 CrossRef CAS PubMed.
- O. Yizhar, L. E. Fenno, T. J. Davidson, M. Mogri and K. Deisseroth, Neuron, 2011, 71, 9–34 CrossRef CAS PubMed.
- I. Diester, M. T. Kaufman, M. Mogri, R. Pashaie, W. Goo, O. Yizhar, C. Ramakrishnan, K. Deisseroth and K. V. Shenoy, Nat. Neurosci., 2011, 14, 387–397 CrossRef CAS PubMed.
- L. Fenno, O. Yizhar and K. Deisseroth, Annu. Rev. Neurosci., 2011, 34, 389–412 CrossRef CAS PubMed.
- A. R. Duke, M. W. Jenkins, H. Lu, J. M. McManus, H. J. Chiel and E. D. Jansen, Sci. Rep., 2013, 3, 2600 CrossRef PubMed.
- H. Hirase, V. Nikolenko, J. H. Goldberg and R. Yuste, J. Neurobiol., 2002, 51, 237–247 CrossRef PubMed.
- K. Oyama, V. Zeeb, Y. Kawamura, T. Arai, M. Gotoh, H. Itoh, T. Itabashi, M. Suzuki and S. Ishiwata, Sci. Rep., 2015, 5, 16661 CrossRef PubMed.
- M. G. Shapiro, K. Homma, S. Villarreal, C.-P. Richter and F. Bezanilla, Nat. Commun., 2012, 3, 736 CrossRef PubMed.
- A. C. Thompson, S. A. Wade, W. G. Brown and P. R. Stoddart, J. Biomed. Opt., 2012, 17, 0750021 CrossRef PubMed.
- S.-R. Tsai and M. R. Hamblin, J. Photochem. Photobiol., B, 2017, 170, 197–207 CrossRef CAS PubMed.
- O. Akhavan, E. Ghaderi, S. Aghayee, Y. Fereydooni and A. Talebi, J. Mater. Chem., 2012, 22, 13773–13781 RSC.
- M. Dong, A. Babalhavaeji, S. Samanta, A. A. Beharry and G. A. Woolley, Acc. Chem. Res., 2015, 48, 2662–2670 CrossRef CAS PubMed.
- J. Hung, M. Chansard, S. S. Ousman, M. D. Nguyen and M. A. Colicos, Brain, Behav., Immun., 2010, 24, 31–40 CrossRef CAS PubMed.
- K. Mathieson, J. Loudin, G. Goetz, P. Huie, L. Wang, T. I. Kamins, L. Galambos, R. Smith, J. S. Harris, A. Sher and D. Palanker, Nat. Photonics, 2012, 6, 391–397 CrossRef CAS PubMed.
- E. Miyako, J. Russier, M. Mauro, C. Cebrian, H. Yawo, C. Ménard-Moyon, J. A. Hutchison, M. Yudasaka, S. Iijima, L. De Cola and A. Bianco, Angew. Chem., Int. Ed., 2014, 53, 13121–13125 CrossRef CAS PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. Sanchez Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- A. Starovoytov, J. Neurophysiol., 2004, 93, 1090–1098 CrossRef PubMed.
- J. Suzurikawa, M. Nakao, Y. Jimbo, R. Kanzaki and H. Takahashi, Sens. Actuators, B, 2014, 192, 393–398 CrossRef CAS.
- I. Tochitsky, A. Polosukhina, V. E. Degtyar, N. Gallerani, C. M. Smith, A. Friedman, R. N. Van Gelder, D. Trauner, D. Kaufer and R. H. Kramer, Neuron, 2014, 81, 800–813 CrossRef CAS PubMed.
- N. Waiskopf, Y. Ben-Shahar, M. Galchenko, I. Carmel, G. Moshitzky, H. Soreq and U. Banin, Nano Lett., 2016, 16, 4266–4273 CrossRef CAS PubMed.
- J. Yong, K. Needham, W. G. A. Brown, B. A. Nayagam, S. L. McArthur, A. Yu and P. R. Stoddart, Adv. Healthcare Mater., 2014, 3, 1862–1868 CrossRef CAS PubMed.
- Y. Yuan, C.-J. Zhang, M. Gao, R. Zhang, B. Z. Tang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 1780–1786 CrossRef CAS PubMed.
- Z. Zhang, L. Wang, J. Wang, X. Jiang, X. Li, Z. Hu, Y. Ji, X. Wu and C. Chen, Adv. Mater., 2012, 24, 1418–1423 CrossRef CAS PubMed.
- G. Lanzani, The photophysics behind photovoltaics and photonics, John Wiley & Sons, 2012 Search PubMed.
- H. E. Elsayed-Ali, T. B. Norris, M. A. Pessot and G. A. Mourou, Phys. Rev. Lett., 1987, 58, 1212–1215 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, Annu. Rev. Phys. Chem., 2003, 54, 331–366 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 8410–8426 CrossRef CAS.
- A. Stella, M. Nisoli, S. De Silvestri, O. Svelto, G. Lanzani, P. Cheyssac and R. Kofman, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 15497 CrossRef CAS.
- B. A. Gregg and M. C. Hanna, J. Appl. Phys., 2003, 93, 3605–3614 CrossRef CAS.
- H. Haug and S. W. Koch, Quantum theory of the optical and electronic properties of semiconductors, World Scientific, New Jersey, NJ, 4 edn, repr., 2005 Search PubMed.
- R. S. Muller, T. I. Kamins and M. Chan, Device Electronics for Integrated Circuits, John Wiley & Sons Inc., 2003 Search PubMed.
- Handbook of photovoltaic science and engineering, ed. A. Luque López and S. Hegedus, Wiley, Chichester, 2 edn, fully rev. and updated., 2011 Search PubMed.
- P. Malý, F. Trojánek, J. Kudrna, A. Hospodková, S. Banáš, V. Kohlová, J. Valenta and I. Pelant, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 7929–7936 CrossRef.
- A. J. Nozik, Annu. Rev. Phys. Chem., 2001, 52, 193–231 CrossRef CAS PubMed.
- Y. Cui, X. Duan, J. Hu and C. M. Lieber, J. Phys. Chem. B, 2000, 104, 5213–5216 CrossRef CAS.
- R. T. Tung, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13509 CrossRef.
- P. Fromherz, ChemPhysChem, 2002, 3, 276–284 CrossRef CAS PubMed.
- R. A. Kaul, N. I. Syed and P. Fromherz, Phys. Rev. Lett., 2004, 92, 038102 CrossRef PubMed.
- C. M. Lieber, MRS Bull., 2011, 36, 1052–1063 CrossRef CAS PubMed.
- G. Zeck and P. Fromherz, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10457–10462 CrossRef CAS PubMed.
- E. H. Bogardus and H. B. Bebb, Phys. Rev., 1968, 176, 993 CrossRef CAS.
- C. Tortiglione, A. Quarta, A. Tino, L. Manna, R. Cingolani and T. Pellegrino, Bioconjugate Chem., 2007, 18, 829–835 CrossRef CAS PubMed.
- M. A. Malvindi, L. Carbone, A. Quarta, A. Tino, L. Manna, T. Pellegrino and C. Tortiglione, Small, 2008, 4, 1747–1755 CrossRef CAS PubMed.
- S. Deka, A. Quarta, M. G. Lupo, A. Falqui, S. Boninelli, C. Giannini, G. Morello, M. De Giorgi, G. Lanzani, C. Spinella, R. Cingolani, T. Pellegrino and L. Manna, J. Am. Chem. Soc., 2009, 131, 2948–2958 CrossRef CAS PubMed.
- B. J. Schwartz, Annu. Rev. Phys. Chem., 2003, 54, 141–172 CrossRef CAS PubMed.
- G. Scarpa, A.-L. Idzko, S. Götz and S. Thalhammer, Macromol. Biosci., 2010, 10, 378–383 CrossRef CAS PubMed.
- Photophysics of molecular materials: from single molecules to single crystals, ed. G. Lanzani, Wiley-VCH, Weinheim, 2006 Search PubMed.
- M. Wohlgenannt, K. Tandon, S. Mazumdar, S. Ramasesha and Z. V. Vardeny, Nature, 2001, 409, 494–497 CrossRef CAS PubMed.
- O. J. Korovyanko, R. Österbacka, X. M. Jiang, Z. V. Vardeny and R. A. J. Janssen, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 235122 CrossRef.
- S. Bellani, D. Fazzi, P. Bruno, E. Giussani, E. V. Canesi, G. Lanzani and M. R. Antognazza, J. Phys. Chem. C, 2014, 118, 6291–6299 CAS.
- L. Luer, Org. Electron., 2004, 5, 83–89 CrossRef CAS.
- K. M. Noone, S. Subramaniyan, Q. Zhang, G. Cao, S. A. Jenekhe and D. S. Ginger, J. Phys. Chem. C, 2011, 115, 24403–24410 CAS.
- G. Barbarella, M. Zangoli and F. Di Maria, Advances in Heterocyclic Chemistry, Elsevier, 2017, vol. 123, pp. 105–167 Search PubMed.
- F. Di Maria, M. Zangoli, I. E. Palamá, E. Fabiano, A. Zanelli, M. Monari, A. Perinot, M. Caironi, V. Maiorano, A. Maggiore, M. Pugliese, E. Salatelli, G. Gigli, I. Viola and G. Barbarella, Adv. Funct. Mater., 2016, 26, 6970–6984 CrossRef CAS.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- J. Janata and M. Josowicz, Nat. Mater., 2003, 2, 19–24 CrossRef CAS PubMed.
- L. Li, F. Zhang, J. Wang, Q. An, Q. Sun, W. Wang, J. Zhang and F. Teng, Sci. Rep., 2015, 5, 9181 CrossRef CAS PubMed.
- J. F. Maya-Vetencourt, D. Ghezzi, M. R. Antognazza, E. Colombo, M. Mete, P. Feyen, A. Desii, A. Buschiazzo, M. Di Paolo, S. Di Marco, F. Ticconi, L. Emionite, D. Shmal, C. Marini, I. Donelli, G. Freddi, R. Maccarone, S. Bisti, G. Sambuceti, G. Pertile, G. Lanzani and F. Benfenati, Nat. Mater., 2017, 16, 681–689 CrossRef CAS PubMed.
- L. H. Rossander, H. F. Dam, J. E. Carlé, M. Helgesen, I. Rajkovic, M. Corazza, F. C. Krebs and J. W. Andreasen, Energy Environ. Sci., 2017, 10, 2411–2419 CAS.
- M. Şenel, Synth. Met., 2011, 161, 1861–1868 CrossRef.
- H. A. Ho, K. Doré, M. Boissinot, M. G. Bergeron, R. M. Tanguay, D. Boudreau and M. Leclerc, J. Am. Chem. Soc., 2005, 127, 12673–12676 CrossRef CAS PubMed.
- H.-H. Han, C.-Z. Wang, Y. Zang, J. Li, T. D. James and X.-P. He, Chem. Commun., 2017, 53, 9793–9796 RSC.
- M. Lan, S. Zhao, Y. Xie, J. Zhao, L. Guo, G. Niu, Y. Li, H. Sun, H. Zhang, W. Liu, J. Zhang, P. Wang and W. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 14590–14595 CAS.
- G. L. Nicolson, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 1451–1466 CrossRef CAS PubMed.
- D. C. Gadsby, Nat. Rev. Mol. Cell Biol., 2009, 10, 344–352 CrossRef CAS PubMed.
- H. Lodish, Molecular cell biology, Macmillan, 2008 Search PubMed.
- D. Ghezzi, M. R. Antognazza, M. Dal Maschio, E. Lanzarini, F. Benfenati and G. Lanzani, Nat. Commun., 2011, 2, 166 CrossRef PubMed.
- M. Voelker and P. Fromherz, Small, 2005, 1, 206–210 CrossRef CAS PubMed.
- S. Bauer and M. Kaltenbrunner, Nature, 2016, 539, 365–367 CrossRef CAS PubMed.
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwödiauer, I. Graz, S. Bauer-Gogonea, S. Bauer and T. Someya, Nature, 2013, 499, 458–463 CrossRef CAS PubMed.
- E. Stavrinidou, P. Leleux, H. Rajaona, D. Khodagholy, J. Rivnay, M. Lindau, S. Sanaur and G. G. Malliaras, Adv. Mater., 2013, 25, 4488–4493 CrossRef CAS PubMed.
- K. Kostarelos, L. Lacerda, G. Pastorin, W. Wu, S. Wieckowski, J. Luangsivilay, S. Godefroy, D. Pantarotto, J.-P. Briand, S. Muller, M. Prato and A. Bianco, Nat. Nanotechnol., 2007, 2, 108–113 CrossRef CAS PubMed.
- S. J. Lim, A. Smith and S. Nie, Curr. Opin. Chem. Eng., 2014, 4, 137–143 CrossRef PubMed.
- N. W. Shi Kam, T. C. Jessop, P. A. Wender and H. Dai, J. Am. Chem. Soc., 2004, 126, 6850–6851 CrossRef PubMed.
- K.-T. Yong, J. Qian, I. Roy, H. H. Lee, E. J. Bergey, K. M. Tramposch, S. He, M. T. Swihart, A. Maitra and P. N. Prasad, Nano Lett., 2007, 7, 761–765 CrossRef CAS PubMed.
- A. Zacheo, A. Quarta, A. Mangoni, P. P. Pompa, R. Mastria, M. C. Capogrossi, R. Rinaldi and T. Pellegrino, IEEE Trans Nanobioscience, 2011, 10, 209–215 CrossRef PubMed.
- A. Zhang and C. M. Lieber, Chem. Rev., 2016, 116, 215–257 CrossRef CAS PubMed.
- R. Parameswaran, J. L. Carvalho-de-Souza, Y. Jiang, M. J. Burke, J. F. Zimmerman, K. Koehler, A. W. Phillips, J. Yi, E. J. Adams, F. Bezanilla and B. Tian, Nat. Nanotechnol., 2018, 13, 260–266 CrossRef CAS PubMed.
- J. F. Zimmerman, R. Parameswaran, G. Murray, Y. Wang, M. Burke and B. Tian, Sci. Adv., 2016, 2, e1601039 Search PubMed.
- J. A. Rogers, T. Someya and Y. Huang, Science, 2010, 327, 1603–1607 CrossRef CAS PubMed.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- G. M. Whitesides, Nat. Biotechnol., 2003, 21, 1161–1165 CrossRef CAS PubMed.
- C. Tortiglione, M. R. Antognazza, A. Tino, C. Bossio, V. Marchesano, A. Bauduin, M. Zangoli, S. V. Morata and G. Lanzani, Sci. Adv., 2017, 3, e1601699 CrossRef PubMed.
- H. Jeon, C. G. Simon and G. Kim, J. Biomed. Mater. Res., Part B, 2014, 102, 1580–1594 CrossRef PubMed.
- P. Roach, T. Parker, N. Gadegaard and M. R. Alexander, Surf. Sci. Rep., 2010, 65, 145–173 CrossRef CAS.
- C. Xie, L. Hanson, W. Xie, Z. Lin, B. Cui and Y. Cui, Nano Lett., 2010, 10, 4020–4024 CrossRef CAS PubMed.
- J.-W. Jeong, G. Shin, S. I. Park, K. J. Yu, L. Xu and J. A. Rogers, Neuron, 2015, 86, 175–186 CrossRef CAS PubMed.
- C. Liao, M. Zhang, M. Y. Yao, T. Hua, L. Li and F. Yan, Adv. Mater., 2015, 27, 7493–7527 CrossRef CAS PubMed.
- G. Schwartz, B. C.-K. Tee, J. Mei, A. L. Appleton, D. H. Kim, H. Wang and Z. Bao, Nat. Commun., 2013, 4, 1859 CrossRef PubMed.
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef CAS PubMed.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef CAS PubMed.
- A. A. Kornyshev, J. Phys. Chem. B, 2007, 111, 5545–5557 CrossRef CAS PubMed.
- E. Mosconi, P. Salvatori, M. Ilenia Saba, A. Mattoni, S. Bellani, F. Bruni, B. Santiago Gonzalez, M. Antognazza, S. Brovelli, G. Lanzani, H. Li, J.-L. Brédas and F. De Angelis, ACS Energy Lett., 2016, 1, 454–463 CrossRef CAS.
- A. Offenhäusser and W. Knoll, Trends Biotechnol., 2001, 19, 62–66 CrossRef.
- R. Weis, B. Müller and P. Fromherz, Phys. Rev. Lett., 1996, 76, 327–330 CrossRef CAS PubMed.
- S. F. Cogan, Annu. Rev. Biomed. Eng., 2008, 10, 275–309 CrossRef CAS PubMed.
- A. Jackson and J. B. Zimmermann, Nat. Rev. Neurol., 2012, 8, 690–699 CrossRef CAS PubMed.
- M. E. J. Obien, K. Deligkaris, T. Bullmann, D. J. Bakkum and U. Frey, Front. Neurosci., 2015, 8, 423 Search PubMed.
- S. M. Wellman, J. R. Eles, K. A. Ludwig, J. P. Seymour, N. J. Michelson, W. E. McFadden, A. L. Vazquez and T. D. Y. Kozai, Adv. Funct. Mater., 2017, 1701269, DOI:10.1002/adfm.201701269.
- P. Fattahi, G. Yang, G. Kim and M. R. Abidian, Adv. Mater., 2014, 26, 1846–1885 CrossRef CAS PubMed.
- R. Kim, S. Joo, H. Jung, N. Hong and Y. Nam, Open Biomed. Eng. Lett., 2014, 4, 129–141 CrossRef.
- M. E. Spira and A. Hai, Nat. Nanotechnol., 2013, 8, 83–94 CrossRef CAS PubMed.
- W. M. Grill, S. E. Norman and R. V. Bellamkonda, Annu. Rev. Biomed. Eng., 2009, 11, 1–24 CrossRef CAS PubMed.
- M. Righi, G. L. Puleo, I. Tonazzini, G. Giudetti, M. Cecchini and S. Micera, Sci. Rep., 2018, 8, 502 CrossRef PubMed.
- A. A. Schendel, K. W. Eliceiri and J. C. Williams, Curr. Opin. Solid State Mater. Sci., 2014, 18, 301–307 CrossRef CAS PubMed.
- M. A. Colicos, J. Exp. Biol., 2006, 209, 2312–2319 CrossRef PubMed.
- D. G. Hafeman, J. W. Parce and H. M. McConnell, Science, 1988, 240, 1182–1185 CAS.
- M. A. Colicos, B. E. Collins, M. J. Sailor and Y. Goda, Cell, 2001, 107, 605–616 CrossRef CAS PubMed.
- P. Fromherz and A. Stett, Phys. Rev. Lett., 1995, 75, 1670 CrossRef CAS PubMed.
- I. Schoen and P. Fromherz, J. Neurophysiol., 2008, 100, 346–357 CrossRef PubMed.
- I. Schoen and P. Fromherz, Biophys. J., 2007, 92, 1096–1111 CrossRef CAS PubMed.
- D. Braun and P. Fromherz, Phys. Rev. Lett., 1998, 81, 5241 CrossRef CAS.
- M. Brittinger and P. Fromherz, Appl. Phys. A: Mater. Sci. Process., 2005, 81, 439–447 CrossRef CAS.
- A. Lambacher and P. Fromherz, J. Opt. Soc. Am. B, 2002, 19, 1435–1453 CrossRef CAS.
- Y. Mandel, G. Goetz, D. Lavinsky, P. Huie, K. Mathieson, L. Wang, T. Kamins, L. Galambos, R. Manivanh, J. Harris and D. Palanker, Nat. Commun., 2013, 4, 1980 Search PubMed.
- L. Bousse, S. Mostarshed, D. Hafeman, M. Sartore, M. Adami and C. Nicolini, J. Appl. Phys., 1994, 75, 4000–4008 CrossRef CAS.
- V. Bucher, J. P. Brugger, D. Kern, G. M. Kim, M. Schubert and W. Nisch, Microelectron. Eng., 2002, 61–62, 971–980 CrossRef CAS.
- J. Suzurikawa, M. Nakao, Y. Jimbo, R. Kanzaki and H. Takahashi, IEEE Trans. Biomed. Eng., 2009, 56, 2660–2665 CrossRef PubMed.
- J. Suzurikawa, H. Takahashi, R. Kanzaki, M. Nakao, Y. Takayama and Y. Jimbo, Appl. Phys. Lett., 2007, 90, 093901 CrossRef.
- R. Chen, A. Canales and P. Anikeeva, Nat. Rev. Mater., 2017, 2, 16093 CrossRef CAS.
- Y. Liang, Y. Wu, D. Feng, S.-T. Tsai, H.-J. Son, G. Li and L. Yu, J. Am. Chem. Soc., 2009, 131, 56–57 CrossRef CAS PubMed.
- B. Weng, J. Diao, Q. Xu, Y. Liu, C. Li, A. Ding and J. Chen, Adv. Mater. Interfaces, 2015, 2, 1500059 CrossRef.
- G. Kaur, R. Adhikari, P. Cass, M. Bown and P. Gunatillake, RSC Adv., 2015, 5, 37553–37567 RSC.
- D. Mawad, K. Gilmore, P. Molino, K. Wagner, P. Wagner, D. L. Officer and G. G. Wallace, J. Mater. Chem., 2011, 21, 5555–5560 RSC.
- M. S. A. Abdou, F. P. Orfino, Y. Son and S. Holdcroft, J. Am. Chem. Soc., 1997, 119, 4518–4524 CrossRef CAS.
- K. Norrman, S. A. Gevorgyan and F. C. Krebs, ACS Appl. Mater. Interfaces, 2009, 1, 102–112 CAS.
- J. Schafferhans, A. Baumann, A. Wagenpfahl, C. Deibel and V. Dyakonov, Org. Electron., 2010, 11, 1693–1700 CrossRef CAS.
- A. Seemann, T. Sauermann, C. Lungenschmied, O. Armbruster, S. Bauer, H.-J. Egelhaaf and J. Hauch, Sol. Energy, 2011, 85, 1238–1249 CrossRef CAS.
- O. V. Kozlov and S. A. Zapunidi, Synth. Met., 2013, 169, 48–54 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- J.-M. Zhuo, L.-H. Zhao, R.-Q. Png, L.-Y. Wong, P.-J. Chia, J.-C. Tang, S. Sivaramakrishnan, M. Zhou, E. C.-W. Ou, S.-J. Chua, W.-S. Sim, L.-L. Chua and P. K.-H. Ho, Adv. Mater., 2009, 21, 4747–4752 CAS.
- V. Benfenati, N. Martino, M. R. Antognazza, A. Pistone, S. Toffanin, S. Ferroni, G. Lanzani and M. Muccini, Adv. Healthcare Mater., 2014, 3, 392–399 CrossRef CAS PubMed.
- F. Lodola, N. Martino, G. Tullii, G. Lanzani and M. R. Antognazza, Sci. Rep., 2017, 1, 8477 CrossRef PubMed.
- V. Gautam, D. Rand, Y. Hanein and K. S. Narayan, Adv. Mater., 2014, 26, 1751–1756 CrossRef CAS PubMed.
- N. Martino, P. Feyen, M. Porro, C. Bossio, E. Zucchetti, D. Ghezzi, F. Benfenati, G. Lanzani and M. R. Antognazza, Sci. Rep., 2015, 5, 8911 CrossRef CAS PubMed.
- G. Tullii, A. Desii, C. Bossio, S. Bellani, M. Colombo, N. Martino, M. R. Antognazza and G. Lanzani, Org. Electron., 2017, 46, 88–98 CrossRef CAS.
- S. Vaquero Morata, C. Bossio, S. Bellani, N. Martino, E. Zucchetti, G. Lanzani and M. R. Antognazza, J. Mater. Chem. B, 2016, 4, 5272–5283 RSC.
- M. Plaksin, E. Shapira, E. Kimmel and S. Shoham, Phys. Rev. X, 2018, 8, 011043 Search PubMed.
- A. Varghese, E. Tenbroek, J. Colesjr and D. Sigg, Prog. Biophys. Mol. Biol., 2006, 90, 26–37 CrossRef CAS PubMed.
- D. Ghezzi, M. R. Antognazza, R. Maccarone, S. Bellani, E. Lanzarini, N. Martino, M. Mete, G. Pertile, S. Bisti, G. Lanzani and F. Benfenati, Nat. Photonics, 2013, 7, 400–406 CrossRef CAS PubMed.
- P. Feyen, E. Colombo, D. Endeman, M. Nova, L. Laudato, N. Martino, M. R. Antognazza, G. Lanzani, F. Benfenati and D. Ghezzi, Sci. Rep., 2016, 6, 22718 CrossRef CAS PubMed.
- V. Gautam, M. Bag and K. S. Narayan, J. Am. Chem. Soc., 2011, 133, 17942–17949 CrossRef CAS PubMed.
- V. Gautam, M. Bag and K. S. Narayan, J. Phys. Chem. Lett., 2010, 1, 3277–3282 CrossRef CAS.
- L. Bareket, N. Waiskopf, D. Rand, G. Lubin, M. David-Pur, J. Ben-Dov, S. Roy, C. Eleftheriou, E. Sernagor, O. Cheshnovsky, U. Banin and Y. Hanein, Nano Lett., 2014, 14, 6685–6692 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- S. G. Bucella, A. Luzio, E. Gann, L. Thomsen, C. R. McNeill, G. Pace, A. Perinot, Z. Chen, A. Facchetti and M. Caironi, Nat. Commun., 2015, 6, 8394 CrossRef CAS PubMed.
- M. R. Antognazza, M. Di Paolo, D. Ghezzi, M. Mete, S. Di Marco, J. F. Maya-Vetencourt, R. Maccarone, A. Desii, F. Di Fonzo, M. Bramini, A. Russo, L. Laudato, I. Donelli, M. Cilli, G. Freddi, G. Pertile, G. Lanzani, S. Bisti and F. Benfenati, Adv. Healthcare Mater., 2016, 5, 2271–2282 CrossRef CAS PubMed.
- M. Sytnyk, M. Jakešová, M. Litviňuková, O. Mashkov, D. Kriegner, J. Stangl, J. Nebesářová, F. W. Fecher, W. Schöfberger, N. S. Sariciftci, R. Schindl, W. Heiss and E. D. Głowacki, Nat. Commun., 2017, 8, 91 CrossRef PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed.
- K. Yang, J. Y. Oh, J. S. Lee, Y. Jin, G.-E. Chang, S. S. Chae, E. Cheong, H. K. Baik and S.-W. Cho, Theranostics, 2017, 7, 4591–4604 CrossRef PubMed.
- E. Colombo, P. Feyen, M. R. Antognazza, G. Lanzani and F. Benfenati, Front. Neurosci., 2016, 10, 105 Search PubMed.
- Y. Wang and L. Guo, Front. Neurosci., 2016, 10, 69 Search PubMed.
- A. T. Young, N. Cornwell and M. A. Daniele, Adv. Funct. Mater., 2017, 1700239 Search PubMed.
- C. H. Thompson, M. J. Zoratti, N. B. Langhals and E. K. Purcell, Tissue Eng., Part B, 2015, 22, 125–135 CrossRef PubMed.
- C. C. Fleischer and C. K. Payne, Acc. Chem. Res., 2014, 47, 2651–2659 CrossRef CAS PubMed.
- P. M. Kelly, C. Åberg, E. Polo, A. O’Connell, J. Cookman, J. Fallon, Ž. Krpetić and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472–479 CrossRef CAS PubMed.
- G. Lanzani, Nat. Mater., 2014, 13, 775–776 CrossRef CAS PubMed.
- T. Repenko, A. Rix, S. Ludwanowski, D. Go, F. Kiessling, W. Lederle and A. J. C. Kuehne, Nat. Commun., 2017, 8, 470 CrossRef PubMed.
- K. Scholten and E. Meng, Lab Chip, 2015, 15, 4256–4272 RSC.
- J. Wells, C. Kao, P. Konrad, T. Milner, J. Kim, A. Mahadevan-Jansen and E. D. Jansen, Biophys. J., 2007, 93, 2567–2580 CrossRef CAS PubMed.
- E. S. Albert, J. M. Bec, G. Desmadryl, K. Chekroud, C. Travo, S. Gaboyard, F. Bardin, I. Marc, M. Dumas, G. Lenaers, C. Hamel, A. Muller and C. Chabbert, J. Neurophysiol., 2012, 107, 3227–3234 CrossRef CAS PubMed.
- G. Bodelón, C. Costas, J. Pérez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzán, Nano Today, 2017, 13, 40–60 CrossRef.
- C. Paviolo and P. Stoddart, Nanomaterials, 2017, 7, 92 CrossRef PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- X. Huang, S. Neretina and M. A. El-Sayed, Adv. Mater., 2009, 21, 4880–4910 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- J. L. Carvalho-de-Souza, J. S. Treger, B. Dang, S. B. H. Kent, D. R. Pepperberg and F. Bezanilla, Neuron, 2015, 86, 207–217 CrossRef CAS PubMed.
- F. Lavoie-Cardinal, C. Salesse, É. Bergeron, M. Meunier and P. De Koninck, Sci. Rep., 2016, 6, 20619 CrossRef CAS PubMed.
- K. Eom, J. Kim, J. M. Choi, T. Kang, J. W. Chang, K. M. Byun, S. B. Jun and S. J. Kim, Small, 2014, 10, 3853–3857 CrossRef CAS PubMed.
- S.-H. Hu and X. Gao, Adv. Funct. Mater., 2010, 20, 3721–3726 CrossRef CAS PubMed.
- C. Paviolo, J. W. Haycock, P. J. Cadusch, S. L. McArthur and P. R. Stoddart, J. Biophotonics, 2014, 7, 761–765 CrossRef CAS PubMed.
- C. Paviolo, J. W. Haycock, J. Yong, A. Yu, P. R. Stoddart and S. L. McArthur, Biotechnol. Bioeng., 2013, 110, 2277–2291 CrossRef CAS PubMed.
- S. Yoo, S. Hong, Y. Choi, J.-H. Park and Y. Nam, ACS Nano, 2014, 8, 8040–8049 CrossRef CAS PubMed.
- S. Yoo, R. Kim, J.-H. Park and Y. Nam, ACS Nano, 2016, 10, 4274–4281 CrossRef CAS PubMed.
- N. Gomez, J. Winter, F. Shieh, A. Saunders, B. Korgel and C. Schmidt, Talanta, 2005, 67, 462–471 CrossRef CAS PubMed.
- K. Lugo, X. Miao, F. Rieke and L. Y. Lin, Biomed. Opt. Express, 2012, 3, 447–454 CrossRef CAS PubMed.
- E. Molokanova, J. A. Bartel, W. Zhao, I. Naasani, M. J. Ignatius, J. A. Treadway and A. Savtchenko, Biophotonics Int., 2008, 26–31 Search PubMed.
- T. C. Pappas, W. M. S. Wickramanyake, E. Jan, M. Motamedi, M. Brodwick and N. A. Kotov, Nano Lett., 2007, 7, 513–519 CrossRef CAS PubMed.
- J. O. Winter, N. Gomez, B. A. Korgel and C. E. Schmidt, Proc. SPIE, 2005, 5705, 235–246 CrossRef.
- J. O. Winter, T. Y. Liu, B. A. Korgel and C. E. Schmidt, Adv. Mater., 2001, 13, 1673–1677 CrossRef CAS.
- A. Ruland, C. Schulz-Drost, V. Sgobba and D. M. Guldi, Adv. Mater., 2011, 23, 4573–4577 CrossRef CAS PubMed.
- Y. Wang and N. Herron, J. Phys. Chem., 1991, 95, 525–532 CrossRef CAS.
- L. Bareket-Keren and Y. Hanein, Int. J. Nanomed., 2014, 65–83 CrossRef PubMed.
- P. Fromherz, Eur. Biophys. J., 1999, 28, 254–258 CrossRef CAS PubMed.
- P. Fromherz, V. Kiessling, K. Kottig and G. Zeck, Appl. Phys. A: Mater. Sci. Process., 1999, 69, 571–576 CrossRef CAS.
- S. Dante, A. Petrelli, E. M. Petrini, R. Marotta, A. Maccione, A. Alabastri, A. Quarta, F. De Donato, T. Ravasenga, A. Sathya, R. Cingolani, R. Proietti Zaccaria, L. Berdondini, A. Barberis and T. Pellegrino, ACS Nano, 2017, 11, 6630–6640 CrossRef CAS PubMed.
- J. D. Marshall and M. J. Schnitzer, ACS Nano, 2013, 7, 4601–4609 CrossRef CAS PubMed.
- R. Tanimoto, T. Hiraiwa, Y. Nakai, Y. Shindo, K. Oka, N. Hiroi and A. Funahashi, Sci. Rep., 2016, 6, 22071 CrossRef CAS PubMed.
- C. E. Rowland, K. Susumu, M. H. Stewart, E. Oh, A. J. Mäkinen, T. J. O’Shaughnessy, G. Kushto, M. A. Wolak, J. S. Erickson, A. L. Efros, A. L. Huston and J. B. Delehanty, Nano Lett., 2015, 15, 6848–6854 CrossRef CAS PubMed.
- L. Yildirimer, N. T. K. Thanh, M. Loizidou and A. M. Seifalian, Nano Today, 2011, 6, 585–607 CrossRef CAS PubMed.
- Nat. Nanotechnol., 2011, 6, 329, DOI:10.1038/nnano.2011.87.
- E. Söderstjerna, P. Bauer, T. Cedervall, H. Abdshill, F. Johansson and U. E. Johansson, PLoS One, 2014, 9, e105359 Search PubMed.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165–172 CrossRef PubMed.
- A. Valizadeh, H. Mikaeili, M. Samiei, S. M. Farkhani, N. Zarghami, A. Akbarzadeh and S. Davaran, Nanoscale Res. Lett., 2012, 7, 480 CrossRef PubMed.
- K.-T. Yong, W.-C. Law, R. Hu, L. Ye, L. Liu, M. T. Swihart and P. N. Prasad, Chem. Soc. Rev., 2013, 42, 1236–1250 RSC.
- A. Ambrosone, L. Mattera, V. Marchesano, A. Quarta, A. S. Susha, A. Tino, A. L. Rogach and C. Tortiglione, Biomaterials, 2012, 33, 1991–2000 CrossRef CAS PubMed.
- A. Ambrosone and C. Tortiglione, Toxicol. Mech. Methods, 2013, 23, 207–216 CrossRef CAS PubMed.
- J. Conde, A. Ambrosone, V. Sanz, Y. Hernandez, V. Marchesano, F. Tian, H. Child, C. C. Berry, M. R. Ibarra, P. V. Baptista, C. Tortiglione and J. M. de la Fuente, ACS Nano, 2012, 6, 8316–8324 CrossRef CAS PubMed.
- V. Marchesano, Y. Hernandez, W. Salvenmoser, A. Ambrosone, A. Tino, B. Hobmayer, J. M. de la Fuente and C. Tortiglione, ACS Nano, 2013, 7, 2431–2442 CrossRef CAS PubMed.
- Reza Fazel-Rezai, Biomedical Engineering – From Theory to Applications, InTech, 2011 Search PubMed.
- M. Palner, K. Pu, S. Shao and J. Rao, Angew. Chem., Int. Ed., 2015, 54, 11477–11480 CrossRef CAS PubMed.
- J. Pecher and S. Mecking, Chem. Rev., 2010, 110, 6260–6279 CrossRef CAS PubMed.
- A. N. Aleshin, A. D. Sokolovskaya, I. P. Shcherbakov, P. N. Brunkov and V. P. Ulin, Phys. Solid State, 2013, 55, 675–680 CrossRef CAS.
- S. Gärtner, A. J. Clulow, I. A. Howard, E. P. Gilbert, P. L. Burn, I. R. Gentle and A. Colsmann, ACS Appl. Mater. Interfaces, 2017, 9, 42986–42995 Search PubMed.
- S. Gärtner, S. Reich, M. Bruns, J. Czolk and A. Colsmann, Nanoscale, 2016, 8, 6721–6727 RSC.
- S. Gärtner, M. Christmann, S. Sankaran, H. Röhm, E.-M. Prinz, F. Penth, A. Pütz, A. E. Türeli, B. Penth, B. Baumstümmler and A. Colsmann, Adv. Mater., 2014, 26, 6653–6657 CrossRef PubMed.
- X. Han, M. Bag, T. S. Gehan, D. Venkataraman and D. Maroudas, J. Phys. Chem. C, 2015, 119, 25826–25839 CAS.
- M. S. Chen, O. P. Lee, J. R. Niskala, A. T. Yiu, C. J. Tassone, K. Schmidt, P. M. Beaujuge, S. S. Onishi, M. F. Toney, A. Zettl and J. M. J. Fréchet, J. Am. Chem. Soc., 2013, 135, 19229–19236 CrossRef CAS PubMed.
- J. Cho, K. H. Cheon, K. H. Park, S.-K. Kwon, Y.-H. Kim and D. S. Chung, Org. Electron., 2015, 24, 160–164 CrossRef CAS.
- J. E. Millstone, D. F. J. Kavulak, C. H. Woo, T. W. Holcombe, E. J. Westling, A. L. Briseno, M. F. Toney and J. M. J. Fréchet, Langmuir, 2010, 26, 13056–13061 CrossRef CAS PubMed.
- J. Cho, S. H. Yu and D. S. Chung, J. Mater. Chem. C, 2017, 5, 2745–2757 RSC.
- K. Pu, N. Chattopadhyay and J. Rao, J. Controlled Release, 2016, 240, 312–322 CrossRef CAS PubMed.
- C. Cui, Z. Yang, X. Hu, J. Wu, K. Shou, H. Ma, C. Jian, Y. Zhao, B. Qi, X. Hu, A. Yu and Q. Fan, ACS Nano, 2017, 11, 3298–3310 CrossRef CAS PubMed.
- G. Feng, Y. Fang, J. Liu, J. Geng, D. Ding and B. Liu, Small, 2017, 13, 1602807 CrossRef PubMed.
- Y. Jiang, P. K. Upputuri, C. Xie, Y. Lyu, L. Zhang, Q. Xiong, M. Pramanik and K. Pu, Nano Lett., 2017, 17, 4964–4969 CrossRef CAS PubMed.
- J. Li, J. Rao and K. Pu, Biomaterials, 2018, 155, 217–235 CrossRef CAS PubMed.
- X. Lin, Y. Wang, X. Chen, R. Yang, Z. Wang, J. Feng, H. Wang, K. W. C. Lai, J. He, F. Wang and P. Shi, Adv. Healthcare Mater., 2017, 6, 1700446 CrossRef PubMed.
- Y. Lyu, D. Cui, H. Sun, Y. Miao, H. Duan and K. Pu, Angew. Chem., Int. Ed., 2017, 56, 9155–9159 CrossRef CAS PubMed.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CAS.
- K. K. Ng and G. Zheng, Chem. Rev., 2015, 115, 11012–11042 CrossRef CAS PubMed.
- X. Xu, R. Liu and L. Li, Chem. Commun., 2015, 51, 16733–16749 RSC.
- M. C. Baier, J. Huber and S. Mecking, J. Am. Chem. Soc., 2009, 131, 14267–14273 CrossRef CAS PubMed.
- S. Ciftci and A. J. C. Kuehne, Macromolecules, 2015, 48, 8389–8393 CrossRef CAS.
- A. J. C. Kuehne, M. C. Gather and J. Sprakel, Nat. Commun., 2012, 3, 1088 CrossRef PubMed.
- H. Li, X. Wu, B. Xu, H. Tong and L. Wang, RSC Adv., 2013, 3, 8645–8648 RSC.
- D. Muenmart, A. B. Foster, A. Harvey, M.-T. Chen, O. Navarro, V. Promarak, M. C. McCairn, J. M. Behrendt and M. L. Turner, Macromolecules, 2014, 47, 6531–6539 CrossRef CAS.
- K. Müller, M. Klapper and K. Müllen, Macromol. Rapid Commun., 2006, 27, 586–593 CrossRef.
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36, 887–913 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- D. Tuncel and H. V. Demir, Nanoscale, 2010, 2, 484 RSC.
- F. Di Maria, A. Zanelli, A. Liscio, A. Kovtun, E. Salatelli, R. Mazzaro, V. Morandi, G. Bergamini, A. Shaffer and S. Rozen, ACS Nano, 2017, 11, 1991–1999 CrossRef CAS PubMed.
- Y.-L. Chang, R. E. Palacios, F.-R. F. Fan, A. J. Bard and P. F. Barbara, J. Am. Chem. Soc., 2008, 130, 8906–8907 CrossRef CAS PubMed.
- C. Wu, C. Szymanski and J. McNeill, Langmuir, 2006, 22, 2956–2960 CrossRef CAS PubMed.
- G. Nagarjuna, M. Baghgar, J. A. Labastide, D. D. Algaier, M. D. Barnes and D. Venkataraman, ACS Nano, 2012, 6, 10750–10758 CrossRef CAS PubMed.
- H. Shimizu, M. Yamada, R. Wada and M. Okabe, Polym. J., 2008, 40, 33–36 CrossRef CAS.
- S. T. Kim, K. Saha, C. Kim and V. M. Rotello, Acc. Chem. Res., 2013, 46, 681–691 CrossRef CAS PubMed.
- X. Feng, Y. Tang, X. Duan, L. Liu and S. Wang, J. Mater. Chem., 2010, 20, 1312–1316 RSC.
- C. Xing, L. Liu, H. Tang, X. Feng, Q. Yang, S. Wang and G. C. Bazan, Adv. Funct. Mater., 2011, 21, 4058–4067 CrossRef CAS.
- M. Zangoli, F. Di Maria, E. Zucchetti, C. Bossio, M. R. Antognazza, G. Lanzani, R. Mazzaro, F. Corticelli, M. Baroncini and G. Barbarella, Nanoscale, 2017, 9, 9202–9209 RSC.
- X. Feng, F. Lv, L. Liu, H. Tang, C. Xing, Q. Yang and S. Wang, ACS Appl. Mater. Interfaces, 2010, 2, 2429–2435 CAS.
- L. P. Fernando, P. K. Kandel, P. C. Ackroyd and K. A. Christensen, Anal. Bioanal. Chem., 2012, 404, 3003–3014 CrossRef CAS PubMed.
- P. Howes, M. Green, J. Levitt, K. Suhling and M. Hughes, J. Am. Chem. Soc., 2010, 132, 3989–3996 CrossRef CAS PubMed.
- C. Wu, S. J. Hansen, Q. Hou, J. Yu, M. Zeigler, Y. Jin, D. R. Burnham, J. D. McNeill, J. M. Olson and D. T. Chiu, Angew. Chem., Int. Ed., 2011, 50, 3430–3434 CrossRef CAS PubMed.
- C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, J. Am. Chem. Soc., 2010, 132, 15410–15417 CrossRef CAS PubMed.
- E. Miceli, M. Kar and M. Calderón, J. Mater. Chem. B, 2017, 5, 4393–4405 RSC.
- W. Li, R. Luo, X. Lin, A. D. Jadhav, Z. Zhang, L. Yan, C.-Y. Chan, X. Chen, J. He, C.-H. Chen and P. Shi, Biomaterials, 2015, 65, 76–85 CrossRef CAS PubMed.
- F. Ye, C. Wu, Y. Jin, Y.-H. Chan, X. Zhang and D. T. Chiu, J. Am. Chem. Soc., 2011, 133, 8146–8149 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- E. Zucchetti, M. Zangoli, I. Bargigia, C. Bossio, F. Di Maria, G. Barbarella, C. D’Andrea, G. Lanzani and M. R. Antognazza, J. Mater. Chem. B, 2017, 5, 565–574 RSC.
- M. L. Capobianco, G. Barbarella and A. Manetto, Molecules, 2012, 17, 910–933 CrossRef CAS PubMed.
- M. Zambianchi, F. D. Maria, A. Cazzato, G. Gigli, M. Piacenza, F. D. Sala and G. Barbarella, J. Am. Chem. Soc., 2009, 131, 10892–10900 CrossRef CAS PubMed.
- Y. Lyu, C. Xie, S. A. Chechetka, E. Miyako and K. Pu, J. Am. Chem. Soc., 2016, 138, 9049–9052 CrossRef CAS PubMed.
- A. Terakita, Genome Biol., 2005, 6, 213 CrossRef PubMed.
- K. Famm, B. Litt, K. J. Tracey, E. S. Boyden and M. Slaoui, Nature, 2013, 496, 159–161 CrossRef CAS PubMed.
- S.-J. Park, M. Gazzola, K. S. Park, S. Park, V. Di Santo, E. L. Blevins, J. U. Lind, P. H. Campbell, S. Dauth, A. K. Capulli, F. S. Pasqualini, S. Ahn, A. Cho, H. Yuan, B. M. Maoz, R. Vijaykumar, J.-W. Choi, K. Deisseroth, G. V. Lauder, L. Mahadevan and K. K. Parker, Science, 2016, 353, 158–162 CrossRef CAS PubMed.
- M. C. Gather and S. H. Yun, Nat. Photonics, 2011, 5, 406–410 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |