
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic functionalities in MOFs: from the framework to the pore†
Guillermo
Mínguez Espallargas
 * and
Eugenio
Coronado
*
* and
Eugenio
Coronado
*
Instituto de Ciencia Molecular (ICMol), Universidad de Valencia, c/Catedrático José Beltrán, 2, 46980 Paterna, Spain. E-mail: guillermo.minguez@uv.es; eugenio.coronado@uv.es
First published on 7th November 2017
Abstract
In this review, we show the different approaches developed so far to prepare metal–organic frameworks (MOFs) presenting electronic functionalities, with particular attention to magnetic properties. We will cover the chemical design of frameworks necessary for the incorporation of different magnetic phenomena, as well as the encapsulation of functional species in their pores leading to hybrid multifunctional MOFs combining an extended lattice with a molecular lattice.
 Guillermo Mínguez Espallargas | Guillermo Mínguez Espallargas is a Ramón a Cajal Fellow at the ICMol of the University of Valencia. He studied at the University of Seville (Spain), and received his PhD from the University of Sheffield (UK). His research interests include metal–organic frameworks (MOFs), molecular magnetism, noncovalent interactions, and solid–gas reactions. He has received the gold medal of the European Young Chemist Award, the RSC Dalton Young Chemist Award and the Young Investigators Award from the Spanish Royal Society of Chemistry (RSEQ), among others. In 2016, he was the recipient of an ERC Consolidator Grant. |
 Eugenio Coronado | Eugenio Coronado is the Director of the Molecular Science Institute (ICMol) at the University of Valencia and the European Institute of Molecular Magnetism (EIMM). His research career has been devoted to the design, synthesis and study of new molecular materials with magnetic, electrical or optical properties. In particular, he has produced new hybrid multifunctional materials (including molecular magnetic conductors and superconductors, and stimuli-responsive magnetic materials) and new molecular nanomagnets. Currently his research interest is focused on the use of these kinds of materials in molecular spintronics and quantum technologies. |
1. Introduction
Metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), have emerged in the last 30 years as revolutionary materials with applications in societally and industrially relevant domains such as storage of fuels (hydrogen and methane), capture of gases (e.g. greenhouse gases), separation, drug delivery and catalysis, among others.1 Recent advances in this area in the past decade have resulted in an explosive growth in their preparation, characterization, and study, with over 5000 publications on this topic in 2016 and more than 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOFs reported.2 The common property of all these open crystalline frameworks, constructed from the assembly of inorganic sub-units (metal centres, clusters, chains…) and organic linkers (carboxylates, phosphonates, azolates, etc.), is their permanent porosity. In a similar way to zeolites, they present large cavities in their structures. However, unlike zeolites and mesoporous silica, the adaptable nature of the organic ligands and their variety of coordination modes, together with the geometry of the constitutive metal ions, provide an assortment of topologies and different architectures that can be accomplished by chemical design. In addition to this structural property, these crystalline solids may also exhibit other properties arising from the diverse manners in which different functionalities can be incorporated into these materials.
000 MOFs reported.2 The common property of all these open crystalline frameworks, constructed from the assembly of inorganic sub-units (metal centres, clusters, chains…) and organic linkers (carboxylates, phosphonates, azolates, etc.), is their permanent porosity. In a similar way to zeolites, they present large cavities in their structures. However, unlike zeolites and mesoporous silica, the adaptable nature of the organic ligands and their variety of coordination modes, together with the geometry of the constitutive metal ions, provide an assortment of topologies and different architectures that can be accomplished by chemical design. In addition to this structural property, these crystalline solids may also exhibit other properties arising from the diverse manners in which different functionalities can be incorporated into these materials.
In particular, electronic properties, i.e. magnetic, electrical or optical properties, can also be introduced into MOFs by adequately choosing the functional nodes, the organic linkers and the way in which they are connected in the solid, or by including functional molecules in the pores. These possibilities, which largely span the applications of these materials, have started to be exploited only very recently. Thus, the introduction of electrical conductivity into a MOF remains an almost unexplored topic since for the most part these materials are insulators.3 A recent review that covers this topic has recently been published.4 As far as luminescent MOFs are concerned, this area has also been extensively reviewed recently for their use in chemical sensing and explosive detection,5 light-harvesting sensitizers,6 and light emitting applications.7
In the present review, we will focus on the molecular design of magnetic MOFs. Among these materials, depending on the magnetic dimensionality within and between the two constituent sublattices (the extended framework and the molecular units), one can distinguish between MOFs exhibiting cooperative properties (e.g., magnetic ordering or cooperative spin crossover), and MOFs exhibiting molecular properties (e.g., single-molecule magnetism). We do not intend to provide a comprehensive collection of the literature, but intend to illustrate with selected examples the current state-of-the-art of the different approaches that have been employed to incorporate this functionality into MOFs.
In the field of molecular magnetism, coordination polymers have been extensively used to generate magnetic materials exhibiting cooperative properties. In this area, a major focus of interest over the last 30 years has been to design high-Tc magnets, i.e., molecule-based magnetic materials exhibiting spontaneous magnetization above room temperature, or at least above liquid nitrogen.8 These materials are highly desirable in magnetism since they can compete with the traditional inorganic magnets, showing at the same time superior properties from the point of view of their chemical versatility (they are prepared using coordination chemistry at low temperatures) and their structural and electronic versatilities (they are transparent and can often incorporate more functional properties). Taking advantage of the latter feature, a current challenge in this field is the incorporation of two or more properties into the same material, i.e. the preparation of a multifunctional magnetic material in which magnetism is co-existing and/or coupled with a second property of interest, like conductivity, superconductivity, luminescence, bistability, porosity, etc.9 Thus, playing with a molecular approach, it has been possible to incorporate into the same material two properties that are difficult, or even impossible, to achieve in a conventional solid-state material. Depending on the way in which the two components are integrated into the coordination material, one can differentiate between “two-network materials”, composed of two independent networks each of them providing a physical property, and “one-network material”, in which the two properties are strongly coupled thus often leading to stimuli-responsive materials. Examples of the first approach have given rise to the combination of magnetism and conductivity, or even superconductivity, in the same crystal,10,11 whereas examples of the second approach can be found in the so-called spin-crossover complexes with a LIESST effect,12 or in Prussian Blue Analogues (PBAs) with photomagnetism (LIESST effect),13 or piezomagnetism.14 In this case, the magnetism is coupled with light or pressure in such a way that by applying the corresponding external stimulus (light irradiation or pressure) the spin state can be tuned.
There are several reasons that justify the extension of this approach to the field of MOFs. On the one hand, the possibility of adding porosity to these magnetic coordination polymers offers an attractive way to generate multifunctional materials in which the magnetism can be tuned by the presence of molecules in the pores. Thus, these systems provide an ideal scenario to unravel the role played by the guest–framework intermolecular interactions, the chemical pressure generated by the guest molecules, or the modifications of the electronic properties in the MOF (i.e. isostructural MOFs with different ligands) in the magnetic behaviour. These features may be of interest for sensing the molecular species trapped in the pores through their magnetic response. On the other hand, the presence of magnetic centres either in the nodes or in the pores of a crystalline MOF opens the possibility of creating organized nanostructures of these magnetic centres, while keeping them well separated in the space. Such a feature may be of potential interest in quantum technologies, as they require a controlled disposition of magnetic moieties in the space. All these possibilities will be illustrated in the present review, which is organized as follows.
The first part is dedicated to MOFs in which the coordination framework is magnetic (Fig. 1a). In these materials, a rational chemical design has been crucial for the appearance of new properties and for controlling the effects of different stimuli on the magnetic properties. This part covers both MOFs showing solid-state properties, such as long-range magnetic order (presenting magnetic cooperativity) or spin-crossover (presenting elastic cooperativity), and MOFs showing molecular properties as a result of the incorporation of functional molecules at the nodes of the framework. In the latter case, the functional molecules – single-molecule magnets, in particular – are isolated by organic spacers (i.e., no cooperative properties are expected). Still, the crystallinity of these porous materials will give rise to long-range arrangements of these functional molecules, providing a useful way to obtain organized nanostructures of these magnetic molecules. The second part presents the inclusion of functional molecules (magnetic in particular) in the channels of the framework to give rise to hybrid functional MOFs combining an extended lattice with a molecular lattice. In this case, one can distinguish between the system formed by a non-magnetic MOF and a functional guest (Fig. 1b), or a magnetic MOF combined with a functional guest (Fig. 1c). In the latter, a judicious choice of the molecule is the key for the appearance of different functionalities. The examples presented here will show that these porous solids provide an ideal platform to create new multifunctional materials covering from the simple co-existence of different electronic properties, provided by the framework and the functional guest, to a synergy between these functionalities as a result of the coupling between the two components.
 | ||
| Fig. 1 Schematic representation of (a) a magnetic MOF with a non-functional guest, (b) a non-magnetic MOF with a functional guest, and (c) a magnetic-MOF with a functional guest. | ||
2. MOFs based on magnetic frameworks
The preparation of MOFs based on magnetic frameworks is an example of a one network multifunctional material where the magnetic property coexists with the presence of porosity. These porous materials offer the possibility of inserting additional molecules into the pores (either gas molecules or other guests), acting as external chemical stimuli that can serve to tune the magnetism of the framework.15 Depending on the origin of the magnetic phenomena, four types of MOFs based on magnetic frameworks can be differentiated (Fig. 2): (a) magnetic MOFs, where magnetic cooperativity results through magnetic exchange via the ligands; (b) spin-crossover MOFs, where the nodes have suitable coordination environments for this phenomenon to exist; (c) MOFs with magnetic relaxation, where the nodes are clusters possessing an anisotropic spin ground state; with single-molecule magnet behaviour; (d) MOFs with a magnetocaloric effect, where the nodes are clusters possessing an isotropic spin ground state. | ||
| Fig. 2 Schematic representation of the different strategies to incorporate magnetic functionalities into MOFs: (a) magnetic exchange via the ligands; (b) spin-crossover at the nodes; (c) MOFs with magnetic clusters in the nodes (anisotropic spin for SMM, isotropic spin for magnetocaloric). | ||
2.1. Magnetic MOFs
The coexistence of magnetism and porosity is a challenging aspect from the point of view of the chemical design, as these two properties are inimical to one another: whereas magnetic exchange interactions require short distances between the metal centres, which are most commonly the spin carriers, porosity is typically favoured with the use of long linkers, which are often too long for magnetic ordering to exist at temperatures much above absolute zero. However, different synthetic routes can be envisaged towards the formation of porous magnetic materials, which will be presented below (Fig. 3). First, the use of short linkers will be described in the preparation of magnetic MOFs (Fig. 3a), with selected examples using formates, cyanides, azolates, diazines, and lactates, and then moving to their combination with longer linkers that can provide large pore apertures. Secondly, a different synthetic route to achieve the preparation of magnetic MOFs will be explained, which consists in using preformed complexes, i.e. a metallo-ligand approach (Fig. 3b). Finally, we will finish this section by showing that exchange coupling and porosity can also be combined through the use of radical ligands (Fig. 3c), and different examples of the use of varied radicals will be described. | ||
| Fig. 3 Schematic representation of the different synthetic routes available for the preparation of magnetic MOFs: (a) use of short linkers; (b) metallo-ligand approach; (c) use of radicals as ligands. | ||
Using this approach, more pronounced cooperative effects were observed in higher dimensional systems and, in particular, in 3D frameworks based on short organic linkers. An example of this kind is provided by ammonium metal formate frameworks. These compounds have shown the coexistence of magnetic and electric orderings in a family of multiferroic three-dimensional frameworks of formula [(CH3)2NH2][M(HCOO)3] and [NH4][M(HCOO)3] (M = Mn, Fe, Co, and Ni), as demonstrated by the groups of Cheetham17 and Gao.18 Specifically, these compounds display paraelectric to ferroelectric phase transitions between 160 and 254 K, which are combined with spin-canted antiferromagnetic ordering (in the range 8 to 36 K), as shown in Fig. 4. The origin of the ferroelectricity is the disorder–order transitions of the ammonium cations and their displacement within the channels of the framework, as demonstrated by single crystal diffraction, whereas the magnetic ordering is caused by the use of the short formate linkers. This type of solid has been extensively investigated in recent years,19 but due to the presence of the cations in the pores no gas sorption has been observed.
 | ||
| Fig. 4 (a) Crystal structure of [NH4][M(HCOO)3]; (b) zero-field-cooled (open symbols) and field-cooled (filled symbols) magnetization plots of the Mn, Fe, Co, and Ni derivatives under 5 or 10 Oe fields. Inset: Temperature dependence of the magnetic susceptibility of the Fe derivative under a 100 Oe field; (c) temperature-dependent traces of the real part of the dielectric permittivities, ε′, for the Mn, Fe, Co, and Zn derivatives, with E‖c at 10 kHz. Inset: Electric hysteresis loops for the four materials at temperatures below and above the transition points, with E‖c. Reprinted with permission from ref. 18, Copyright 2011 American Chemical Society. | ||
An approach that has been developed to introduce porosity in the above family consists of using bulky amines instead of ammonium. This yields a related family of three-dimensional isostructural porous formates of formula M3(HCOO)6 (M = Mn, Fe, Co, Ni), in which the bulky amine acts as a structure directing agent and is not incorporated into the solids. This family of porous magnets, discovered independently by three groups, consists of a diamond net combining octahedral and tetrahedral nodes with the pores occupied by a great variety of solvent molecules, which can be removed upon heating (Fig. 5).20–26 The magnetic behaviour of these magnetic MOFs depends on the type of metal ion: the iron and manganese formates are ferromagnets, with critical temperatures Tc of 16.1 and 8.0, whereas the cobalt compound is a spin-canted antiferromagnet below 1.8 K and the nickel derivative displays 3D long-range ferromagnetic ordering at 2.7 K (see Fig. 5b). The permanent porosity of these MOFs has been proven by gas adsorption, revealing a BET surface area of 360 m2 g−1. Furthermore, the available space has been confirmed by successfully inserting over 40 different guests into the pores, which also modulate Tc possibly due to subtle changes in the M–O–M angles and/or host–guest hydrogen-bond interactions.
 | ||
| Fig. 5 (a) Crystal structure of M3(HCOO)6; (b) temperature dependence of susceptibility for [M3(HCOO)6] with M = Mn, Fe, Co, and Ni between 2 and 40 K. Note that the χT scale is logarithmic for easy representation of the large difference in χT values for different compounds. Adapted from ref. 26, with permission from Wiley. | ||
The smallest bridging ligand that has been used to design magnetic coordination polymers is cyanide. An extensive family of three-dimensional (3D) bimetallic frameworks based on this linker, referred to as Prussian blue analogues,27 has been reported. In these coordination polymers the small size of the cyanide, which provides suitable pathways for magnetic superexchange, together with the 3D character of the magnetic lattice, leads to magnetic ordering at relatively high temperatures. Many laboratories have been very active on this type of material since the pioneering works of Verdaguer and Hashimoto, including the groups of Ohkoshi, Sieklucka, Dunbar, Girolami, Sato, Oshio, Coronado, Clerac, Long, Mallah, Larionova and Miller.28 One compound of this family, Co3[Co(CN)5]2, was the first material in which long-range magnetic ordering and microporosity were rigorously demonstrated to coexist.29 Thus, despite the use of such a short linker, Long and co-workers reported magnetic ordering at 38 K with a BET surface area of 480 m2 g−1, determined by N2 adsorption. Some years later, it was demonstrated on two related compounds of formula CsNi[Cr(CN)6] and Cr3[Cr(CN)6]2·6H2O (Fig. 6) that sorption of paramagnetic O2 molecules causes an increase in the ordering temperature, explained by the authors as a result of ferromagnetic coupling between the gas and the [Cr(CN)6]3− units of the frameworks, as diamagnetic N2 does not cause any shift that could originate from a structural transition.30 More specifically, CsNi[Cr(CN)6] is a ferromagnet with an ordering temperature of 75 K, and upon O2 sorption the [Cr(CN)6]3− units couple ferromagnetically with the spin of the adsorbed O2 molecules. Differently, Cr3[Cr(CN)6]2·6H2O orders at 219 K, and the sorption of O2 molecules causes a decrease in the magnetic moment of the system, as well as a reduction in the coercivity from 110 to 10 Oe and the remanent magnetization from 1200 to 440 emu G mol−1.
 | ||
| Fig. 6 (a) Crystal structure of CsNi[Cr(CN)6] (left) and Cr3[Cr(CN)6]2·6H2O (right); (b) nitrogen adsorption isotherms of CsNi[Cr(CN)6] (red squares) and Cr3[Cr(CN)6]2·6H2O (blue circles), as measured at 77 K; (c) magnetic hysteresis loops, measured at 5 K, of Cr3[Cr(CN)6]2·6H2O (blue) and Cr3[Cr(CN)6]2·6H2O sealed in a quartz tube containing 2.9 molecules of O2 per formula unit (red); (d) magnetic hysteresis loops, measured at 2 K of CsNi[Cr(CN)6] (blue) and CsNi[Cr(CN)6] sealed in a quartz tube containing 1.8 molecules of O2 per formula unit (red). Reproduced from ref. 30, Copyright 2008 American Chemical Society. Further permissions related to this material should be directed to the ACS. | ||
Another family of short organic ligands that have also been employed for the preparation of MOFs is that provided by azolates, which can form strong and directional coordination bonds serving as bridges between metal ions,31,32 and therefore are appealing candidates for the preparation of magnetic MOFs. For example, zeolitic imidazolate frameworks (ZIFs) are a subfamily of MOFs consisting of tetrahedral metal centers (ZnII and CoII primarily) connected by bent imidazolate-derived organic ligands, with geometrical features analogous to those of zeolites.33 Thus, despite the use of a short linker such as imidazolate, highly porous solids of different topologies can be obtained, akin to zeolites, which in the case of CoII are also magnetic. The term ZIF was coined in 2006,34 but some years earlier Gao and You reported the structures and magnetic properties of seven polymorphs of Co(imidazolate)2, indicating structural features similar to that of silica, although no studies on gas sorption were performed at that moment (Fig. 7).35–37 The robustness of the porous framework was nevertheless proven in one of these polymorphs by exchanging the included template with EtOH and removing it later by vacuum treatment.35 Although imidazolates transmit the antiferromagnetic coupling between the CoII ions in these structures, the uncompensated antiferromagnetic couplings arising from the non-centrosymmetric structures have often led to the appearance of weak ferromagnets (also known as canted-antiferromagnets) as a consequence of spin-canting.
 | ||
| Fig. 7 (a) Crystal structure of Co(imidazolate)2; (b) plots of temperature dependence of χM and χMT for Co(imidazolate)2 measured at a field of 10 kOe (left) and temperature dependence of AC susceptibility χ′ (top) and χ′′ (bottom) obtained at a field of 20 Oe. Adapted from ref. 36, with permission from Wiley. | ||
Functionalized imidazolates with additional coordination groups have also been used for the formation of magnetic MOFs. For example, Fig. 8 shows the crystal structure of a Co(II)–imidazolate-4-amide-5-imidate based MOF, which possesses 1D hexagonal channels and a BET surface area of 649 m2 g−1.38 The cobalt centres are penta-coordinated by the imidazolate–amidate–imidate linkers to form a distorted trigonal–bipyramidal geometry, with antiferromagnetic coupling between the metal centres.
 | ||
| Fig. 8 Crystal structure of Co(imidazolate-4-amide-5-imidate)·0.5DMF. Adapted from ref. 38, with permission from The Royal Society of Chemistry. | ||
Another interesting example of the use of a functionalized azolate is provided by a magnetic MOF that undergoes multiple changes in magnetic properties upon desolvation/resolvation, as shown in Fig. 9.39 The parent compound, [Cu(tzc)(dpp)]·2H2O, can lose one equivalent of water to form the monohydrate phase, [Cu(tzc)(dpp)]·H2O, or can also transform into an anhydrous phase [Cu(tzc)(dpp)], which can exist in three different polymorphs. The dihydrate phase shows antiferromagnetic exchange interactions, whereas ferromagnetic properties are observed for the trimorphic anhydrate system.
 | ||
| Fig. 9 Crystal structures of the hydrates [Cu(tzc)(dpp)]·2H2O (A), [Cu(tzc)(dpp)]·H2O (B) and its polymorphic anhydrous modifications [Cu(tzc)(dpp)] (C-I, C-II, C-III) viewed as stacked chains in a single unit cell. Arrows and labels indicate the directions and conditions, respectively, for phase transitions occurring between these phases. Reprinted with permission from ref. 39, Copyright 2013 American Chemical Society. | ||
A related family of heterocyclic ligands that can also promote magnetic exchange is provided by diazines, which have given rise to the first example of a gas-responsive magnetic material. The Cu-based MOF of formula [Cu(F-pymo)2(H2O)1.25]n (F-pymo = 5-fluoropyrimidin-2-olate), shown in Fig. 10, has helical channels of ca. 2.9 Å diameter which are filled with water molecules that can be removed upon heating.40 The as-synthesized solid shows antiferromagnetic interactions, mediated by the pyrimidine ligands, and it orders as a canted antiferromagnet below TN = 24 K (22 K upon activation). Different gas molecules can be incorporated into the empty channels, like CO2, whose presence in the pores causes an increase in the Neel temperature from 22 to 29 K. The authors attribute this increase to a structural perturbation exerted by the CO2 guests which affect the exchange interaction pathways through the ligands, albeit it cannot be confirmed due to lack of structural information after gas sorption.
 | ||
| Fig. 10 (a) Crystal structure of [Cu(F-pymo)2(H2O)1.25]n; (b) CO2 adsorption isotherm of activated sample of [Cu(F-pymo)2]n at 273 K (circles) and 298 K (squares), with the open symbols denoting desorption; (c) effect of CO2 sorption on the magnetic behavior of [Cu(F-pymo)2]n at an external magnetic field of 100 Oe. Reprinted with permission from ref. 40, Copyright 2008 American Chemical Society. | ||
An unusual ligand recently employed by Zeng and co-workers for the formation of a porous magnet is lactate. [CoII3(lac)2(pybz)2]·3DMF is the magnetic analogue of the double-walled framework [ZnII3(lac)2(pybz)2]·3DMF. It is formed by a square rod of Co-lactate and the double wall connection between them (Fig. 11).41 The structure contains M–O–M connectivity within the well-separated square chains, which provides a short pathway for magnetic exchange. Interestingly, the solvent molecules that fill the pores can be exchanged with several guests without affecting the integrity of the structure, including gases (N2, H2 and CH4) and volatile guests (methanol, ethanol, propanol, benzene). However, upon exposure to water molecules or iodine, a structural transformation takes place with the coordination of a water molecule or the oxidation of a CoII centre, yielding respectively a hydrated and a partially oxidized MOF. The nature of different solvents in the channels modifies the magnetic properties, with a complex magnetic phenomenology observed in this MOF, with four different ground states: canted antiferromagnets for [CoII3(pybz)2(lac)2]·xsolvent and iodine-loaded samples due to the antiferromagnetic interchain coupling (J′ < 0) via the solvent or iodine molecules, single-chain magnet for the desolvated [CoII3(pybz)2(lac)2] as J′ ≈ 0, ferrimagnet for [CoII3(pybz)2(lac)2(H2O)2]·7H2O as J′ > 0, and ferromagnet for the partially oxidized [CoIIICoII2(pybz)2(lac)2(H2O)2]I·2H2O·1.5DMSO as J′ > 0.
 | ||
| Fig. 11 (a) Crystal structure of [CoII3(pybz)2(lac)2(H2O)2]; (b) N2 sorption isotherm of [CoII3(pybz)2(lac)2(H2O)2] at 77 K; (c) post-synthetic modifications and resulting magnetic ground states and changes in coordination and valence of cobalt ions. Reprinted with permission from ref. 41, Copyright 2014 American Chemical Society. | ||
All the above examples have been based on the use of short linkers to create a dense metallic packing, while keeping some porosity. A different approach to improve the porosity in these magnetic solids consists in the combination of a short ligand and a considerably longer one. In this case the short ligand may organize the metal centres in infinite chains (or layers), while the long organic links are orthogonal to those chains (or layers) providing large pore apertures. With this approach, a strong magnetic interaction between the metal centers can be envisaged, although with the limitation of occurring in one (or two) dimensions, which also limits the occurrence of magnetic ordering to low temperatures. A successful example of this methodology is illustrated by the group of MOFs known as MOF-74 or CPO-27, which display Brunauer–Emmett–Teller surface areas over 1000 m2 g−1 and have been identified as among the most promising MOFs for CO2 capture. These materials, of formula M2(dhtp)(H2O)2 (dhtp = 2,5-dihydroxyterephthalate), have been prepared from several transition metals including MgII, ZnII and the magnetic MnII, FeII, CoII, NiII, CuII. The crystal structure is formed by helical chains of cis-edge connected metal oxygen octahedral which are each linked by the organic ligand with three adjacent chains, resulting in a honeycomb distribution of one-dimensional channels 11 Å wide (Fig. 12). The coordinated water molecule can be removed, leaving an open coordination site accessible to incoming adsorbate molecules. The first magnetic member of this family to be reported was the CoII derivative,42 whose magnetic study revealed a metamagnetic-like behaviour, with antiferromagnetic ordering below 8 K and a field-induced transition to a ferromagnetic-like ordered state upon application of magnetic fields stronger than ca. 2 T. The superexchange pathway in the metal–oxygen chain suggests that the magnetic moments couple ferromagnetically in the chains, while the antiferromagnetic long-range order, observed at low magnetic fields, is a consequence of the antiparallel alignment of the spins of adjacent chains. The FeII derivative Fe-MOF-74 (or CPO-27-Fe) was also reported.43 This compound shows a change of the magnetic exchange along the chains upon exposure to 1 bar of different hydrocarbons, which can be related to the strength of the interaction with the framework (Fig. 12c). Thus, weakly interacting adsorbates (methane, ethane, and propane) slightly diminish the strength of the ferromagnetic exchange, whereas those that interact more strongly (propylene, ethylene, and acetylene) reverse the nature of the intrachain coupling from ferromagnetic to antiferromagnetic. Computational studies reveal that in this structure the FeII centers are always in a high-spin state.44–47
 | ||
| Fig. 12 (a) Crystal structure of MOF-74; (b) gas adsorption isotherms for methane, ethane, ethylene, and acetylene in Fe-MOF-74 at 318 K; (c) variable-temperature magnetic susceptibility data in an applied field of 1 kOe for samples of Fe-MOF-74 in a vacuum (bare) and under 1 bar of the indicated hydrocarbon (black lines represent magnetic fits). Adapted from ref. 43, with permission from the AAAS. | ||
A successful example of the metallo-ligand approach for the synthesis of magnetic MOFs is the use of oxamato-based oligonuclear complexes, as recently reviewed by Ferrando-Soria and Pardo.49 For instance, oxamato-based dinuclear CuII2 metallacyclic complexes, with weak ferromagnetic coupling between the CuII ions, can coordinate MnII ions through the free carbonyl-oxygen atoms, yielding a 3D MOF of formula [Na(H2O)3.25]4{Mn4[Cu2(Me3mpba)2(H2O)3.33]3}·37H2O [Me3mpba4− = N,N′-2,4,6-trimethyl-1,3-phenylenebis(oxamate)].50 The structure consists of an extended parallel array of anionic, oxamato-bridged Mn4Cu6 layers that are further interconnected through two m-phenylene spacers among the CuII ions (Fig. 13). 3D ferromagnetic ordering is observed, which results from the antiferromagnetic coupling between the high-spin MnII (S = 5/2) and CuII (S = 1/2) ions through the oxamato bridges (2D ferrimagnetic behaviour), and interlayer ferromagnetic interaction across the double 2,4,6-trimethylphenylenebis(amidate) bridges. The magnetic behaviour depends on the solvent molecules that fill the pores, although gas molecules (CO2 or CH4) do not interact with the magnetic framework, maybe due to the amorphization process caused upon activation.
 | ||
| Fig. 13 (a) Example of an oxamate-based dinuclear metalloligand; (b) crystal structure of [Na(H2O)3.25]4{Mn4[Cu2(Me3mpba)2(H2O)3.33]3}·37H2O, viewed along the crystallographic c axis showing the pillared square/octagonal layer architecture. Copper and manganese atoms are represented by green and purple octahedra respectively. Free water molecules and Na+ counterions have been omitted for clarity; (c) temperature dependence of the product of the direct current (dc) molar magnetic susceptibility and temperature (χMT) of the activated MOF (green), with methanol molecules filling the pores (red), and with water molecules filling the pores (blue). Reprinted with permission from ref. 50, Copyright 2012 American Chemical Society. | ||
 | ||
| Fig. 14 (a) Schematic representation of the ptmtc radical ligand; (b) structure of {[Cu3(ptmtc)2(py)6(EtOH)2(H2O)]} showing the open-framework; (c) value of χT as a function of the temperature for MOROF-1 (orange filled circle, MOROF-1; open circle, evacuated MOROF-1). Adapted from ref. 52, with permission from the Nature Publishing Group. | ||
A ubiquitous organic radical in the field of functional molecular materials is the radical anion tetracyanoquinodimethane (TCNQ˙−), as well as the related tetracyanoethylene (TCNE˙−). Through the use of TCNQ and TCNE as building units of MOFs, the functional properties of these non-innocent ligands can be exploited to develop charge transfer frameworks which may exhibit electrical conductivity as well as interesting magnetic properties, although the formation of non-porous coordination polymers is rather common. Remarkably, the compound [{Ru2(O2CPh-o-Cl)4}2TCNQ(MeO)2]·CH2Cl2 (o-ClPhCO2− = o-chlorobenzoate; TCNQ(MeO)2 = 2,5-dimethoxy-7,7,8,8-tetracyanoquinodimethane), reported by Miyasaka and co-workers, consists of two-dimensional layers where Ru2 paddlewheels are linked through the TCNQ ligands, yielding antiferromagnetic ordering at TN = 75 K (Fig. 15). Interstitial CH2Cl2 molecules are located in the void spaces between the layers, which can be lost at room temperature to form a dried sample, which orders ferromagnetically at Tc = 56 K.55 This process is reversible upon exposure to CH2Cl2 vapor for 72 h. The magnetic change occurs as a result of slight structural modifications caused by the ordering/disordering of ligand orientation upon the extrusion of the CH2Cl2 molecules.
 | ||
| Fig. 15 (a) Reversible CH2Cl2 extrusion/uptake in [{Ru2(O2CPh-o-Cl)4}2TCNQ(MeO)2]·CH2Cl2 provokes subtle changes that affect the pendant ligand (the CH2Cl2 molecules are represented by a yellow CPK model); (b) effects of the desolvation on the magnetic properties of the Ru coordination polymer: variation of 1 Hz ac susceptibilities χ′′ (zero dc field and 3 Oe ac oscillating field) of the solvated and desolvated compounds. Reprinted with permission from ref. 55, Copyright 2010 American Chemical Society. | ||
The radical derived from chloranilic acid, 2,5-dichloro-3,6-dihydroxy-1,4-benzoquinone (Cl2dhbq), has been combined with FeIII yielding a crystalline solid of formula (Me2NH2)2[Fe2(Cl2dhbq)3]·2H2O·6DMF, where the iron centres are bridged by the radical form of the chloranilic acid forming two-dimensional honeycomb-like anionic layers, with Me2NH2+ serving as countercations (Fig. 16).56 The one-dimensional hexagonal channels are filled with solvent molecules, but these can be removed upon activation with a slight structural distortion of the framework, yielding a microporous solid with a surface area of 885 m2 g−1. Magnetic susceptibility measurements indicate the presence of strong magnetic coupling at room temperature, with spontaneous magnetization occurring below T = 100 K, which is reduced to 30 K upon activation of the material. The ordering temperatures of both solvated and unsolvated MOFs have been accurately determined by AC susceptibility, revealing that Tc = 80 K and Tc = 26 K, respectively.
 | ||
| Fig. 16 (a) X-ray crystal structure of (Me2NH2)2[Fe2(Cl2dhbq)3]·2H2O·6DMF (cations and solvent molecules omitted for clarity) viewed along the crystallographic c axis (top) and b axis (bottom). Orange = Fe, green = Cl, red = O, and grey = C. (b) Thermal dependence of the field-cooled magnetization for as-synthesized (Me2NH2)2[Fe2(Cl2dhbq)3]·2H2O·6DMF (blue) and activated (Me2NH2)2[Fe2(Cl2dhbq)3] (red), collected under an applied dc field of 10 Oe. Inset: Field dependence of the magnetization for (Me2NH2)2[Fe2(Cl2dhbq)3]·2H2O·6DMF at 60 K (blue) and (Me2NH2)2[Fe2(Cl2dhbq)3] at 10 K (red). Reprinted with permission from ref. 56, Copyright 2015 American Chemical Society. | ||
To summarize this section, the different approaches investigated so far for the formation of porous magnets have both advantages and disadvantages. For example, the use of short linkers permits a large magnetic coupling between the metal centres, but limits the possibility of porosity, whereas the combination with other larger units that provide porosity reduces the dimensionality of the framework (from a magnetic point of view) thus resulting in low Tc values. In a different approach, the use of radical ligands seems to be more attractive, as the coupling is favoured even with large distances between the metallic nodes, but the highest limitation resides in their stability. Finally, the use of metallo-ligands seems to be the most convenient approach for the preparation of magnetic MOFs with high Tc, although there are a limited number of metallo-ligands capable of promoting magnetic exchange.
2.2. Spin-crossover MOFs
Spin-crossover (SCO) is a phenomenon in which electronic configurations of a transition metal ion can be switched between high-spin (HS) and low-spin (LS) states in response to external stimuli (temperature, pressure, light irradiation, magnetic field, electric field, guest sorption), producing changes in magnetism, colour and structure.57,58 Therefore, due to its molecular origin, no exchange interactions between neighbouring magnetic centres are required, and it is only necessary to design a MOF with first row transition metals in a suitable coordination environment that provides an appropriate ligand field for the transition between the HS and LS states. Thus, there is no limitation on the length of the ligands, nor the connectivity between the metal centres, which in principle should facilitate the inclusion of this magnetic property in MOFs. However, cooperative effects are still necessary to make this phenomenon useful in order to have an abrupt crossover, and this arises from the elastic forces present in the solid. In these systems, such a cooperativity is favoured by the polymeric nature of the framework, which keeps the spin-crossover centers connected even if they are not very close. Thanks to this cooperativity this type of MOF does show high sensitivity to the subtle structural changes occurring in metal coordination environments upon inclusion of guest species within the porous framework. Thus, from a multifunctional point of view, one can classify this material as a two-network material in which one network is composed of the framework itself, whereas the second network is provided by the guest molecules. As a consequence, one would expect a weak influence of the molecular network (guests) on the framework. This effect can be strongly enhanced by diminishing the size of the pore, as will be discussed in this section, in which physisorption of gases can even affect the temperature of the spin transition. Moreover, we will focus on unusual MOFs in which the spin-crossover properties are tuned via post-synthetic modification of the framework. The effects of guests and small molecules in SCO-MOFs have been recently reviewed by Ohtani and Hayami,59 and therefore will not be analysed here.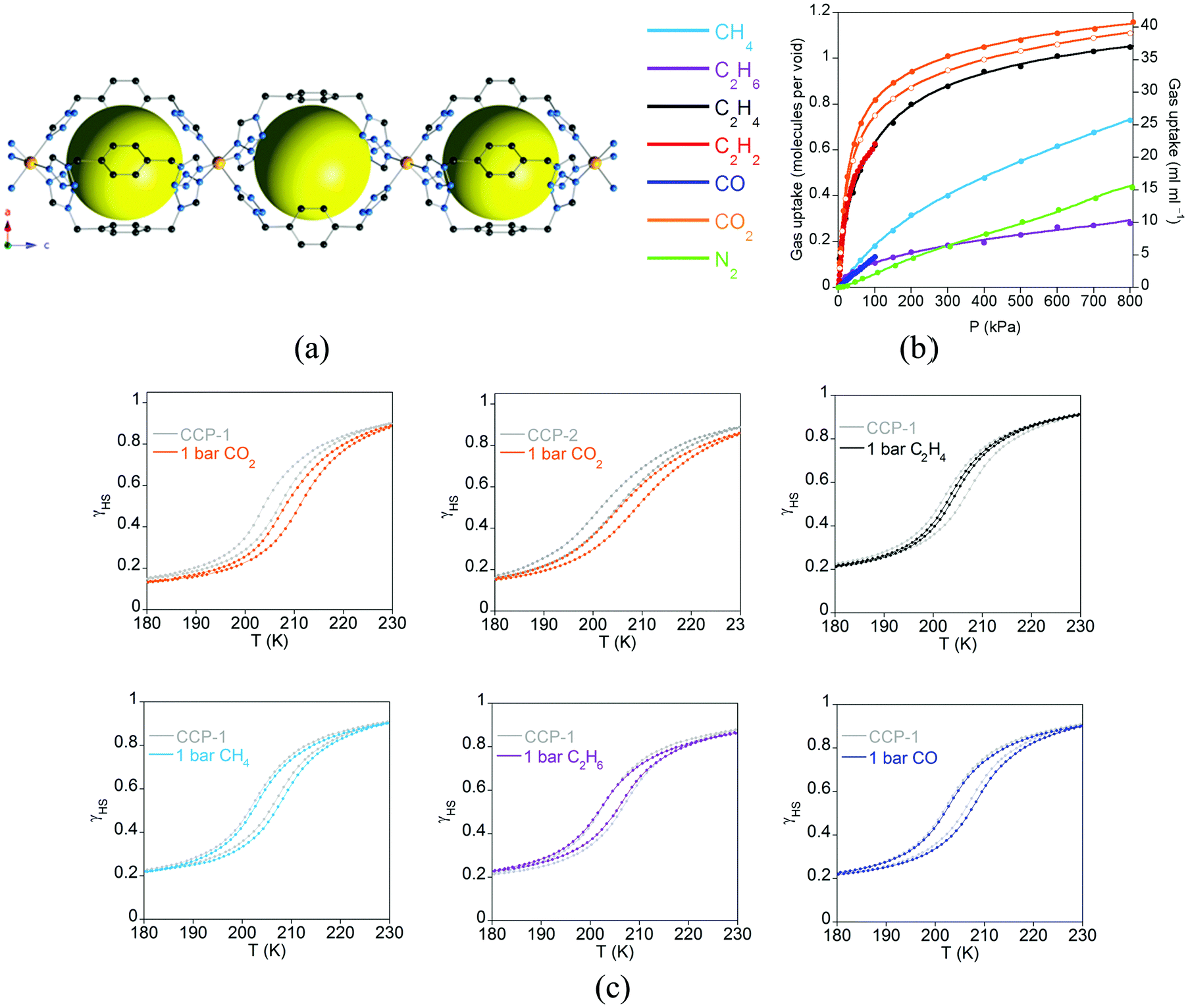 | ||
| Fig. 17 (a) Crystal structure of the compartmentalized coordination polymers CCP-1 and CCP-2 emphasizing the internal cavities (as yellow spheres) formed by the connection of FeII centres to three bistetrazol-p-xylene ligands in the syn conformation. Key: Fe, orange; C, gray; N, blue; H, white; counteranions (ClO4− and BF4−) are omitted for clarity; (b) gas adsorption isotherms at 298 K of CCP-1 (closed symbols) and CCP-2 (open symbols) of different gases (lines correspond to the best fits); (c) temperature dependence of the high spin fraction (γHS) for CCP-1 and CCP-2 before (grey) and after inclusion of CO2 (orange), ethylene (black), methane (sky blue), ethane (purple) and CO (dark blue). Adapted from ref. 63, with permission from The Royal Society of Chemistry. | ||
The increase in the size of the internal voids has also been examined by extending the length of the flexible organic ligand with the purpose of augmenting the gas sorption capacity (Fig. 18).64 Through this ligand design, isostructural analogues of these compartmentalized coordination polymers have been prepared, with formula [Fe(btzbp)3](ClO4)2 (CCP-3) and [Fe(btzbp)3](BF4)2 (CCP-4) (btzbp = 4,4′-bis((1H-tetrazol-1-yl)methyl)-1,1′-biphenyl), which present discrete voids of 257 Å3. Gas sorption measurements show that two molecules of CO2 can be loaded in each void at 1 bar and 298 K. Both compounds present spin transitions centred at 195 and 199 K respectively, similar to those observed in CCP-1 and CCP-2, although different magnetic behaviour has been observed for CCP-3 and CCP-4 depending on the number of CO2 molecules that are physisorbed. Upon inclusion of one molecule of CO2 in each internal void, a shift of T1/2 from 199 K to 206 K has been observed (Fig. 19). Thus, physisorption of CO2 stabilizes the LS state due to the interaction between CO2 and the cationic framework. However, upon additional CO2 loading (two molecules in each void), a reduction of T1/2 occurs, reaching 202 K. This behaviour could be caused by gas–gas interaction inside the void of the compartmentalized coordination polymer.
 | ||
| Fig. 18 Chemical structures of the ligands, btzx (top) and btzbp (bottom). The dashed arrows represent the distances between the two coordinating nitrogens. | ||
 | ||
| Fig. 19 (a) Crystal structure of CCP-4 viewed along the b-axis. The BF4− anions and hydrogen atoms have been removed for clarity. Key: Fe, orange; C, gray; N, blue. The yellow ellipsoids are placed in the structure to represent the empty space of the internal voids; (b) gas sorption isotherms at 298 K for CO2, CH4 and N2 for CCP-4; (c) temperature dependence of the product of the magnetic susceptibility and temperature (χmT) for activated CCP-4 (black) and CCP-4 loaded with one molecule of CO2 in each void (red line). (d) Temperature dependence of the product of the magnetic susceptibility and temperature (χmT) for activated CCP-4 (black) and CCP-4 loaded with two molecules of CO2 in each void (blue line). Adapted from ref. 64, with permission from The Royal Society of Chemistry. | ||
Another important family of SCO MOFs able to trap molecules is the so-called Hofmann clathrates. This family has been extensively studied for the co-existence of SCO phenomena and porosity along 1D channels where different guests can be incorporated affecting the transition temperature.65 More recently, it has also been demonstrated that adsorption of SO2 molecules can modify the SCO properties.66 The adsorption isotherm at 293 K displays a type I behaviour featuring a sharp SO2 uptake at low pressures (below 50 mbar) and practically reaches saturation at 0.20 bar, which corresponds to ca. 1.3 equivalents of SO2. However, this sorption is not completely reversible, and after vacuuming for 4 h at 258 K, ca. 0.5 equivalents of SO2 remain trapped in the pores. The sorbed SO2 molecules coordinate the PtII centres through the sulfur atom (Fig. 20), stabilizing the low spin state of the FeII ions and causing an increase of 8 K in the transition temperature.
 | ||
| Fig. 20 (a) Fragment of the structure of {Fe(pz)[PtII(CN)4]}·SO2. Colour code: Fe (orange), Pt (light pink), S (yellow), N (blue), C (grey). (b) SO2 sorption–desorption isotherms for {Fe(pz)[PtII(CN)4]} at 293 K (○) and at 258 K (●). Red and blue colour codes refer to the first and second sorption–desorption cycles, respectively. (c) Magnetic properties of {Fe(pz)[PtII(CN)4]} (black line) and {Fe(pz)[PtII(CN)4]}·SO2 (red line). Reprinted with permission from ref. 66, Copyright 2013 American Chemical Society. | ||
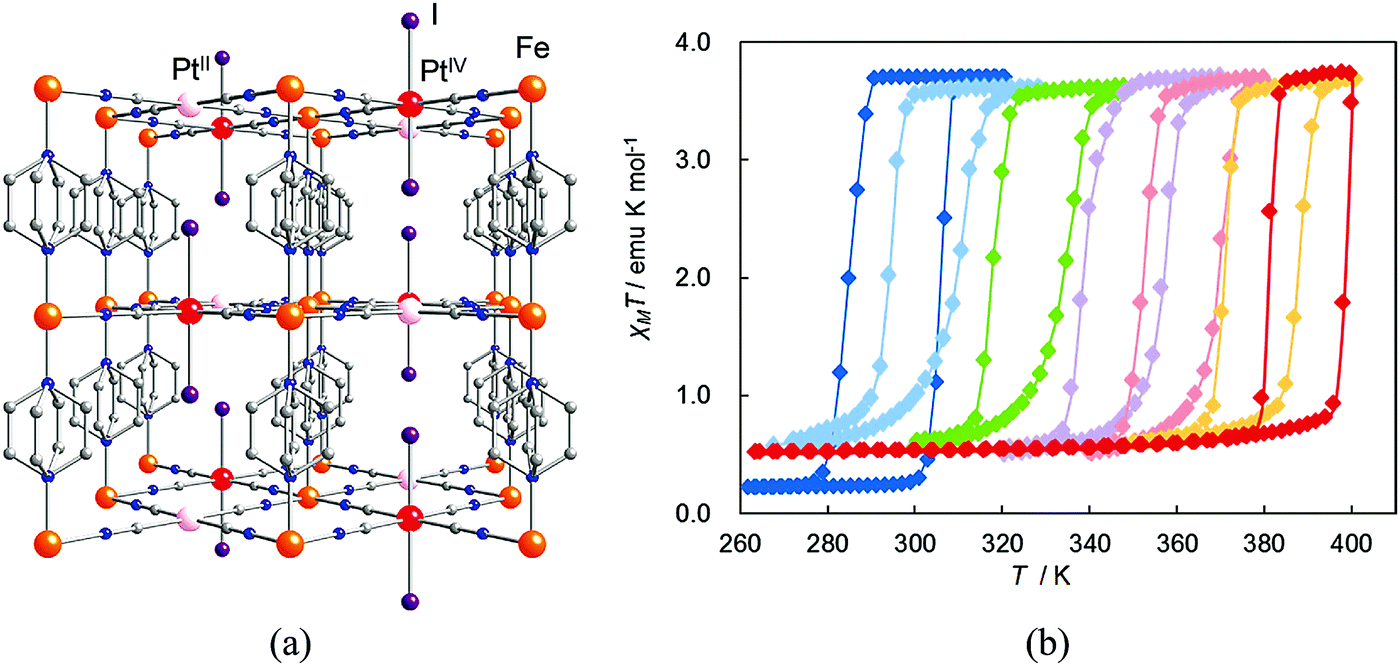 | ||
| Fig. 21 (a) Crystal structure of {Fe(pz)[PtII/IV(CN)4(I)]}. Atoms: Fe (orange), PtII (pink), PtIV (red), I (purple), C (gray), N (blue). (b) Magnetic behavior of {Fe(pz)[Pt(CN)4(I)n]} n = 0.0 (blue); 0.1 (sky blue); 0.3 (green); 0.5 (violet); 0.7 (pink); 0.9 (orange); 1.0 (red). Reprinted with permission from ref. 68, Copyright 2011 American Chemical Society. | ||
A covalent post-synthetic modification of a SCO MOF has been reported by Kepert and co-workers in a Hofmann clathrate of formula [Fe(bipytz)(Au(CN)2)2] through the incorporation of an organic linker, 3,6-bis(4-pyridyl)-1,2,4,5-tetrazine (bipytz), that readily undergoes an inverse-electron-demand Diels–Alder reaction.69 The octahedral FeII centers are bridged equatorially by four linear [Au(CN)2]− linkers forming a 2D layer which is further extended into 3D through bipytz units that serve as pillars (Fig. 22). Upon this modification, the SCO behaviour differs from the pristine material in three ways: it is less abrupt; occurs at lower temperature (T1/2 = 178 K vs. T1/2= 275 K); and does not show a thermal hysteresis behaviour.
 | ||
| Fig. 22 (a) Structural representations of [Fe(bipytz)(Au(CN)2)2] viewed down the b axis with solvent and interpenetration removed for clarity; (b) inverse-electron-demand Diels–Alder reaction of 3,6-bis(4-pyridyl)-1,2,4,5-tetrazine (bipytz) with 2,5-norbornadiene to form 3,6-bis(4-pyridyl)-1,2-diazine (bipydz); (c) magnetic susceptibility data for [Fe(bipytz)(Au(CN)2)2] and post-synthetically modified [Fe(bipydz)(Au(CN)2)2]. Adapted from ref. 69, with permission from John Wiley and Sons. | ||
2.3. MOFs with single-molecule magnets at the inorganic nodes
Single-molecule magnets are magnetic molecules that exhibit slow relaxation of the magnetization at low temperatures. Usually, they are formed by polynuclear magnetic clusters with a large spin value and high magnetic anisotropy. Recently, a second generation of SMMs based on mononuclear metal complexes composed of highly anisotropic magnetic ions has been reported, which are simpler to design from a chemical point of view. This latter class of SMM have additional interest in quantum technologies, as they can provide ideal examples of quantum bits (qubits), which are the basic units in quantum computing. Since the ground state of these molecules is a spin doublet, it can be seen as a spin qubit, as far as its quantum coherence is large enough to have time to perform a quantum operation. One important requirement for maximizing the quantum coherence is to minimize the magnetic dipolar interactions between qubits, which can be achieved by separating these magnetic units in space. In this sense, MOF can be ideal platforms to reach this goal since they provide spatial separation at will.The linkage of single-molecule magnets based on polynuclear cluster-type metal complexes by bridging organic ligands has been exploited in the past for the isolation of crystalline coordination polymers.70 This approach has been useful to investigate the interplay between the single-molecule magnetic behaviour and the new (cooperative) properties that may appear when these molecular nanomagnets are magnetically coupled. Still, this has most commonly resulted in the preparation of systems with lower dimensionality, chains being the most common.71 The first 3D coordination network of SMMs was prepared by Clérac and co-workers using Mn4 clusters as nodes.72 In these networks, subtle modifications in the synthetic strategies influence enormously and unpredictably the dimensionality of the network,73 and in some other cases the formation of the extended system causes quenching in the magnet-like properties of the metal clusters.74 Although these solids typically lack permanent porosity, Kou and co-workers have recently reported the combination of porosity and SMM behaviour in a MOF based on Mn6 clusters.75
With the recent discovery of the second generation of single-molecule magnets, i.e. the so-called single-ion magnets, or SIMs, new coordination polymers based on these mononuclear lanthanoid complexes have been isolated. These SIMs are easier to assemble than the cluster-type SMMs and therefore they can allow circumventing the problems observed for the preparation of SMM-MOFs. The first family of SIM-MOF has been recently reported.76 It can be formulated as Ln(bipyNO)4(TfO)3 (bipyNO = 4,4′-bypyridyl-N,N′-dioxide, TfO = triflate). Its structure consists of a 3D coordination network containing lanthanoid SIMs in the nodes of a porous cationic framework formed by the long bipyNO as the bridging ligand (Fig. 23). The pores of this framework are filled by triflate anions. Interestingly, this family shows the capability of exchanging the anions placed in its pores, while maintaining the magnetic behaviour. This feature has been exploited to incorporate into the pores bulky anions such as polyoxometalates (POMs) and other smaller anions such as [AuCl4]− by post-synthetic modification of the magnetic MOF (Fig. 24). In these cases, the anion exchange process has occurred in a single-crystal-to-single-crystal manner thus providing structural evidence for the location of the encapsulated species.77 The interest in these SIM-MOFs is twofold. On one hand, the controlled organization of the SIMs is an important issue for their possible application in quantum computing since each SIM can behave as a quantum bit. Indeed, the ability to tune the spatial arrangement of more than one non-identical SIM qubit (in the present example three structurally distinguishable SIMs are present) is precisely what is currently needed to advance in the global control paradigm of quantum computing.78 On the other hand, the incorporation of functional molecular species into the pores can provide a convenient way for introducing a second property into these hybrid materials. After this first report on the preparation of a SIM-MOF, many other examples have been reported based either on lanthanoids,79–81 or on transition metals.82 A very remarkable result in this context has been recently reported by Freedman and co-workers83 with the demonstration of atomic clock-like transitions in the MOF of formula [(TCPP)Co0.07Zn0.93]3[Zr6O4(OH)4(H2O)6]2 (TCCP = 5,10,15,20-tetrakis(carboxyphenyl)porphyrin), where the cobalt(II) porphyrin units, diluted within a diamagnetic network, possess lifetimes of 13.7 μs at 5 K (1.8 μs at 15 K), observed with pulsed EPR spectroscopy, which is a modest improvement over the 8.4 μs obtained in the POM [Ho(W5O18)2]9−.84
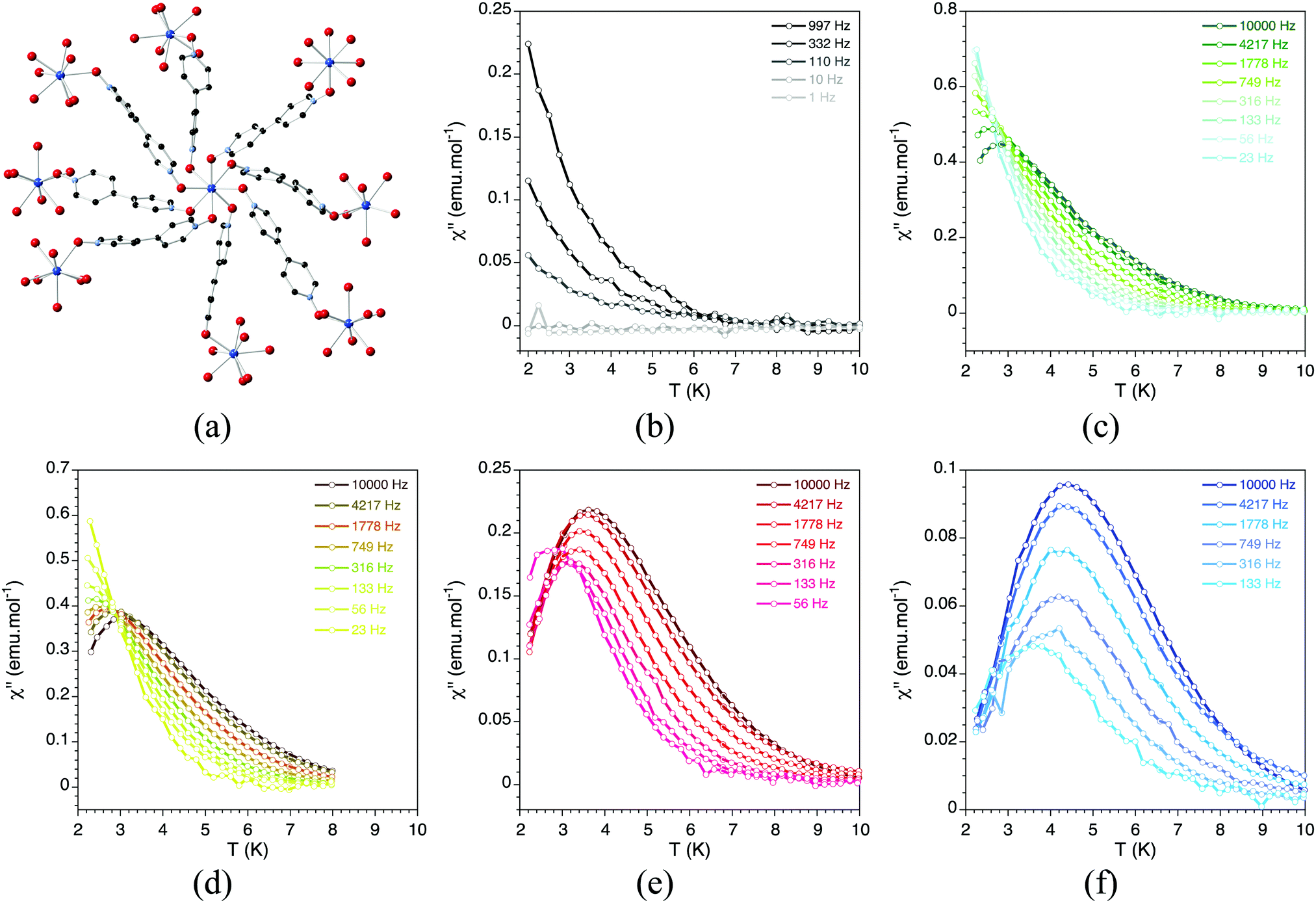 | ||
| Fig. 23 (a) Crystal structure of Ln(bipyNO)4(TfO)3. Out-of-phase dynamic susceptibility under different external dc magnetic fields: (b) 4 G; (c) 1000 G; (d) 2000 G; (e) 5000 G; and (f) 10000 G. Solid lines are eye guides. Adapted from ref. 76, with permission from John Wiley and Sons. | ||
 | ||
| Fig. 24 Crystal structures after anion exchange in Ln(bipyNO)4(TfO)3 with different anions: (a) [W6O19]2−; (b) [Mo6O19]2−; (c) [AuCl4]−; and (d) the mixed system [W6O19]2−, and [AuCl4]−. [W6O19]2− [Mo6O19]2−, [AuCl4]− and Cl− anions are shown in blue, purple, orange and green, respectively. | ||
2.4. MOFs for magnetic refrigeration
A different magnetic phenomenon that is of high interest for cooling applications is that of magnetic refrigeration (Fig. 25), which is based on the magnetocaloric effect (MCE). This effect is the consequence of a change in the magnetic entropy (ΔSm) and related adiabatic temperature (ΔTad) in response to a change in the applied magnetic field (ΔB). Although the MCE is intrinsic to any magnetic material, only in a few cases are the changes sufficiently large to make them suitable for applications. In order to maximize the change in the magnetic entropy, the magnetic material should have a large spin ground state S (the maximum magnetic entropy amounts to Rln(2S + 1)), high spin degeneracy or low lying spin ground state, negligible magnetic anisotropy, and a high magnetic density. In this sense, gadolinium and manganese based molecular compounds have been largely studied because of their large spin-only magnetic moment (SGd = 7/2, SMn = 5/2) and quasi-isotropic character, showing great promise as low temperature magnetic refrigerants based on molecular clusters that can rival traditional materials such as lanthanide alloys and magnetic nanoparticles.
 | ||
| Fig. 25 The magnetocaloric cooling cycle consists of four steps: (1) first, the magnetic material (part “a” of the figure) is exposed to a magnetic field, B (represented by a red arrow), resulting in heating of the material (part “b” of the figure) as a consequence of the reduction in magnetic entropy; (2) the heat excess caused by the alignment of all the spins is expelled to the surroundings, thus yielding the magnetic material at the same temperature as in the beginning, but with the spins aligned (part “c” of the figure); (3) the removal of the magnetic field causes cooling of the refrigerant due to the increase of magnetic entropy caused by the spin relaxation (part “d” of the figure); (4) heat can be absorbed from the cooling compartment. The orange colour represents an increase in temperature of the magnetic material, and the dark blue colour represents a decrease in temperature of the magnetic material. The grey arrows represent the large spin of the magnetic material, whose orientation can be modified upon application of a magnetic field (represented by red arrows). | ||
The use of magnetic MOFs is restricted to ultra-low temperature applications, whereas magnetic refrigerators near room temperature are dominated by lanthanoid-based alloys. In this context, the use of magnetic MOFs for magnetic refrigerators is very appealing since they can be designed to consist of isolated paramagnetic centers (i.e. with weak superexchange interactions) that favors a large MCE, combined with higher thermal and solvent stabilities than their discrete molecular cluster analogues. However, there are two important obstacles that should be taken into account: MOFs are typically poor thermal conductors (which is important to dissipate the heat), although their facile deposition on metallic surfaces can overcome this problem; and the density of magnetic centres is very low, which is a consequence of the presence of porosity.
There have been numerous examples in the literature of the use of magnetic coordination polymers for MCE since the initial reports on Prussian blue analogues,85–88 which are mainly based on Gd or Mn centres for similar reasons to their interest in molecular clusters mentioned above. Most of these coordination polymers are dense materials, such as [Gd(HCOO)(bdc)] (H2bdc = terephthalic acid),89 which present a higher density of magnetic centres. For example, this compound presents values of −ΔSm = 47.0 J kg−1 K−1 for ΔB = 9 T. In other cases, the materials present channels or voids which are filled with solvent molecules, as in Gd2(fum)3(H2O)4·3H2O (with MCE values of ΔSm = 20.7 J kg−1 K−1 for ΔB = 5 T),90 but the sorption of gases has not been demonstrated. However, the combination of porosity and MCE is very scarce, and has only been successfully combined in a couple of examples presented below.
The Gd-based MOF of formula [Gd2(pam)3(DMF)2(H2O)2]n·nDMF (H2pam = pamoic acid) forms a 3D framework with channels of sizes ca. 17 Å × 15 Å in which the organic ligands adopts different conformations (Fig. 26).91 CO2 sorption at 195 K shows an uptake of 8.4 wt% (43.5 cm3 g−1). The MCE values are not very high, as expected for a porous system, with calculated values of −ΔSm, obtained through magnetization data, of 17.25 J kg−1 K−1 at 3 K for ΔB = 7 T.
 | ||
| Fig. 26 (a) Structural view of [Gd2(pam)3(DMF)2(H2O)2]n; (b) ΔSm calculated by using the magnetization data at different fields and temperatures. Reprinted with permission from ref. 91, Copyright 2014 American Chemical Society. | ||
These low values of ΔSm have been improved, in combination with porosity, by using lanthanoid clusters as nodes of MOFs. Specifically, two MOFs based on Ln5 clusters, {[Ln5Zn(BPDC)3-H2O]10(μ3-OH)6(CO3)0.5(NO3)4·mH2O}n (BPDC = 4,4′-dicarboxylate-2,2′-dipyridine anion; Ln = Gd, m = 12; Dy, m = 10) were investigated.92 These compounds are cationic heterometal–organic framework composed of [Ln5Zn(BPDC)3(H2O)10(OH)6]5+ units, which are formed by pentanuclear clusters of formula [Ln5(H2O)10(OH)6]9+ which are further linked by [Zn(BPDC)3]4− units, forming a three-dimensional structure with triangular channels with a diameter of ca. 5.2 Å along the c direction (Fig. 27). The argon isotherm of the dehydrated Gd derivative shows an uptake of 38.79 cm3 g−1 at approximately 1 bar and 87 K, thus showing the presence of porosity. The MCE presents maximum values of −ΔSm of 30.7 J kg−1 K−1 and 10.8 J kg−1 K−1 for the Gd (at 3 K) and the Dy (at 4 K) derivatives, respectively (for ΔB = 7 T), thus showing the successful combination of porosity with the MCE effect.
 | ||
| Fig. 27 (a) 3D framework and channel along the c direction and schematic representation of the topology: black and white represent [Ln5(H2O)10(OH)6]9+ clusters and [Zn(BPDC)3]4− units, respectively; (b) calculated ΔSm for the pentanuclear-based MOFs (Gd derivative shown on top, Dy derivative shown at the bottom) at various fields (1–7 T). Adapted from ref. 92, with permission from John Wiley and Sons. | ||
To sum up, although some MOFs presenting MCE have been reported, the values are still far from those of other current state-of-the-art molecule-based materials. For example, the dense layered material Gd2(OH)5Cl1.5H2O has −ΔSm of 51.9 J kg−1 K−1 for ΔB = 7 T.93 The low values found in MOFs is a consequence of the intrinsic characteristics of these materials, in particular their low magnetic density. Thus, despite their high chemical stability, which is the major advantage of using MOFs for magnetic refrigeration, their real use is not very promising.
3. Hybrid MOFs incorporating functional molecules in the channels
The presence of a periodic array of empty channels in MOFs can structurally control the positioning of multiple functional guests. In this sense, porous materials capable of absorbing and orienting guest molecules through the use of networked cages have been recently developed as a new strategy for the structural determination of exotic molecules otherwise unachievable.94 Furthermore, MOFs have also been exploited for the encapsulation of active species,95 which can be as varied as chromophores,96–98 drugs,99 CO releasing molecules,100 catalysts,101–105 or nanoparticles.106–108 Using this hybrid approach two-network solids can be prepared through the self-assembly of different molecular fragments (organic, inorganic, or organometallic) used as starting building blocks, or using a post-synthetic method in which a molecular guest is inserted into a pre-formed extended network acting as the host lattice (Fig. 28). As pointed out in the introduction, when the MOF is also functional this hybrid approach can afford the isolation of multifunctional materials (Fig. 1c). In this section, we will focus on these multifunctional hybrids, in which each network furnishes a solid with distinct properties. As guest molecules we will show the insertion of magnetic molecules (spin-crossover complexes and single-molecule magnets) and electro-active molecules (electron donors and acceptors). | ||
| Fig. 28 Schematic representation of different functional magnetic guests that can be incorporated into MOFs: (a) spin-crossover complexes, and (b) single-molecule magnets. | ||
3.1. SCO@MOFs
Oxalate-based bimetallic coordination polymers of general formula [MIIMIII(ox)3]− are archetypical examples of 2D and 3D magnetic networks, which can host a wide variety of functional cations to give rise to hybrid materials exhibiting multifunctional properties. Our group has extensively exploited this feature to combine cooperative magnetism with other properties such as electrical conductivity, paramagnetism, single-molecule magnetism and spin crossover.109 The synthetic approach to obtain these hybrid materials involves a self-assembly method that consists in growing the extended network from its molecular precursors, [MIII(ox)3]3− and M2+(aq), in the presence of the functional cations, which act as templates to stabilize the dimensionality of the network (2D or 3D). Using this method many examples of hybrid materials with coexistence of magnetic ordering and spin-crossover have been successfully prepared,110 as well as using diamagnetic frameworks.111 The most interesting properties have been observed in the 2D compounds [FeIII(sal2-trien)][MnIICrIII(ox)3]·X (X = CHBr3, CHCl3, CH2Br2 and CH2Cl2) which, apart from ferromagnetism and spin-crossover, also exhibit a photo-induced spin transition (i.e., a light-induced excited spin trapping (LIEEST) effect).112,113Another approach that has also been successfully used to incorporate spin-crossover complexes within a magnetic MOF involves the so-called post-synthetic method (already pointed out in Section 2.3). The first example was obtained through a solid-state incorporation of the [FeIII(sal2-trien)]+ complex into the large pores (ca. 2.2 nm) of a MnIICuII 3D MOF of formula Na4{Mn4[Cu2(Me3pba)2]3}·60H2O (Fig. 29).114 Interestingly, the magnetic properties of the MnIICuII MOF change upon insertion of the spin-crossover complex (the critical temperature increases from 14 to 19 K) as a consequence of the strengthening of the antiferromagnetic coupling MnIICuII, which is likely associated with the changes induced in the crystal lattice by the exchange of Na+ cations with the [FeIII(sal2-trien)]+ complex.
 | ||
| Fig. 29 (a) Schematic representation of the SC to SC cation exchange process leading to the hybrid Fe(sal2-trien)@MnIICuII MOF. (b) χMT versus T plot for MnIICuII MOF (blue) and Fe(sal2-trien)@MnIICuII MOF (red). The inset shows the minima and the high temperature region in detail, emphasising the thermal hysteresis loop. The full and empty circles represent the Fe(sal2-trien)@MnIICuII MOF data recorded in the heating and cooling modes respectively. Adapted from ref. 114, with permission from The Royal Society of Chemistry. | ||
3.2. SMM@MOFs
Similar to spin-crossover complexes, metal complexes exhibiting SMM behaviour have been inserted into magnetic coordination polymers. Magnetic bimetallic oxalates have also been used to reach this goal. An appealing example of this kind is provided by the hybrid compound [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·(MeOH)·(CH3CN)2, which is formed by the SMM [MnIII(salen)(H2O)]22+ (salen2− = N,N′-ethylenebis-(salicylideneiminate)) inserted into a ferromagnetic 3D oxalate network [MnIICrIII(ox)3]−.115 Notice that in the two-network compounds containing a magnetic oxalate network the electronic coupling between the two sublattices is usually very small and therefore a lack of interplay between the two properties has always been observed. Interestingly, this compound is an exception to this rule. Thus, the magnetic coupling between the two spin sublattices leads to an antiparallel arrangement between the magnetization of the oxalate lattice and that of the inserted SMM. This coupling affects the magnetic relaxation of the SMM, but more interestingly it also affects the magnetic behavior of the ferromagnetic lattice. In fact, in contrast to the rest of the hybrid materials containing the MnCr oxalate lattice, which behave as soft magnets, the coupling of this soft magnetic lattice with the highly anisotropic SMM leads to a drastic enhancement of the coercive field of the hybrid (from 10 to ca. 800 G), which behaves as a permanent magnet below 5 K (Fig. 30). Although limited to low temperatures, this situation resembles that found in alloys of 3d metals and lanthanide ions, like NdFe14B, that are among the best permanent magnets known to date, but has the advantage of using SMMs based on d-transition metal complexes instead of lanthanide ions as a source of magnetic anisotropy. | ||
| Fig. 30 (a) Crystal structure of [MnIII(salen)(H2O)]2[MnIICrIII(ox)3]2·(MeOH)3·(CH3CN). Hydrogen atoms and disordered solvent molecules have been omitted for clarity. (b) Magnetization hysteresis loops of the hybrid SMM@MnCr (blue), the SMM in a diamagnetic network (black) and MnCr (red) measured at T = 0.43 K. The sweeping field rate was 6 mT s−1. Adapted from ref. 115, with permission from John Wiley and Sons. | ||
The above example uses a self-assembly method to form the hybrid material. As pointed out above, the porosity and stability of some MOFs can be useful for loading molecules into their pores using post-synthetic procedures. These methods have also been used to incorporate SMMs within both magnetic and non-magnetic MOFs. In the former case, the presence of a MnIII porphyrin, which is a magnetic molecule behaving as a SIM, in a MnIICuII 3D magnetic MOF leads to an interplay of the magnetic properties.116 Thus, the internal magnetic field created by the long-range magnetic ordering of the MOF seems to have an influence on the slow magnetic relaxation of the SIM leading to an attenuation of the quantum tunneling of the magnetization in the mononuclear complex. Non-magnetic MOFs have also been used to accommodate the magnetic molecules with the aim of creating isolated nanostructures of these nanomagnets. An example has been the incorporation of Mn12 acetate into the mesoporous aluminium-based MOF [Al(OH)(SDC)]n (H2SDC = 4.4′-stilbenedicarboxylic acid).117 The chemically robust magnetic polyoxometalate [(FeW9O34)2Fe4(H2O)2]10− has been inserted into both diamagnetic and antiferromagnetic MOFs (UiO-67 and MIL-101(Cr), respectively).118 In the diamagnetic matrix the SMM behavior is retained because the magnetic anisotropy of the POMs is not altered by their interaction with the host. Differently, in the antiferromagnetic matrix the SMM character is clearly reduced because the magnetic interactions lead to faster relaxation of the magnetization.
To conclude this part we can say that magnetic molecules exhibiting a SMM behavior can be incorporated into the pores of a MOF using both self-assembly and post-synthetic methods. By using a diamagnetic MOF this approach has allowed keeping the SMMs isolated, leading to controlled nanostructures that retain the SMM behavior of the pure compound. In contrast, when the MOF matrix is magnetic, some changes in the SMM behavior are observed due to the weak interactions established between the two networks. However, with one exception, these interactions are too weak to affect the properties of the magnetic MOF.
3.3. Electroactive molecules@MOFs
Polyoxometalates (POMs) form a class of molecular anions that have been incorporated into MOFs. These metal-oxide clusters are robust species with unique electronic properties owing to their ability to act as electron reservoirs or to accommodate magnetic centers. In previous sections we have already shown some examples that illustrate these possibilities.67,68,97 In view of the ability of POMs to act as heterogeneous catalysts, their insertion and dispersion into the pores of a MOF can be beneficial in order to increase the active surface area of the POM and consequently its catalytic properties, while enhancing also the stability of the resulting POM-MOF material thanks to the host–guest interactions established between the two sublattices. In this context MOFs have shown to be more suitable to disperse POMs than other host matrices such as silica, activated carbon, ion-exchange resins and mesoporous molecular sieves, which often lead to low POM loading, POM leaching, agglomeration of POM particles and ill-defined solids. Some examples that illustrate this concept are the following: (i) the immobilization of the protonated Keggin POMs [HnXM12O40]m− (X = Si, Ge, P, As; M = Mo, W) into the MOF Cu-BTC (BTC = benzenetricarboxylate) for hydrolysis of esters;119 (ii) the incorporation of the Keggin POM [PW12O40]3−, used as a template, in a sodalite-type MOF. This POM-MOF has displayed its potential application in the removal and decomposition of the nerve gas dimethyl methylphosphonate;120 (iii) the incorporation of [CuPW11O39]5− into the pores of HKUST-1 for air-based oxidations;121 (iv) the incorporation of POMs K4[PW11VO40], H3[PW12VO40] and K4[SiW12VO40] in the MOF MIL-101 for selective adsorption of cationic dyes;122 (v) the encapsulation of POMs within the large pores of the Zr(IV) biphenyldicarboxylate UiO-67 MOF.123 These examples illustrate the use of electronically innocent MOFs for dispersing electroactive molecules in their pores (Fig. 1b). In this case, the MOFs exclusively play a structural role.Another electroactive molecule that has been incorporated into the MOFs is the TCNQ, which is a well-known organic acceptor able to form low dimensional chain structures showing conductive properties. This molecule has been incorporated into the channels of the HKUST-1 MOF to give rise to a hybrid material showing an enhanced electrical conductivity from 10−6 S m−1 for the as-synthesised material to 7 S m−1 for the TCNQ loaded MOF.124
4. Concluding remarks
In this review we have discussed the new opportunities that can appear in the crossroad between molecular magnetism and MOFs. Thus, the different types of multifunctional materials that can be encountered when combining magnetism with porosity have been discussed. In the first part, we have shown with three different classes of magnetic MOFs that the incorporation of magnetic centers into the coordination framework can result in the appearance of new properties in the crystalline MOF. In particular, this concept has provided a new generation of stimuli-responsive materials in which the magnetism can be tuned by the presence of molecular species in the pores (see the first and second class of MOFs, vide infra), or the possibility of keeping the magnetic centers well insulated in a crystalline framework (see the third class of MOFs, vide infra).The first class of magnetic MOFs shows cooperative magnetic properties below a given critical temperature, Tc. The challenge with these materials is to shift Tc to temperatures as high as possible, a goal which is difficult to achieve since porosity and cooperative magnetism are two inimical properties that require opposite structural features (large porosity requires the use of long linkers between the metal centres, while strong exchange interactions require the use of short ones). In fact, this “magnetic exchange” approach is seriously hindered by the relatively low temperatures at which the cooperative magnetism appears (typically much below 100 K).
To overcome this limitation, a second class of magnetic MOFs based on “spin-crossover complexes” has been proposed as the spin transition typically occurs near room temperature. In that case, cooperativity is controlled by elastic forces in the lattice, which can be maintained at long metal–metal distances thanks to the polymeric nature of the MOF lattice. In fact, it has been shown that the structural changes occurring upon uptake/release of a guest molecule can be detected in these spin-crossover MOFs, while the cooperativity of this phenomenon is maintained for small pore sizes (typically in the nanometer range). Notice that this molecular confinement has been shown to be beneficial to enhance the interaction between adsorbed gas molecules and the framework. This feature has resulted in a shift in the spin transition temperature upon gas sorption, which, in some cases, can be accompanied by selectivity in gas sorption.
A third class of magnetic MOFs that has been reported contains magnetic centres at the nodes of the framework. In that case, the interest is to generate magnetic nanostructures which are well organized and well insulated in space thanks to the structural features of the MOF. Since these magnetic centres may behave as single-molecule magnets, their controlled organization in space could find an application in quantum computing since each magnetic centre may be used as a quantum bit. In addition, the good magnetic insulation provided by the MOF has shown to be useful in obtaining isotropic paramagnetic materials which can be of interest as magnetic coolers.
All the examples reported in this part have exploited the possibility of incorporating the magnetism at the nodes of the framework. Still, the porosity offered by these coordination polymers can also allow us to fill the pores with functional molecules. This possibility is discussed in the second part of the review where hybrid functional MOFs combining an extended lattice with a molecular lattice are presented. These hybrids have been used as an ideal platform to create new multifunctional materials covering from the simple co-existence of different properties, provided by the framework and the functional guest, to a synergy between these functionalities as a result of the coupling between the two components. Thus, in that part different types of functional molecules have been introduced into the channels, including magnetic molecules (spin-crossover and single-molecule magnets) as well as electro-active molecules (electron donors and acceptors, for example). The result of such a combination has given rise in some cases to an improvement in the properties of the hybrid MOFs, making them of interest in magnetism (lanthanide-free hard magnets), heterogeneous catalysis (catalysts with enhanced surface areas) or electronics (MOFs with semiconducting properties).
5. Perspectives
To finish we should mention some of the current challenges and perspectives in the field of magnetic MOFs. The incorporation of magnetic functionality into a MOF is quite recent, so that at this stage most of the studies in this area are focused on the synthesis of materials in bulk, with an analysis of the crystal structures and properties. The field has to move on to the processing of these materials towards applications, including, among others, their use as sensors, in electronic applications, in magnetic refrigeration, or in quantum computing. For example, the study of the quantum coherence in SMM-MOFs is at a very early stage, and the integration of these qubits into superconducting circuits would be the next stage in order to manipulate the spin state. For some of these applications it is crucial to process the materials, and to not only use single crystals, or polycrystalline materials, which are currently the goal of chemists in order to get unequivocal information regarding the structural aspects of the new materials. Thus, magnetic MOFs should be processed as films, nanoparticles, or nanostructures, while retaining the properties.The initial studies on this matter have already been done in films of coordination polymers based on spin-crossover, where thick films have been prepared.125,126 Despite their roughness, these films can be used as sensors, but their application is unfeasible in nano- and micro-electronics (including spintronics) as these applications require films of higher quality. In this context, a current trend in this area is that of integrating these MOFs as ultrathin films onto solid surfaces (SURMOFs). The first generation of SURMOFs has given rise to the emergence of new MOF-based devices of interest as separation membranes, catalytic coatings, or sensors that exploit their porosity as an active function.127 In the context of MOFs with electronic functionalities, an appealing issue is that of exploiting not only their porosity but also these electronic functionalities in order to use these films as active interfaces in electronic molecular-based devices. Most properties of these devices are crucially determined by these interfaces so that an active manipulation of these interfaces is the key to improve the device performance. This second generation of SURMOFs is chemically much more demanding than the first one because it requires the organization of continuous, high-quality and oriented ultrathin films (below 10–20 nm) of these materials on surfaces. Furthermore, it is also very demanding from the electronic point of view since it requires tailoring the degree of interaction between the SURMOF and the inorganic surface as that generally impacts the electronic properties of the hybrid interface and the functioning of the entire device. Still, the accomplishment of these challenging goals can open new perspectives in the use of MOFs in molecular electronics and spintronics. Here, the transport of charge carriers as well as the spin injection across the MOF interface could be manipulated by loading of molecular guests in the porous scaffold. In addition, the open structures of these crystalline materials display higher surface areas for direct contact with the molecules than those offered by a normal 2D surface, thus improving the interactions between these molecules and the inorganic surface. These complex hybrid interfaces, integrated by an open framework and molecular guests, are linked to a new idea of assembled 3D interfaces with addressable functionality, which may generate conceptually new hybrid devices. Along this line, the possibility of incorporating molecules with different functionalities (magnetism, bistability, luminescence, etc.) inside the MOF, or increasing the intrinsic conductivity of the framework could enlarge the versatility of these complex interfaces.
A recent result that exemplifies the fabrication of these electronically active MOF interfaces has been recently reported.128 The approach involves a sequential layer-by-layer transfer of 2D preassembled MOF nanosheets, organized by using the Langmuir–Blodgett method, to SAM-functionalized substrates. As a model system, a layered MOF built from the sequential stacking of 2D networks of Co(II) tetracarboxylate porphyrin units interconnected by Cu(II) ions (NAFS-1) has been used (Fig. 31). In contrast to previous methodologies, SAM-assisted transfer enables the fabrication of homogeneous, highly oriented, ultrathin films across millimeter-scale areas, regardless of the substrate. This approach has allowed transfer of NAFS-1 onto a nonconventional, ferromagnetic substrate such as permalloy. Owing to the high quality and low-thickness (<10 nm) of these SURMOFs, it has been possible to investigate their electrical properties and evaluate the effect of the thickness of the MOF on the conductance using Hg drop micrometric electrode junctions. This result illustrates that this liquid-phase layer-by-layer method can be a useful approach to fabricate electronic interfaces based on magnetic SURMOFs. This kind of approach has also shown to be useful in preparing thin films of good optical quality based on Hofmann clathrates exhibiting spin crossover.124,125,129
 | ||
| Fig. 31 NAFS-1 structure (a) and Langmuir–Blodgett (LB) transfer process (b). Reprinted with permission from ref. 128, Copyright 2016 American Chemical Society. | ||
A different approach that can be foreseen to improve the quality of these ultrathin films is based on the deposition of two-dimensional (2D) crystals on electronic/magnetic surfaces which can provide very clean interfaces of direct application in electronics/spintronics, as has already been shown with graphene and graphene-like 2D materials.130 This goal can be achieved either by using chemical vapour deposition (CVD) methods, or by direct micromechanical exfoliation of the bulk materials and subsequent transfer of the 2D layer onto the substrate. The first approach (CVD method), although very promising, has been exploited only very recently in MOF chemistry for the preparation of ZIF-8.131 This archetypical MOF has served to demonstrate the feasibility of CVD but its electronic properties are rather limited. The second approach (micromechanical exfoliation method) has not yet been reported in MOF chemistry. The only examples are limited to the use of liquid exfoliation methods,132,133 which lack the high quality requirements for electronic applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish MINECO (Unit of Excellence María de Maeztu MDM-2015-0538, CTQ2014-59209-P and MAT2014-56143-R), the Generalitat Valenciana (Prometeo programme), the EU (ERC-2016-CoG 724681-S-CAGE) and the VLC/Campus Program is gratefully acknowledged. G. M. E. acknowledges Spanish MINECO for a Ramón y Cajal fellowship.References
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed
.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS
- S. S. Park, E. R. Hontz, L. Sun, C. H. Hendon, A. Walsh, T. Van Voorhis and M. Dinca, J. Am. Chem. Soc., 2015, 137, 1774–1777 CrossRef CAS PubMed
- L. Sun, M. G. Campbell and M. Dinca, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
- L.-J. Xu, G.-T. Xu and Z.-N. Chen, Coord. Chem. Rev., 2014, 273–274, 47–62 CrossRef CAS
- Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273–274, 76–86 CrossRef CAS
-
(a) J. M. Manriquez, G. T. Yee, R. S. McLean, A. J. Epstein and J. S. Miller, Science, 1991, 252, 1415–1417 CAS
- M. Clemente-León, E. Coronado, C. Martí-Gastaldo and F. M. Romero, Chem. Soc. Rev., 2011, 40, 473–497 RSC
- E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and V. Laukhin, Nature, 2000, 408, 447–449 CrossRef CAS PubMed
- E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419–5448 CrossRef CAS PubMed
- P. Guetlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef CAS PubMed
- S. Ohkoshi and H. Tokoro, Acc. Chem. Res., 2012, 45, 1749–1758 CrossRef CAS PubMed
- T. Nuida, T. Matsuda, H. Tokoro, S. Sakurai, K. Hashimoto and S. Ohkoshi, J. Am. Chem. Soc., 2005, 127, 11604–11605 CrossRef CAS PubMed
- E. Coronado and G. Mínguez Espallargas, Chem. Soc. Rev., 2013, 42, 1525–1539 RSC
- E. Coronado, M. Giménez-Marqués, G. Mínguez Espallargas and L. Brammer, Nat. Commun., 2012, 3, 828 CrossRef PubMed
- P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625 CrossRef CAS PubMed
- G.-C. Xu, W. Zhang, X.-M. Ma, Y.-H. Chen, L. Zhang, H.-L. Cai, Z.-M. Wang, R.-G. Xiong and S. Gao, J. Am. Chem. Soc., 2011, 133, 14948–14951 CrossRef CAS PubMed
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed
- Z. M. Wang, B. Zhang, H. Fujiwara, H. Kobayashi and M. Kurmoo, Chem. Commun., 2004, 416 RSC
- B. Zhang, Z.-M. Wang, M. Kurmoo, S. Gao, K. Inoue and H. Kobayashi, Adv. Funct. Mater., 2007, 17, 577–584 CrossRef CAS
- Z.-M. Wang, B. Zhang, M. Kurmoo, M. A. Green, H. Fujiwara, T. Otsuka and H. Kobayashi, Inorg. Chem., 2005, 44, 1230–1237 CrossRef CAS PubMed
- M. Viertelhaus, P. Adler, R. Clérac, C. E. Anson and A. K. Powell, Eur. J. Inorg. Chem., 2005, 692–703 CrossRef CAS
- D. N. Dybtsev, H. Chun, S. H. Yoon, D. Kim and K. Kim, J. Am. Chem. Soc., 2004, 126, 32–33 CrossRef CAS PubMed
- Z. Wang, Y. Zhang, T. Liu, M. Kurmoo and S. Gao, Adv. Funct. Mater., 2007, 17, 1523–1536 CrossRef CAS
- Z. Wang, K. Hu, S. Gao and H. Kobayashi, Adv. Mater., 2010, 22, 1526–1533 CrossRef CAS PubMed
-
M. Verdaguer and G. S. Girolami, Magnetic Prussian Blue Analogs, in Magnetism: Molecules to Materials V, ed. J. S. Miller and M. Drillon, Wiley-VCH, 2005, p. 283 Search PubMed
-
M. Shatruk, S. Gómez-Coca and K. R. Dunbar, Molecular Magnetism, in Molecular Magnetic Materials: Concepts and Applications, ed. B. Sieklucka and D. Pinkowicz, Wiley-VCH, 2016 Search PubMed
- L. G. Beauvais and J. R. Long, J. Am. Chem. Soc., 2002, 124, 12096–12097 CrossRef CAS PubMed
- S. S. Kaye, H. J. Choi and J. R. Long, J. Am. Chem. Soc., 2008, 130, 16921–16925 CrossRef CAS PubMed
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed
- C. Pettinari, A. Tabacaru and S. Galli, Coord. Chem. Rev., 2016, 307, 1–31 CrossRef CAS
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed
- Y.-Q. Tian, C.-X. Cai, Y. Ji, X.-Z. You, S.-M. Peng and G.-H. Lee, Angew. Chem., Int. Ed., 2002, 41, 1384–1386 CrossRef CAS PubMed
- Y.-Q. Tian, C.-X. Cai, X.-M. Ren, C.-Y. Duan, Y. Xu, S. Gao and X.-Z. You, Chem. – Eur. J., 2003, 9, 5673–5685 CrossRef CAS PubMed
- Y.-Q. Tian, Z.-X. Chen, L.-H. Weng, H.-B. Guo, S. Gao and D. Y. Zhao, Inorg. Chem., 2004, 43, 4631–4635 CrossRef CAS PubMed
- S. S. Mondal, A. Bhunia, S. Demeshko, A. Kelling, U. Schilde, C. Janiak and H.-J. Holdt, CrystEngComm, 2014, 16, 39–42 RSC
- M. Wriedt, A. A. Yakovenko, G. J. Halder, A. V. Prosvirin, K. R. Dunbar and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 4040–4050 CrossRef CAS PubMed
- J. A. R. Navarro, E. Barea, A. Rodríguez-Diéguez, J. M. Salas, C. O. Ania, J. B. Parra, N. Masciocchi, S. Galli and A. Sironi, J. Am. Chem. Soc., 2008, 130, 3978–3984 CrossRef CAS PubMed
- M.-H. Zeng, Z. Yin, Y.-X. Tan, W.-X. Zhang, Y.-P. He and M. Kurmoo, J. Am. Chem. Soc., 2014, 136, 4680–4688 CrossRef CAS PubMed
- P. D. C. Dietzel, Y. Morita, R. Blom and H. Fjellvåg, Angew. Chem., Int. Ed., 2005, 44, 6354–6358 CrossRef CAS PubMed
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606 CrossRef CAS PubMed
- J. Park, H. Kim and Y. Jung, J. Phys. Chem. Lett., 2013, 4, 2530–2534 CrossRef CAS
- Q. Zhang, B. Li and L. Chen, Inorg. Chem., 2013, 52, 9356 CrossRef CAS PubMed
- H. Wu, J. M. Simmons, G. Srinivas, W. Zhou and T. Yildirim, J. Phys. Chem. Lett., 2010, 1, 1946–1951 CrossRef CAS
- D. Yu, A. O. Yazaydin, J. R. Lane, P. D. C. Dietzel and R. Q. Snurr, Chem. Sci., 2013, 4, 3544–3556 RSC
- M. C. Das, S. Xiang, Z. Zhang and B. Chen, Angew. Chem., Int. Ed., 2011, 50, 10510–10520 CrossRef CAS PubMed
- T. Grancha, J. Ferrando-Soria, M. Castellano, M. Julve, J. Pasán, D. Armentano and E. Pardo, Chem. Commun., 2014, 50, 7569–7585 RSC
- J. Ferrando-Soria, P. Serra-Crespo, M. de Lange, J. Gascon, F. Kapteijn, M. Julve, J. Cano, F. Lloret, J. Pasán, C. Ruiz-Pérez, Y. Journaux and E. Pardo, J. Am. Chem. Soc., 2012, 134, 15301–15304 CrossRef CAS PubMed
- T. B. Faust and D. M. D'Alessandro, RSC Adv., 2014, 4, 17498–17512 RSC
- D. Maspoch, D. Ruiz-Molina, K. Wurst, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190–195 CrossRef CAS PubMed
- N. Roques, D. Maspoch, I. Imaz, A. Datcu, J.-P. Sutter, C. Rovira and J. Veciana, Chem. Commun., 2008, 3160–3162 RSC
- A. Datcu, N. Roques, V. Jubera, D. Maspoch, X. Fontrodona, K. Wurst, I. Imaz, G. Mouchaham, J.-P. Sutter, C. Rovira and J. Veciana, Chem. – Eur. J., 2012, 18, 152–162 CrossRef CAS PubMed
- N. Motokawa, S. Matsunaga, S. Takaishi, H. Miyasaka, M. Yamashita and K. R. Dunbar, J. Am. Chem. Soc., 2010, 132, 11943–11951 CrossRef CAS PubMed
- I.-R. Jeon, B. Negru, R. P. Van Duyne and T. D. Harris, J. Am. Chem. Soc., 2015, 137, 15699–15702 CrossRef CAS PubMed
-
Spin-crossover materials properties and applications, ed. M. Halcrow, Wiley, 2013 Search PubMed
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC
- R. Ohtani and S. Hayami, Chem. – Eur. J., 2017, 23, 2236–2248 CrossRef CAS PubMed
- P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef CAS PubMed
- M. Ohba, K. Yoneda, G. Agustí, M. C. Muñoz, A. B. Gaspar, J. A. Real, M. Yamasaki, H. Ando, Y. Nakao, S. Sakaki and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4767–4771 CrossRef CAS PubMed
- E. Coronado, M. Giménez-Marqués, G. Mínguez Espallargas, F. Rey and I. J. Vitórica-Yrezábal, J. Am. Chem. Soc., 2013, 135, 15986–15989 CrossRef CAS PubMed
- M. Giménez-Marqués, N. Calvo Galve, M. Palomino, S. Valencia, F. Rey, G. Sastre, I. J. Vitórica-Yrezábal, M. Jiménez-Ruiz, J. A. Rodríguez-Velamazán, M. A. González, J. L. Jordá, E. Coronado and G. Mínguez Espallargas, Chem. Sci., 2017, 8, 3109–3120 RSC
- N. Calvo Galve, M. Giménez-Marqués, M. Palomino, S. Valencia, F. Rey, G. Mínguez Espallargas and E. Coronado, Inorg. Chem. Front., 2016, 3, 808–813 RSC
- M. C. Muñoz and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068–2093 CrossRef
- Z. Arcís-Castillo, F. J. Muñoz-Lara, M. C. Muñoz, D. Aravena, A. B. Gaspar, J. F. Sánchez-Royo, E. Ruiz, M. Ohba, R. Matsuda, S. Kitagawa and J. A. Real, Inorg. Chem., 2013, 52, 12777–12783 CrossRef PubMed
- G. Agustí, R. Ohtani, K. Yoneda, A. B. Gaspar, M. Ohba, J. F. Sánchez-Royo, M. C. Muñoz, S. Kitagawa and J. A. Real, Angew. Chem., Int. Ed., 2009, 48, 8944–8947 CrossRef PubMed
- R. Ohtani, K. Yoneda, S. Furukawa, N. Horike, S. Kitagawa, A. B. Gaspar, M. C. Muñoz, J. A. Real and M. Ohba, J. Am. Chem. Soc., 2011, 133, 8600–8605 CrossRef CAS PubMed
- J. E. Clements, J. R. Price, S. M. Neville and C. J. Kepert, Angew. Chem., Int. Ed., 2014, 53, 10164–10168 CrossRef CAS PubMed
-
A. D. Katsenis, E. K. Brechin and G. S. Papaefstathiou, Metal-Organic Frameworks from Single-Molecule Magnets, in Metal-Organic Framework Materials – Encyclopedia of Inorganic and Bioinorganic Chemistry, 2014, pp. 245–258 Search PubMed
- I.-R. Jeon and R. Clerac, Dalton Trans., 2012, 41, 9569 RSC
- H. Miyasaka, K. Nakata, K.-i. Sugiura, M. Yamashita and R. Clérac, Angew. Chem., Int. Ed., 2004, 43, 707 CrossRef CAS PubMed
- H. Miyasaka, K. Nakata, L. Lecren, C. Coulon, Y. Nakazawa, T. Fijisaki, K.-i. Sugiura, M. Yamashita and R. Clérac, J. Am. Chem. Soc., 2006, 128, 3770 CrossRef CAS PubMed
- M. Murrie, S. J. Teat, H. Stoeckli-Evans and H. Güdel, Angew. Chem., Int. Ed., 2003, 42, 4653 CrossRef CAS PubMed
- X. Jiang, C.-M. Liu and H.-Z. Kou, Inorg. Chem., 2016, 55, 5880–5885 CrossRef CAS PubMed
- J. J. Baldoví, E. Coronado, A. Gaita-Ariño, C. Gamer, M. Giménez-Marqués and G. Mínguez Espallargas, Chem. – Eur. J., 2014, 20, 10695–10702 CrossRef PubMed
- J. López-Cabrelles, G. Mínguez Espallargas and E. Coronado, Polymers, 2016, 8, 171 CrossRef
-
J. M. Clemente-Juan, E. Coronado and A. Gaita Ariño, in Lanthanides and Actinides in Molecular Magnetism, ed. R. Layfield and M. Murugesu, 2015, pp. 27–60 Search PubMed
- I. Oyarzabal, B. Fernández, J. Cepeda, S. Gómez-Ruiz, A. J. Calahorro, J. M. Seco and A. Rodríguez-Diéguez, CrystEngComm, 2016, 18, 3055–3063 RSC
- Q. Chen, J. Li, Y.-S. Meng, H.-L. Sun, Y.-Q. Zhang, J.-L. Sun and S. Gao, Inorg. Chem., 2016, 55, 7980–7987 CrossRef PubMed
- K. Liu, H. Li, X. Zhang, W. Shi and P. Cheng, Inorg. Chem., 2015, 54, 10224–10231 CrossRef CAS PubMed
- J. Vallejo, F. R. Fortea-Pérez, E. Pardo, S. Benmansour, I. Castro, J. Krzystek, D. Armentano and J. Cano, Chem. Sci., 2016, 7, 2286–2293 RSC
- J. M. Zadrozny, A. T. Gallagher, T. D. Harris and D. E. Freedman, J. Am. Chem. Soc., 2017, 139, 7089–7094 CrossRef CAS PubMed
- M. Shiddiq, D. Komijani, Y. Duan, A. Gaita-Ariño, E. Coronado and S. Hill, Nature, 2016, 531, 348–351 CrossRef CAS PubMed
- M. Evangelisti, E. Manuel, M. Affronte, M. Okubo, C. Train and M. Verdaguer, J. Magn. Magn. Mater., 2007, 316, e569–e571 CrossRef CAS
- E. Manuel, M. Evangelisti, M. Affronte, M. Okubo, C. Train and M. Verdaguer, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 172406 CrossRef
- N. Sharma, S. M. Yusuf, A. Kumar and J. V. Yakhmi, AIP Conf. Proc., 2008, 1003, 8 CrossRef CAS
- S. M. Yusuf, A. Kumar and J. V. Yakhmi, Appl. Phys. Lett., 2009, 95, 182506 CrossRef
- R. Sibille, T. Mazet, B. Malaman and M. François, Chem. – Eur. J., 2012, 18, 12970–12973 CrossRef CAS PubMed
- L. Sedláková, J. Hanko, A. Orendácová, M. Orendác, C.-L. Zhou, W.-H. Zhu, B.-W. Wang, Z.-M. Wang and S. Gao, J. Alloys Compd., 2009, 487, 425–429 CrossRef
- S. Biswas, H. S. Jena, S. Goswami, S. Sanda and S. Konar, Cryst. Growth Des., 2014, 14, 1287–1295 CAS
- P.-F. Shi, Y.-Z. Zheng, X.-Q. Zhao, G. Xiong, B. Zhao, F.-F. Wan and P. Cheng, Chem. – Eur. J., 2012, 18, 15086–15091 CrossRef CAS PubMed
- G. Abellán, G. Mínguez Espallargas, G. Lorusso, M. Evangelisti and E. Coronado, Chem. Commun., 2015, 51, 14207–14210 RSC
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 CrossRef CAS PubMed
- J. Juan-Alcañiz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102–10118 RSC
- Y.-Q. Lan, H.-L. Jiang, S.-L. Li and Q. Xu, Inorg. Chem., 2012, 51, 7484–7491 CrossRef CAS PubMed
- P. Mahato, N. Yanai, M. Sindoro, S. Granick and N. Kimizuka, J. Am. Chem. Soc., 2016, 138, 6541–6549 CrossRef CAS PubMed
- C.-Y. Sun, W.-P. To, X.-L. Wang, K.-T. Chan, Z.-M. Su and C.-M. Che, Chem. Sci., 2015, 6, 7105–7111 RSC
- M. Giménez-Marqués, T. Hidalgo, C. Serre and P. Horcajada, Coord. Chem. Rev., 2016, 307, 342–360 CrossRef
- F. J. Carmona, S. Rojas, P. Sánchez, H. Jeremias, A. R. Marques, C. C. Romão, D. Choquesillo-Lazarte, J. A. R. Navarro, C. R. Maldonado and E. Barea, Inorg. Chem., 2016, 55, 6525–6531 CrossRef CAS PubMed
- R. C. Klet, S. Tussupbayev, J. Borycz, J. R. Gallagher, M. M. Stalzer, J. T. Miller, L. Gagliardi, J. T. Hupp, T. J. Marks, C. J. Cramer, M. Delferro and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 15680–15683 CrossRef CAS PubMed
- A. Grigoropoulos, G. F. S. Whitehead, N. Perret, A. P. Katsoulidis, F. M. Chadwick, R. P. Davies, A. Haynes, L. Brammer, A. S. Weller, J. Xiao and M. J. Rosseinsky, Chem. Sci., 2016, 7, 2037–2050 RSC
- D. Yang, S. O. Odoh, T. C. Wang, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and B. C. Gates, J. Am. Chem. Soc., 2015, 137, 7391–7396 CrossRef CAS PubMed
- T. Kajiwara, M. Fujii, M. Tsujimoto, K. Kobayashi, M. Higuchi, K. Tanaka and S. Kitagawa, Angew. Chem., Int. Ed., 2016, 55, 2697–2700 CrossRef CAS PubMed
- A. Chołuj, A. Zieliński, K. Grela and M. J. Chmielewski, ACS Catal., 2016, 6, 6343–6349 CrossRef
- C. R. Kim, T. Uemura and S. Kitagawa, Chem. Soc. Rev., 2016, 45, 3828–3845 RSC
- C. Rösler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC
- P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev., 2016, 307, 237–254 CrossRef CAS
- M. Clemente-León, E. Coronado, C. Martí-Gastaldo and F. M. Romero, Chem. Soc. Rev., 2011, 40, 473–497 RSC
- M. López-Jordà, M. Giménez-Marqués, C. Desplanches, G. Mínguez Espallargas, M. Clemente-León and E. Coronado, Eur. J. Inorg. Chem., 2016, 2187–2192 CrossRef
- T. Zhao, I. Boldog, V. Spasojevic, A. Rotaru, Y. Garcia and C. Janiak, J. Mater. Chem. C, 2016, 4, 6588–6601 RSC
- M. Clemente-León, E. Coronado, M. López-Jordà, C. Desplanches, S. Asthana, H. Wang and J.-F. Létard, Chem. Sci., 2011, 2, 1121–1127 RSC
- M. Clemente-León, E. Coronado, M. López-Jordà, J. C. Waerenborgh, C. Desplanches, H. Wang, J.-F. Létard, A. Hauser and A. Tissot, J. Am. Chem. Soc., 2013, 135, 8655–8667 CrossRef PubMed
- A. Abhervé, T. Grancha, J. Ferrando-Soria, M. Clemente-León, E. Coronado, J. C. Waerenborgh, F. Lloret and E. Pardo, Chem. Commun., 2016, 52, 7360–7363 RSC
- M. Clemente-León, E. Coronado, C. J. Gómez-García, M. López-Jordà, A. Camón, A. Repollés and F. Luis, Chem. – Eur. J., 2014, 20, 1669–1676 CrossRef PubMed
- M. Mon, A. Pascual-Álvarez, T. Grancha, J. Cano, J. Ferrando-Soria, F. Lloret, J. Gascon, J. Pasán, D. Armentano and E. Pardo, Chem. – Eur. J., 2016, 22, 539–545 CrossRef CAS PubMed
- D. Aulakh, J. B. Pyser, X. Zhang, A. A. Yakovenko, K. R. Dunbar and M. Wriedt, J. Am. Chem. Soc., 2015, 137, 9254–9257 CrossRef CAS PubMed
- W. Salomon, Y. Lan, E. Rivière, S. Yang, C. Roch-Marchal, A. Dolbecq, C. Simonnet-Jégat, N. Steunou, N. Leclerc-Laronze, L. Ruhlmann, T. Mallah, W. Wernsdorfer and P. Mialane, Chem. – Eur. J., 2016, 22, 6564–6574 CrossRef CAS PubMed
- C.-Y. Sun, S.-X. Liu, D.-D. Liang, K.-Z. Shao, Y.-H. Ren and Z.-M. Su, J. Am. Chem. Soc., 2009, 131, 1883–1888 CrossRef CAS PubMed
- F.-J. Ma, S.-X. Liu, C.-Y. Sun, D.-D. Liang, G.-J. Ren, F. Wei, Y.-G. Chen and Z.-M. Su, J. Am. Chem. Soc., 2011, 133, 4178–4181 CrossRef CAS PubMed
- J. Song, Z. Luo, D. K. Britt, H. Furukawa, O. M. Yaghi, K. I. Hardcastle and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 16839–16846 CrossRef CAS PubMed
- A.-X. Yan, S. Yao, Y.-G. Li, Z.-M. Zhang, Y. Lu, W.-L. Chen and E.-B. Wang, Chem. – Eur. J., 2014, 20, 6927–6933 CrossRef CAS PubMed
- W. Salomon, C. Roch-Marchal, P. Mialane, P. Rouschmeyer, C. Serre, M. Haouas, F. Taulelle, S. Yang, L. Ruhlmann and A. Dolbecq, Chem. Commun., 2015, 51, 2972–2975 RSC
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed
- S. Cobo, G. Molnár, J. A. Real and A. Bousseksou, Angew. Chem., Int. Ed., 2006, 45, 5786–5789 CrossRef CAS PubMed
- C. Bartual-Murgui, L. Salmon, A. Akou, C. Thibault, G. Molnár, T. Mahfoud, Z. Sekkat, J. A. Real and A. Bousseksou, New J. Chem., 2011, 35, 2089–2094 RSC
- B. Liu, J. Mater. Chem., 2012, 22, 10094–10101 RSC
- V. Rubio-Giménez, S. Tatay, F. Volatron, F. J. Martínez-Casado, C. Martí-Gastaldo and E. Coronado, J. Am. Chem. Soc., 2016, 138, 2576–2584 CrossRef PubMed
- S. Sakaida, K. Otsubo, O. Sakata, C. Song, A. Fujiwara, M. Takata and H. Kitagawa, Nat. Chem., 2016, 8, 377–383 CrossRef CAS PubMed
- D. Jariwala, T. Marks and M. C. Hersam, Nat. Mater., 2017, 16, 170–181 CrossRef CAS PubMed
- I. Stassen, M. Styles, G. Grenci, H. Van Gorp, W. Vanderlinden, S. De Feyter, P. Falcaro, D. De Vos, P. Vereecken and R. Ameloot, Nat. Mater., 2016, 5, 304–310 Search PubMed
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356–1359 CrossRef CAS PubMed
- J. A. Foster, S. Henke, A. Schneemann, R. A. Fischer and A. K. Cheetham, Chem. Commun., 2016, 52, 10474–10477 RSC
Footnote |
| † Themed issue of Chemical Society Reviews on “Metal–organic frameworks and porous polymers – current and future challenges”. |
| This journal is © The Royal Society of Chemistry 2018 |