
The life of proteins under mechanical force
 ac
and
Raul
Perez-Jimenez
*ac
ac
and
Raul
Perez-Jimenez
*ac
Abstract
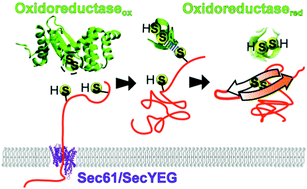
Although much of our understanding of protein folding comes from studies of isolated protein domains in bulk, in the cellular environment the intervention of external molecular machines is essential during the protein life cycle. During the past decade single molecule force spectroscopy techniques have been extremely useful to deepen our understanding of these interventional molecular processes, as they allow for monitoring and manipulating mechanochemical events in individual protein molecules. Here, we review some of the critical steps in the protein life cycle, starting with the biosynthesis of the nascent polypeptide chain in the ribosome, continuing with the folding supported by chaperones and the translocation into different cell compartments, and ending with proteolysis in the proteasome. Along these steps, proteins experience molecular forces often combined with chemical transformations, affecting their folding and structure, which are measured or mimicked in the laboratory by the application of force with a single molecule apparatus. These mechanochemical reactions can potentially be used as targets for fighting against diseases. Inspired by these insightful experiments, we devise an outlook on the emerging field of mechanopharmacology, which reflects an alternative paradigm for drug design.
- This article is part of the themed collection: Peptide and protein nanotechnology
 Please wait while we load your content...
Please wait while we load your content...

