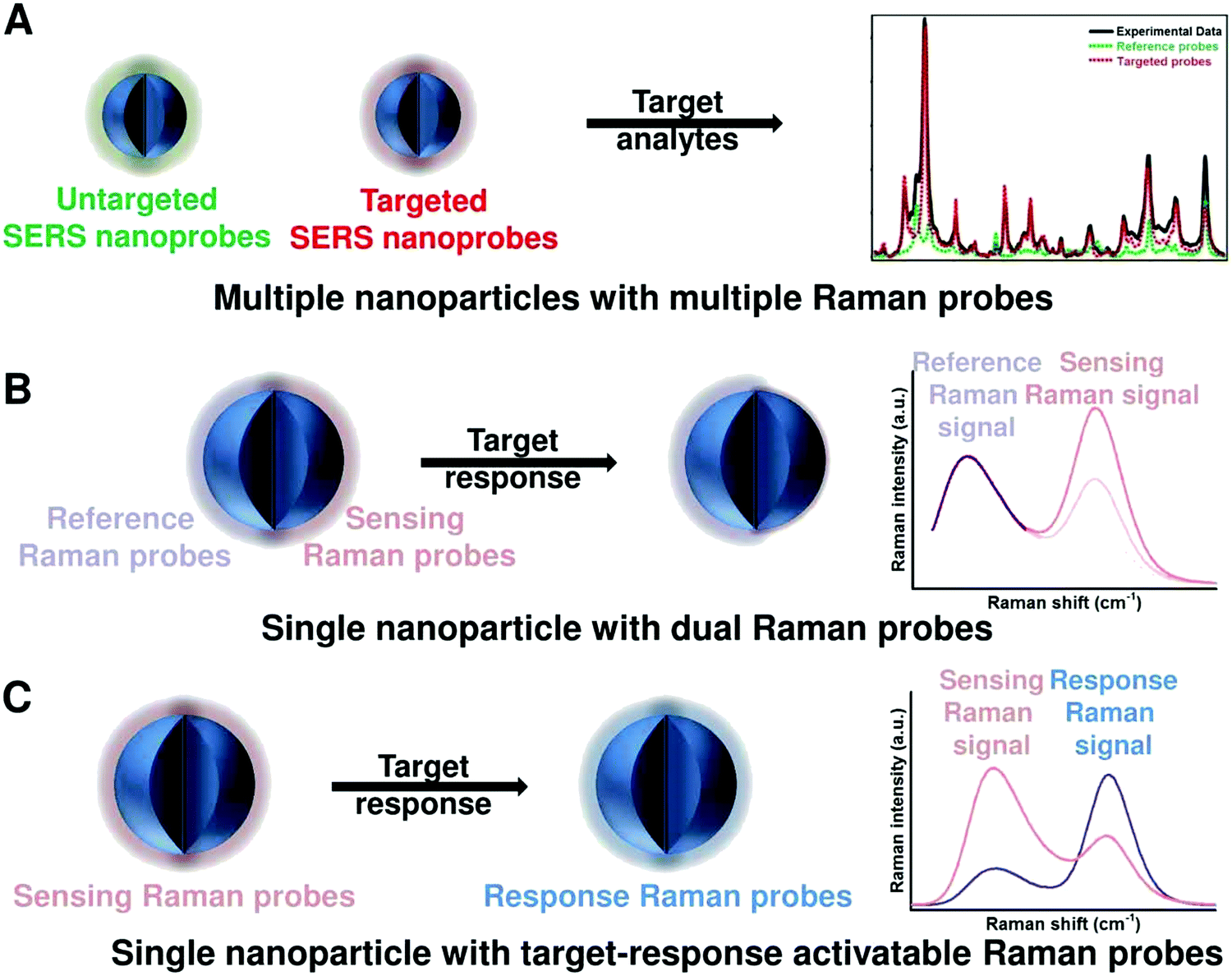
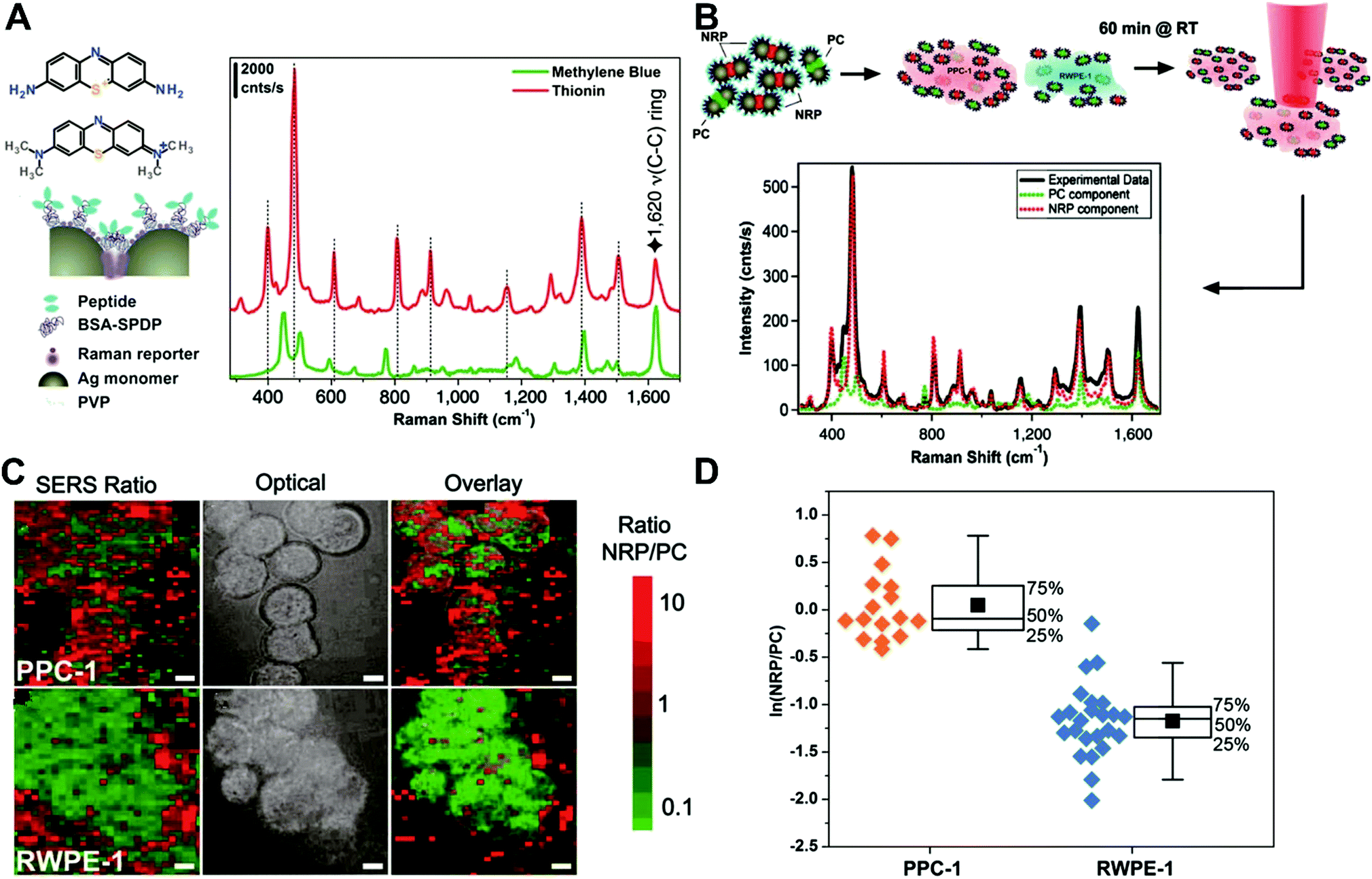
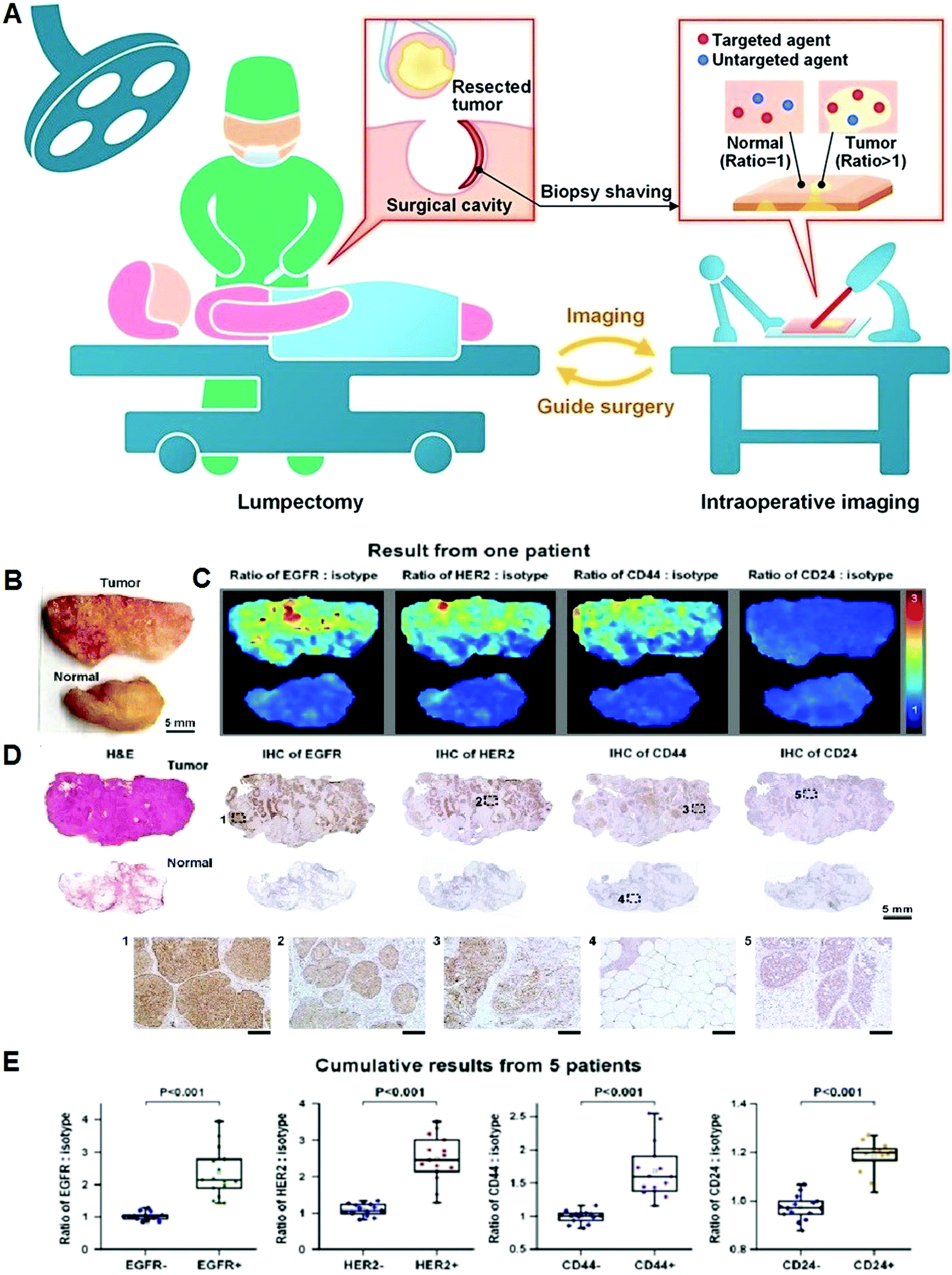
DOI:
10.1039/C7CS00612H
(Review Article)
Chem. Soc. Rev., 2018,
47, 2873-2920
Ratiometric optical nanoprobes enable accurate molecular detection and imaging
Received
26th November 2017
First published on 23rd March 2018
Abstract
Exploring and understanding biological and pathological changes are of great significance for early diagnosis and therapy of diseases. Optical sensing and imaging approaches have experienced major progress in this field. Particularly, an emergence of various functional optical nanoprobes has provided enhanced sensitivity, specificity, targeting ability, as well as multiplexing and multimodal capabilities due to improvements in their intrinsic physicochemical and optical properties. However, one of the biggest challenges of conventional optical nanoprobes is their absolute intensity-dependent signal readout, which causes inaccurate sensing and imaging results due to the presence of various analyte-independent factors that can cause fluctuations in their absolute signal intensity. Ratiometric measurements provide built-in self-calibration for signal correction, enabling more sensitive and reliable detection. Optimizing nanoprobe designs with ratiometric strategies can surmount many of the limitations encountered by traditional optical nanoprobes. This review first elaborates upon existing optical nanoprobes that exploit ratiometric measurements for improved sensing and imaging, including fluorescence, surface enhanced Raman scattering (SERS), and photoacoustic nanoprobes. Next, a thorough discussion is provided on design strategies for these nanoprobes, and their potential biomedical applications for targeting specific biomolecule populations (e.g. cancer biomarkers and small molecules with physiological relevance), for imaging the tumor microenvironment (e.g. pH, reactive oxygen species, hypoxia, enzyme and metal ions), as well as for intraoperative image guidance of tumor-resection procedures.

Xiaolin Huang
| Xiaolin Huang received his BS degree in laboratory medicine from Nanchang University in 2012. Then, he joined the laboratory of Prof. Yonghua Xiong at Nanchang University to pursue his PhD degree in Food Science and Engineering. In 2016, he joined the Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH), as a pre-doctoral fellow under the supervision of Dr Xiaoyuan (Shawn) Chen. His research focuses on the design, development, and applications of highly sensitive nanosensors for clinical early diagnosis and food safety analysis towards his PhD thesis. |

Jibin Song
| Jibin Song obtained his PhD degree in Chemical and Biomedical Engineering at Nanyang Technological University, Singapore, in 2014. His research was focused on creating novel nanomaterials for drug delivery, bioimaging and cancer therapy, including polymeric and plasmonic assemblies, quantum dots, nano- and micro-particles. Since 2014, he has been working with Prof. Xiaoyuan (Shawn) Chen as a postdoctoral fellow at National Institutes of Health (NIH). His research interests include synthesis of biocompatible nanoparticles for biomarker detection, in vivo imaging, and drug/gene delivery. Recently he became a faculty at Fuzhou University. |

Bryant C. Yung
| Bryant C. Yung obtained his BS (2009) in Chemistry from the University of Cincinnati. Later on, he received his PhD (2014) in Pharmaceutics from The Ohio State University. His dissertation research under the direction of Robert J. Lee evaluated several novel polymer and lipid based nanoparticles for cancer gene therapy. In 2015, he joined the National Institute of Biomedical Imaging and Bioengineering (NIBIB) as a member of the Laboratory of Molecular Imaging and Nanomedicine (LOMIN). His current research interests include the design of nanoparticles for cancer specific targeting, biomarker detection, and drug/gene delivery. |

Xiaohua Huang
| Xiaohua Huang received her BSc from Jilin University in 1996, MSc from Peking University in 2001, and PhD from Georgia Institute of Technology in 2006 with Mostafa A. El-Sayed. After being a postdoctoral fellow at Georgia Institute of Technology in Mostafa A. El-Sayed's lab and Emory University in Shuming Nie's lab, she joined the faculty at the University of Memphis in 2010. Her current research interests are bioconjugated nanoparticles for cancer detection and treatment. |

Yonghua Xiong
| Yonghua Xiong obtained his PhD degree in food science and technology from the Nanchang University, China in 2004. He joined the Nanchang University as an Assistant Professor of Food Science in 1992, and then was promoted to a Professor in 2007. During 2007 to 2008, he joined the Institute of Poultry Science, University of Arkansas as a visiting scholar. Then, he became a steady member in State Key Laboratory of Food Science and Technology, Nanchang University, in 2012. During 2015 to 2016, he joined the Department of Chemistry, University of Massachusetts as a visiting scholar. His current research concerns the fabrication of high-sensitivity nanosensors for biomarker detection and food safety analysis. |

Xiaoyuan Chen
| Xiaoyuan (Shawn) Chen received his PhD in Chemistry from the University of Idaho in 1999. He joined the University of Southern California as an Assistant Professor of Radiology in 2002. He then moved to Stanford University in 2004 and was promoted to Associate Professor in 2008. In the summer of 2009, he joined the Intramural Research Program of the NIBIB as a tenured Senior Investigator and Chief of the LOMIN. He has published over 650 papers and numerous books and book chapters. He is the founding editor-in-chief of the journal Theranostics. He is interested in developing molecular imaging tools for early diagnosis of disease, monitoring therapy response, and guiding nanodrug discovery/development. |
1. Introduction
Exploring and understanding biological and pathological changes are of primary importance for early diagnosis and therapy of diseases, as well as for basic biological and medical research.1,2 To this end, various non-invasive molecular sensing and imaging technologies including optical,3,4 magnetic,5–7 and electrochemical8,9 methods have been widely proposed. Among these developed methods, optical molecular sensing and imaging (OMSI) technologies have obtained increasing attention because of their unique advantages.10,11 OMSI technologies can enable the direct, real-time, and dynamic visualization of biomolecules of interest or molecular events at different levels of organization in molecules, cells, tissues, and even organs in living organisms.12,13 In addition, OMSI techniques exhibit high analytical sensitivity, excellent specificity, rapidity, technical simplicity, multiplexing, and multimodal capabilities.14
Recently, the emergence of various optical molecular probes including fluorescence, surface enhanced Raman scattering (SERS), and photoacoustic probes have led to significant advances in the field of OMSI in vitro and in vivo. Especially with the rapid development of materials science and nanotechnology, the design and fabrication of various optical nanomaterial-based probes (nanoprobes) has played a key role in improving OMSI techniques. Compared with conventional small molecule-based probes, nanoprobes can effectively improve the sensitivity, specificity, targeting ability, as well as multiplexing and multimodal abilities of OMSI because of their intrinsic optical and physicochemical properties.15–18 First, nanoprobes with relatively small dimensions below 100 nm are typically smaller than the pore and opening sizes of human vasculature and tissues, permitting them to freely traverse the whole body via systemic circulation. Second, nanoprobes exhibit long blood circulation time,19–21 ensuring that they can efficiently accumulate in the neovasculature of tumors, which offers great potential for the delivery of nanoprobes into disease sites. Third, the optical activities of nanoprobes can be easily and controllably manipulated for diverse applications through engineering of their composition, size, shape, and surface functionalization. Fourth, the large specific surface area of nanoprobes can be used for conjugation of targeting molecules such as antibodies, peptides, or nucleic acids, which can ensure the specificity of the nanoprobes. Fifth, nanoprobes can serve as vehicles for various sensing and signal-generating molecules, which can significantly improve their stability in biological environments, as well as enhance the sensitivity of molecular sensing and imaging due to their high payload capacity which results in high signal intensity. Sixth, nanoprobes offer the possibility of multiplexed sensing and imaging of diverse target molecules through the use of different signaling and/or sensing molecules. Finally, nanoprobes can be employed for multimodal molecular imaging based on multiplexed signaling/imaging modalities engineered into each nanoparticle.
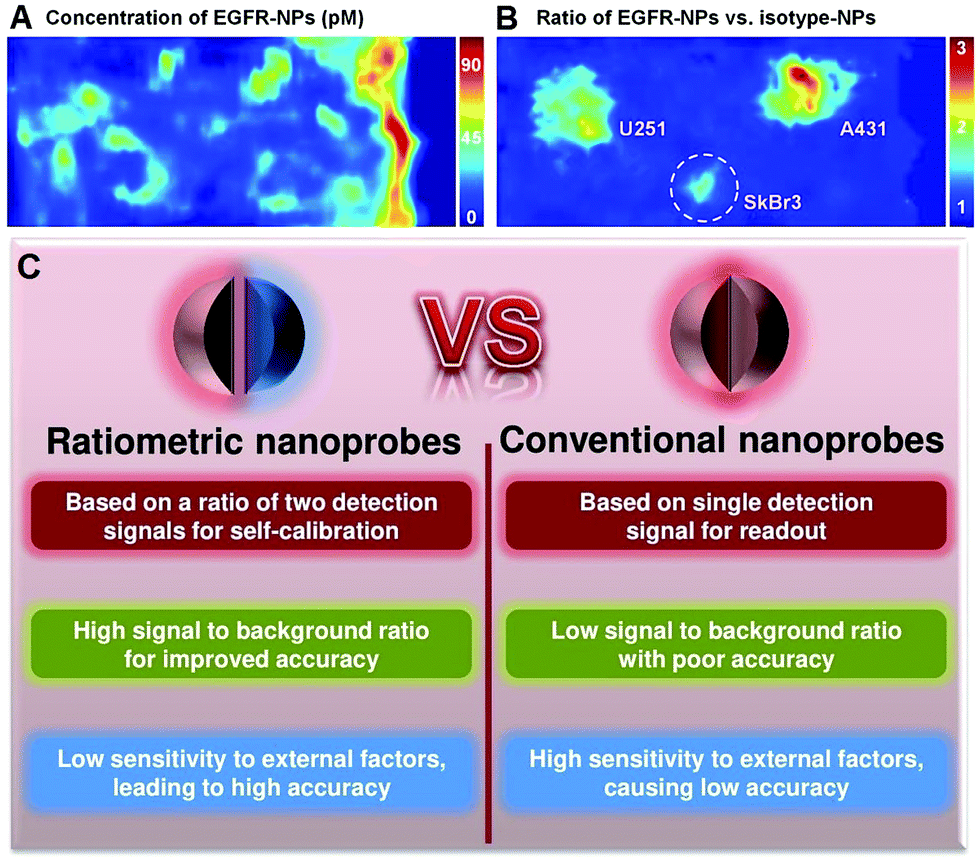
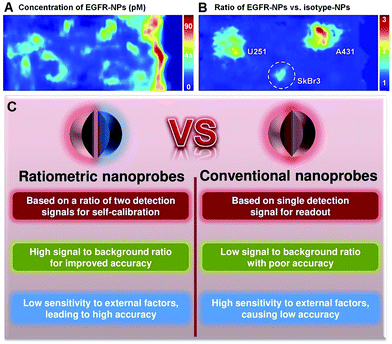
Although the use of nanoprobes as alternatives to small molecule-based probes can significantly improve the performance of OMSI technologies, there remain several significant challenges. Conventional nanoprobes for OMSI technologies mainly depend on signal transduction modes that are “always on” or “always off” to sense and image analyte concentrations and molecular events. Unfortunately, absolute intensity-dependent signal acquisition from a single targeted nanoprobe may be influenced by target concentration-independent experimental or physiological factors, including: (1) uneven delivery and poor washout; (2) variations in tissue mechanical properties of permeability and retention between benign and diseased tissues; (3) instrumentation variables such as detection working distance and illumination angle; (4) off-target chemical binding or trapping of nanoprobe in tissues.22–25 Such factors can cause nonspecific and misleading images, and can further result in an increase in false positives, which has become a major impediment in conventional molecular sensing and imaging when using only a single targeted probe. For example, as shown in Fig. 1A, strong nonspecific background signals with high variability in nanoprobe concentrations are seen across the tissues, which cause the three stained tumors (U251, SkBr3 and A431) to be poorly defined when only single EGFR-targeted nanoprobes (EGFR-NPs) are applied.26 Thus, imaging based on the absolute signal intensity of a single targeted molecular probe can fail to accurately quantify the difference in the true concentration of target analytes.
 |
| | Fig. 1
Ex vivo imaging of the modified rat esophagus. (A) An image showing the measured concentration of EGFR nanoprobes (EGFR-NPs in pM), which is ambiguous due to uneven delivery and nonspecific retention. (B) A ratiometric image for mitigating these confounding effects by imaging the concentration ratio of EGFR-NPs versus isotype nanoprobes (isotype-NPs). Reproduced with permission from ref. 26. Copyright 2015 Optical Society of America. (C) Comparison of convention optical nanoprobes and ratiometric optical nanoprobes. | |
In response to these challenges, various strategies including kinetic modeling,27,28 dual-tracer background subtraction,29 and paired-agent methods,30 have been designed and proposed to mitigate the effects of nonspecific background. Among these methods, one simple way to minimize nonspecific effects is to utilize a ratiometric strategy rather than absolute intensity-dependent signal readout. Ratiometric measurement is based on the self-calibration of signal intensity via recording two or more analyte-induced signal fluctuations, where one signal can act as a reference factor for normalizing the other signals. Ratiometric measurement is independent of local nanoprobe concentration and various analyte-independent confounding factors, which can facilitate more accurate and reliable quantitation.31–33 As shown in Fig. 1B, ratiometric signal processing with the ratio between targeted EGFR-NPs vs. isotype control (isotype-NPs) provides a means to normalize away the background signal, which can enable improved image interpretation for accurately identifying the tumor location compared to the image using a single targeted nanoprobe. As such, this particular ratiometric optical approach allows one to quantify the specific binding against the nonspecific binding of exogenously applied molecular probes to enable accurate contrast (Fig. 1C).34,35 Thus, optimizing nanoprobe designs by exploiting ratiometric measurements is a valuable approach for improving the ability to monitor physiological or pathological processes, improving the effectiveness of diagnosis and treatments of diseases, and accelerating clinical translation. In the past several decades, we have witnessed great progress in designing various ratiometric optical probes, especially fluorescence-based probes, and several pivotal reviews can be found elsewhere.31,36–39 However, all of these published reviews have mainly focused on the design and fabrication of small molecule-based ratiometric fluorescence probes, and none of them have emphasized the design of ratiometric fluorescence probes based on nanomaterials. Additionally, other types of ratiometric optical nanoprobes including surface enhanced Raman scattering (SERS) and photoacoustics have also been proposed to enhance the sensitivity and reliability of traditional optical sensing and imaging probes, which have not been previously reviewed. Thus, in this review, we comprehensively and systematically summarize recent advances for designing and applying various optical nanoprobes including fluorescence, SERS, and photoacoustic nanoprobes for ratiometric targeting of specific biomolecules (e.g. cancer biomarkers and small molecules of physiological importance), for imaging the changes in the tumor microenvironment (e.g. pH, reactive oxygen species (ROS), hypoxia, enzyme and metal ions), and for intraoperative image guidance of tumor-resection procedures (Scheme 1). For each type of nanoprobe, we elaborate upon the design strategies that enhance ratiometry, and also describe representative applications. Finally, we discuss the potential challenges and the further directions of this field.
 |
| | Scheme 1 Schematic representation of ratiometric optical nanoprobes for molecular sensing and imaging in vitro and in vivo. | |
2. General principles for designing ratiometric optical nanoprobes
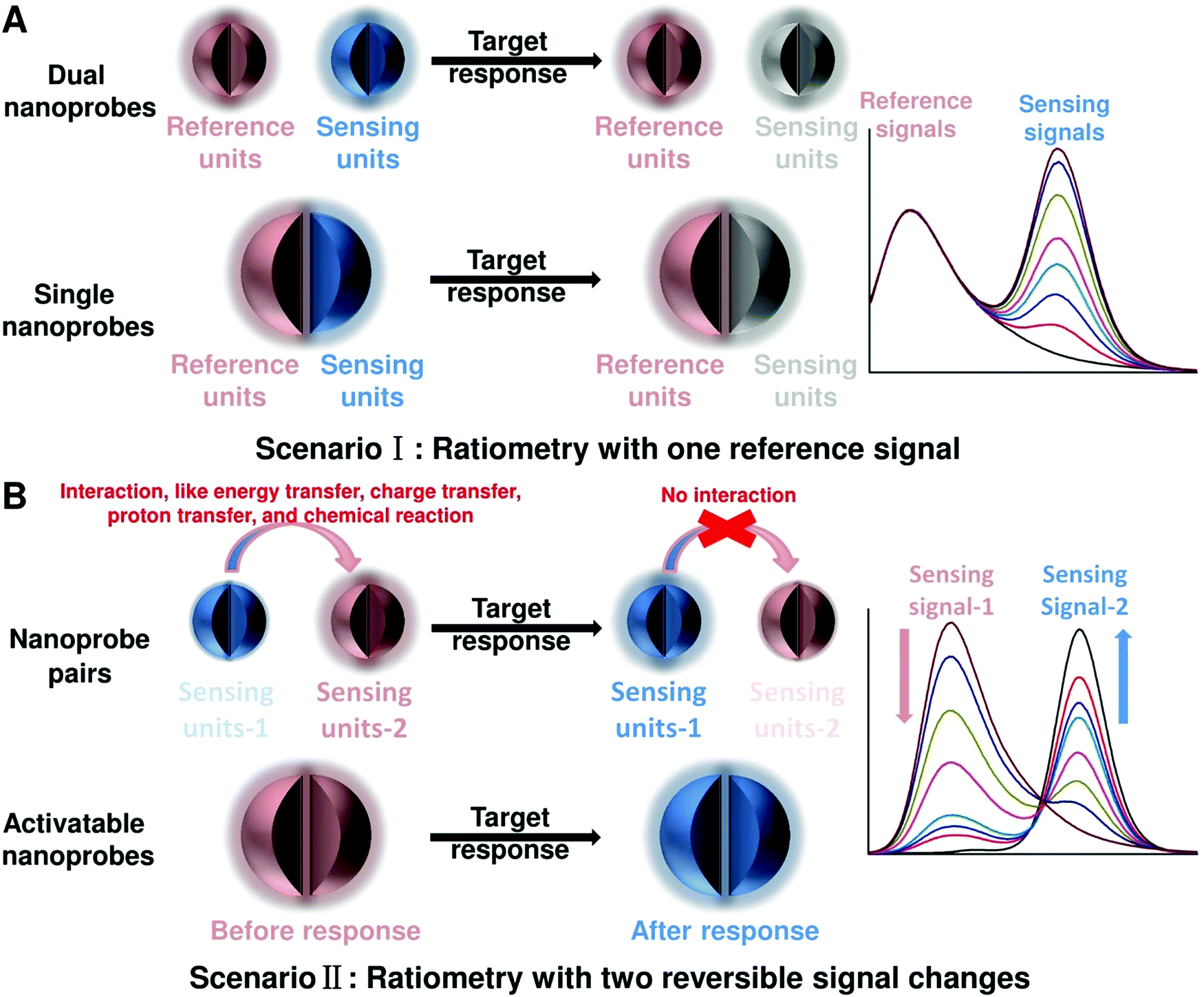
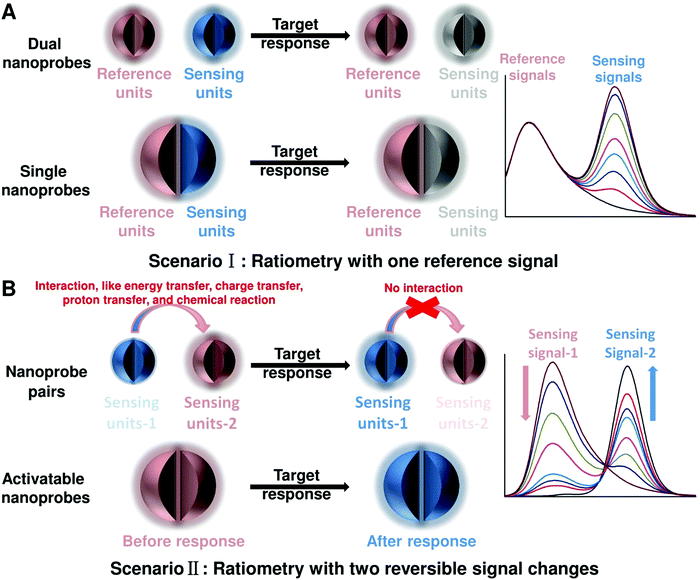
To achieve ratiometric detection, there are basically two universal design strategies for the fabrication of ratiometric optical nanoprobes: one strategy is to introduce the second signal as a reference that is target-insensitive; the other strategy is to apply two target-responsive reversible signal changes that enable the ratiometry.
2.1 Ratiometry with one reference signal
As shown in Fig. 2A, the first scenario of ratiometric detection depends on the action of two entirely independent signals from each sensor entity. In this case, one signal is target-sensitive and can specifically respond to targets or molecular events, whereas the other one is target-insensitive and acts as a reference signal to allow the primary signal to be normalized. In general, the simplest way to achieve this ratiometry is through physical incorporation of two independent nanoprobes (Fig. 2A). However, the requirement of two independent nanoprobes can make the ratiometric detection complicated. For example, heterogeneous and unequal distributions of these two nanoprobes can accumulate in cells, tissues, or organs if the reference probes are not ideal, which can result in false imaging results. Single nanoprobes with dual detection signals for ratiometry exhibit advantages for eliminating the errors associated with variations in nanoprobe concentration (Fig. 2A). Generating two independent signals from a single nanoprobe requires preconjugation or preassembly by physical or chemical methods. By using this strategy, the designed ratiometric sensing and imaging nanoprobes are often more refined and reliable, further accelerating the corresponding applications.
 |
| | Fig. 2 General principles for designing ratiometric optical nanoprobes. (A) Ratiometry with one reference signal. (B) Ratiometry with two reversible signal changes. | |
2.2 Ratiometry with two reversible signal changes
The second scenario of ratiometric detection in Fig. 2B presents a dynamic mechanism of two interrelated detection signals that exhibit reversible changes. For example, the presence of analytes can specifically induce the increase of one signal, accompanied with the decrease of the other, thereby producing a large change in the ratio between these two detection signals. The commonly used strategy for this ratiometry is to prepare nanoprobe pairs with two or more different signal precursors to trigger analyte-binding-driven optical phenomena, such as energy transfer, charge transfer, proton transfer, or chemical reaction (Fig. 2B). With the presence of molecular targets or events, the analyte binding can perturb the resultant nanoprobe pairs to cause reversible variation of two or more different signals, thus allowing for ratiometry. The signal changes can be achieved through physical absorption, chemical coupling, nucleic acid hybridization, or antigen–antibody interaction, making this approach versatile. Additionally, the stability and reproducibility of these designed nanoprobe pairs in actual sensing and imaging applications should be considered. Another strategy for this type of ratiometry is through designing stimuli-responsive activatable nanoprobes, where the presence of molecular targets or events can induce the disappearance of one detection signal along with the emergence of a new one, thereby establishing a ratiometric detection scheme (Fig. 2B). Compared with the former, this strategy for ratiometry is simpler and possesses a lower background noise and higher signal to background ratio (SBR),40–42 which has been widely proposed for the design of reversible ratiometric optical sensing and imaging nanoprobes.
3. Ratiometric fluorescence nanoprobes
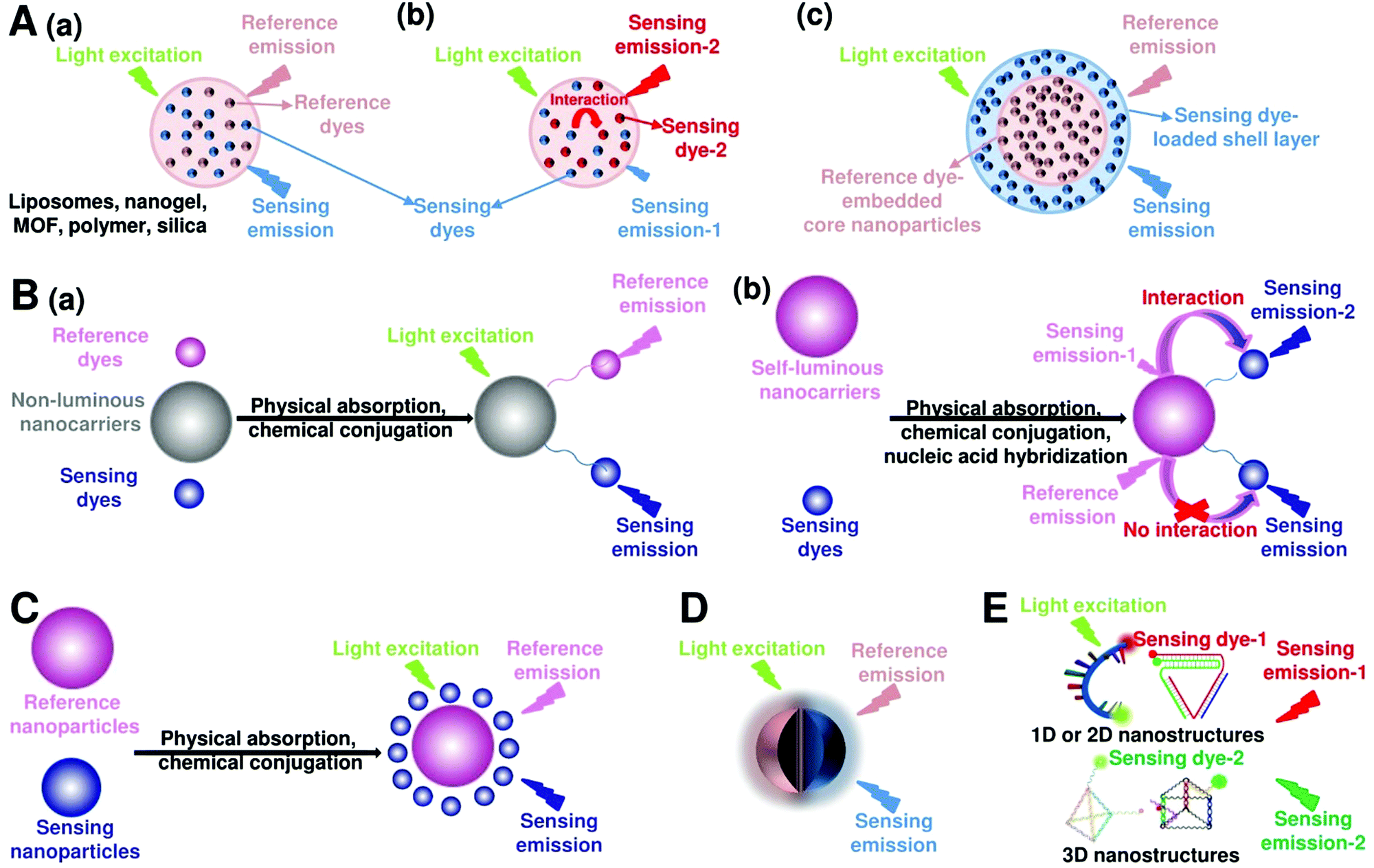
Fluorescent nanoprobes have become one of the most powerful tools for molecular sensing and imaging.1,43,44 Fluorescent nanoprobes provide the opportunity for direct visualization and real-time monitoring of changes in the local microenvironment and targeting of analyte concentrations.3,45,46 However, in traditional fluorescence sensing and imaging, the absolute-intensity-dependent signal acquisition from single fluorescence nanoprobe is sometimes inaccurate because of the existence of various analyte-independent confounding factors, including instrumental parameters (e.g. excitation and emission source fluctuation, changes in detector working distance, changes in angle of detection, etc.), background light scattering from the complex sample matrix, microenvironmental variations that affect the nanoprobes, as well as local fluctuations in the concentration of the nanoprobes due to uneven delivery or poor washout of the nanoparticles, rather than differences in chemical binding or specific signal generation.37,47 These analyte-independent factors can cause absolute signal intensities to fluctuate significantly, thereby causing false-positive and false-negative imaging results. Ratiometric fluorescence nanoprobes can effectively overcome these issues by introducing another fluorescence emission band to achieve ratiometric signal readouts.31,36–39 Ratiometric fluorescence nanoprobes enable more accurate imaging contrast, which often leads to higher detection sensitivity. In this section, we will review the latest developments in designing and applying ratiometric nanoprobes for fluorescence sensing and imaging. Although there exists two models for realizing ratiometric fluorescence detection of dual excitation and dual emission, this review mainly focuses on dual-emission fluorescence-based ratiometric strategies, which can be classified into five categories: (i) two-dye-embedded nanoparticles, (ii) nanoparticle–dye nanoconjugates with dyes attached to the surface, (iii) hybrid nanoparticles, (iv) single nanoparticles with intrinsic dual emission, and (v) DNA nanostructures (Fig. 3).
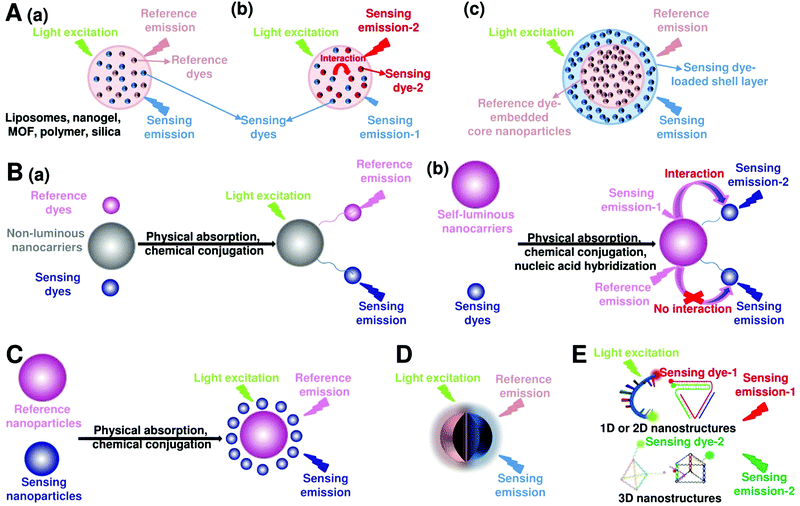 |
| | Fig. 3 Design strategies for ratiometric fluorescence nanoprobes. (A) Two-dye-embedded nanoparticles: nanoparticles with dyes randomly distributed in the interior without or with interaction (a and b), and nanoparticles with dyes located within the core and shell (c). (B) Nanoparticle–dye nanoconjugates with dyes attached to the surface: non-luminous nanocarriers with two dyes (a), and self-luminous nanocarriers with one dye (b). (C) Hybrid nanoparticles. (D) Single nanoparticles with intrinsic dual emission. (E) Dual-emission DNA nanostructures. | |
3.1 Two-dye-embedded nanoparticles with dual emission
3.1.1 Nanoparticles with randomly distributed dyes.
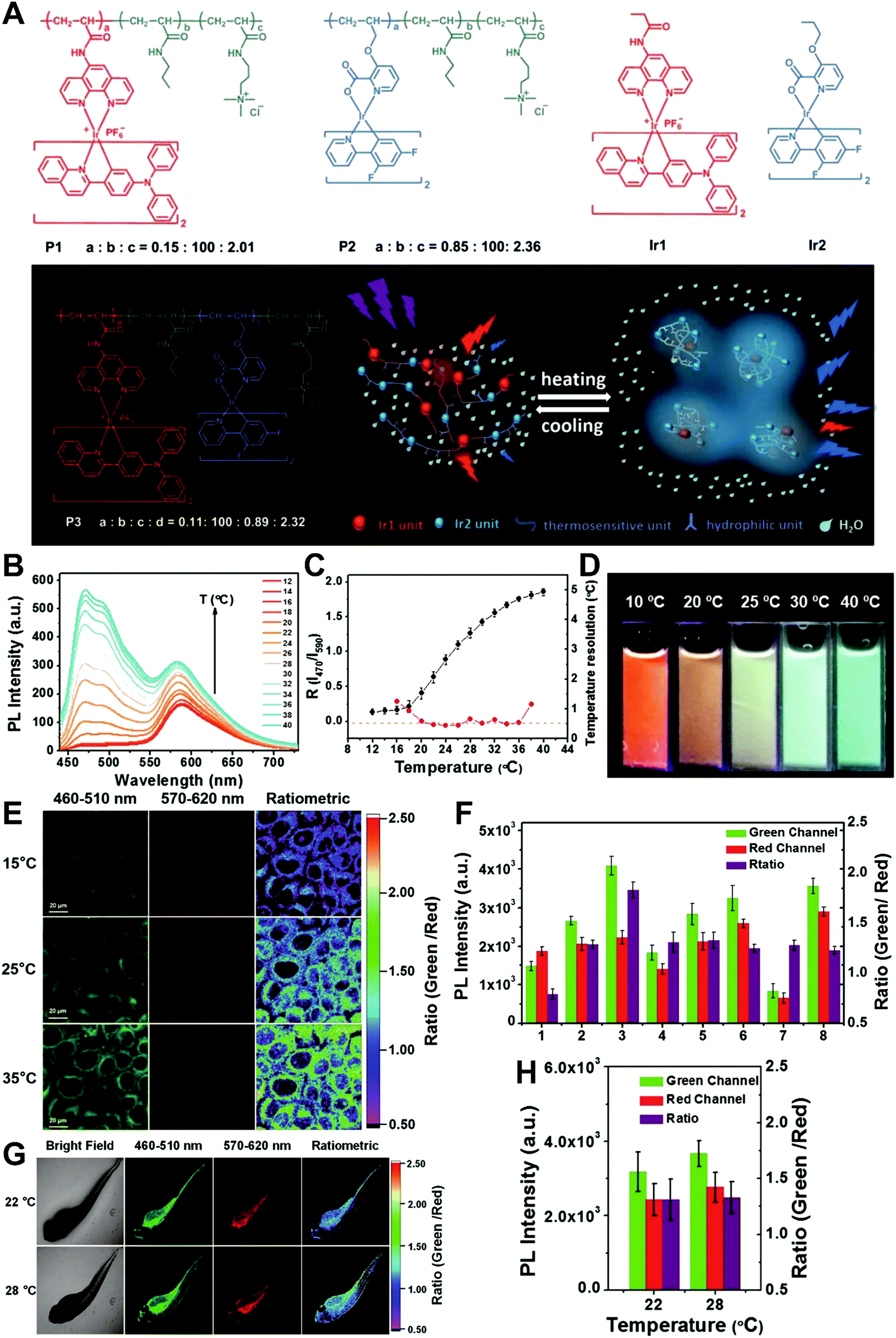
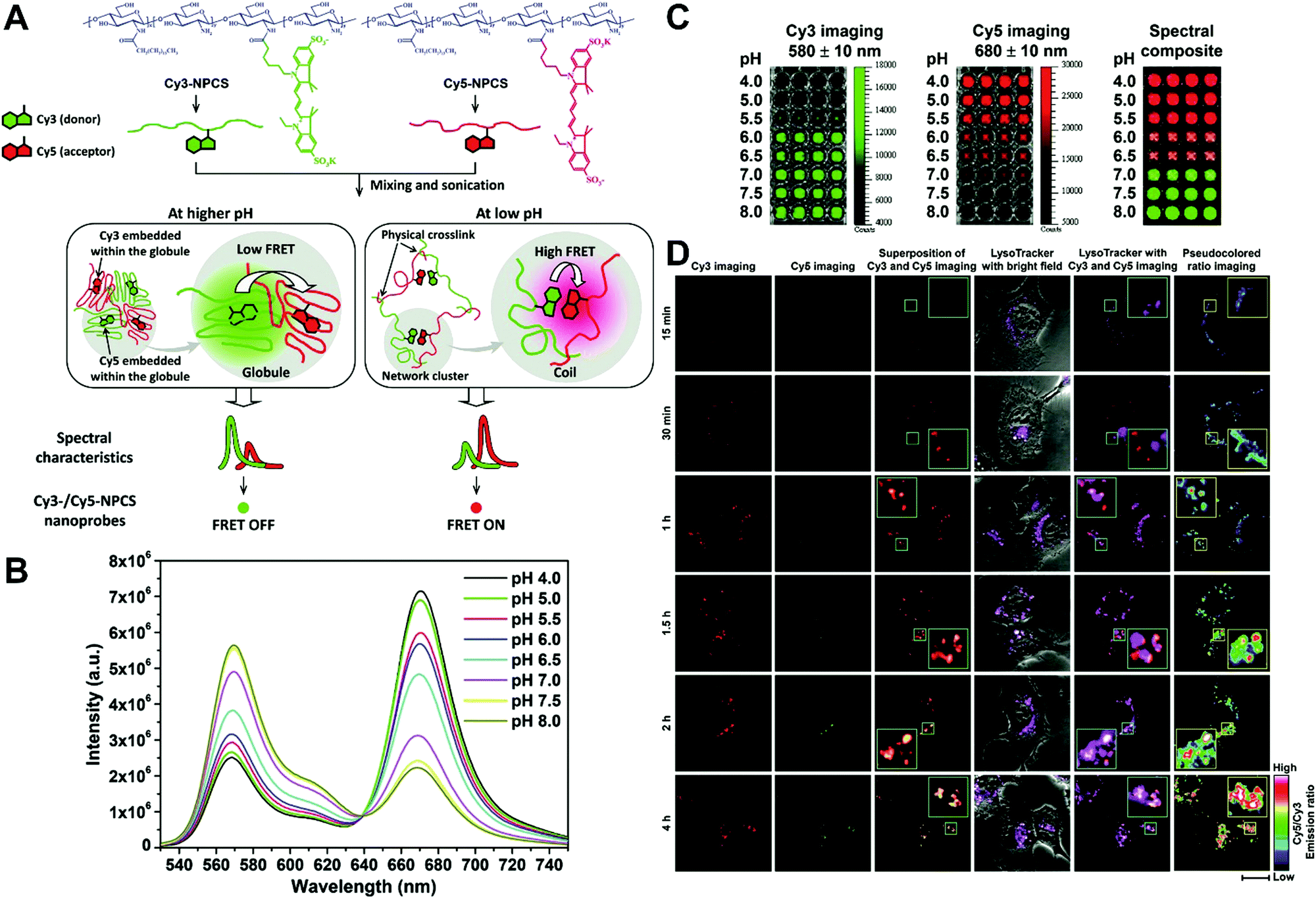
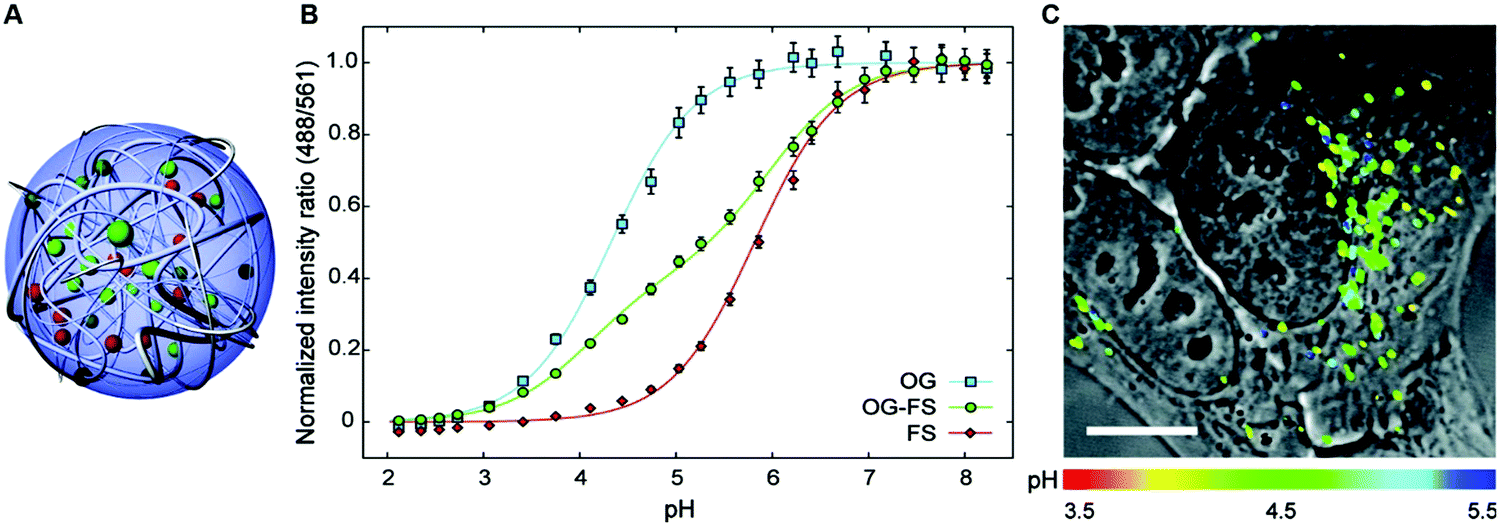
In general, dual-emission nanoprobes for ratiometric fluorescence sensing are achieved by incorporating two or more fluorescent dyes with different emission bands into one nanoparticle, in which one fluorophore serves as the reference and the other acts as a response molecule for ratiometric signal output. After being entrapped into a single nanoparticle matrix, these dyes can interact with each other through fluorescence resonance energy transfer (FRET) processes inside the nanoparticle. Pioneering work was performed by Kopelman and colleagues in 1999,48 who first prepared a so-called PEBBLE (probes encapsulated by biologically localized embedding) nanosensor for ratiometric detection of intracellular pH by incorporating a pH-sensitive fluorescent indicator and a pH-insensitive internal standard into an acrylamide polymeric matrix. Inspired by this work, various dual-emission PEBBLE nanoparticles have been designed and synthesized by simply switching the nanoparticle matrix to silica,49–53 liposome,54 polymer,55–67 nanogel,68 and metal–organic frameworks (MOF).69–74 For example, Huang and colleagues synthesized a dual-emission phosphorescent nanoprobe for ratiometric temperature sensing by incorporating two iridium(III) complexes, Ir1 and Ir2, into an acrylamide-based thermosensitive polymer matrix (Fig. 4A).75In vitro emission spectrum analysis demonstrated that the emission from Ir2 at 470 nm remarkably enhanced with increasing temperature, whereas that from Ir1 at 590 nm as a reference was virtually unchanged (Fig. 4B). By using the ratio of I470nm/I590nm as the signal output, an 18.2-fold signal increase was obtained with the solution color changing from orange to white and then cyan as the temperature increased from 10 °C to 40 °C (Fig. 4C and D). In addition, live cell imaging for temperature measurement was demonstrated via confocal laser scanning microscopy at an excitation wavelength of 405 nm, in which the signals from the green channel (460–510 nm) and orange channel (570–620 nm) were recorded at the same time. As shown in Fig. 4E, with the temperature increase from 15 °C to 30 °C, the signal intensity from the green channel obviously rose, whereas the red channel was minimally changed, thereby effecting a significant enhancement of the ratio from 0.82 to 1.79 (Fig. 4F). Further in vivo ratiometric images were collected in live zebrafish larva, in which ratiometric mappings at 22 °C and 28 °C are shown in Fig. 4G and H. These results revealed that this polymer nanoprobe can be applied for ratiometric sensing of temperature in living cells and in vivo. In this case, there was no interaction such as FRET between the two phosphorescent iridium(III) complexes. Recently, Sung et al. designed a FRET-based dual-emission nanoprobe for sensing and imaging intracellular pH. This nanoprobe, named Cy3-/Cy5-labeled NPCS, was prepared through using associating polyelectrolyte, namely, N-palmitoyl chitosan (NPCS), conjugated with a donor (Cy3) or an acceptor (Cy5) moiety (Fig. 5A).76 The NPCS exhibits a pH-responsive conformational transition for modulating the FRET efficiency. FRET spectrum measurements indicated that increases in pH were concomitant with decreases at 670 nm from Cy5 emission and increases at 570 nm from Cy3 emission due to the increased FRET efficiency between both (Fig. 5B), thus favoring an increase in the Cy5/Cy3 ratio with pH. Ratiometric imaging in Fig. 5C showed an evident color change from red to orange and then green over the pH range from 4.0 to 8.0, indicating that this nanoprobe could effectively distinguish the pH changes in the local environment. The potential for intracellular ratiometric pH imaging was further evaluated with HT1080 human fibrosarcoma cells. As presented in Fig. 5D, the designed nanoprobe is well-suited for ratiometric tracking and mapping of the environmental pH changes in living cells, especially in acidic organelles, such as endosomes and lysosomes. A large number of similar PEBBLE nanoprobes with dual-emission properties have been introduced for ratiometric sensing and imaging of microenvironmental changes such as pH,77–80 temperature,81,82 hypoxia,83–88 ROS,89–91 and biologically important molecules.92–95 Nevertheless, a broad dynamic measurement range is very difficult to achieve by using traditional dual-emission PEBBLE nanosensors. To address this limitation, a triple-labeled ratiometric PEBBLE nanosensor was proposed recently.96–99 Andresen et al. developed a triple-labeled PEBBLE nanosensor for intracellular pH detection by embedding two pH-sensitive dyes, Oregon green (OG) and fluorescein (FS), and a pH-insensitive reference dye, rhodamine B (RhB), into an acrylamide-crosslinked matrix (Fig. 6).100 By using a ratiometric readout, the triple-labeled pH nanosensor exhibited a broad detection dynamic range of up to 4 pH units, which was superior to the conventional dual-labeled nanosensor (Fig. 6B). Similarly, this proposed triple-labeled ratiometric nanosensor was also employed for hypoxia101 and temperature102 imaging in live cells.
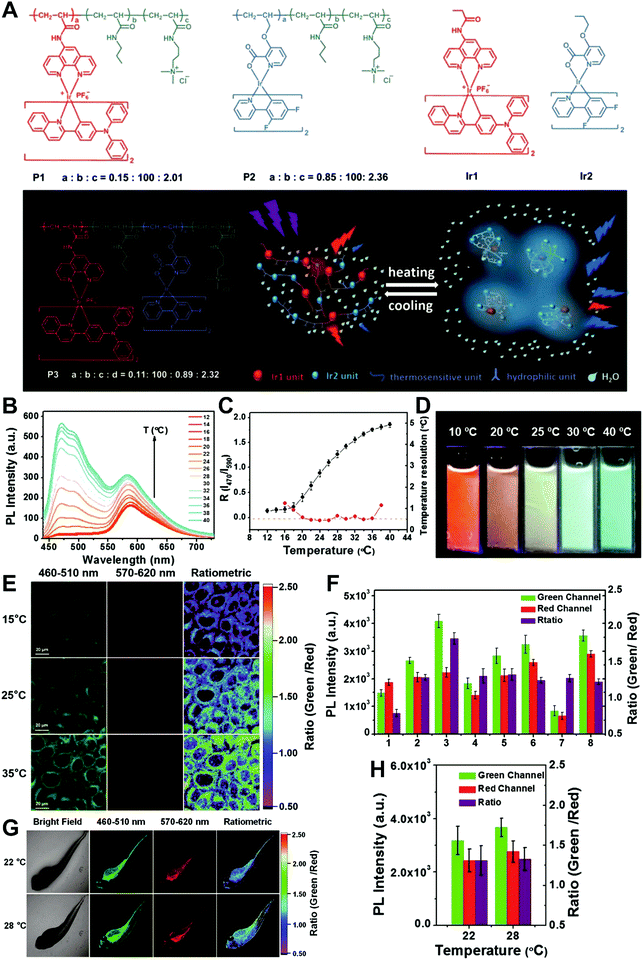 |
| | Fig. 4 (A) Schematic diagram and chemical structures of polymers and iridium(III) complexes. (B) Emission spectra of P3 in phosphate buffered saline (PBS) at various temperatures. (C) Temperature-dependent ratio of phosphorescence intensity at 470 and 590 nm (black, left axis) and temperature resolution (red, right axis). (D) Photographs of P3 in aqueous solution at different temperatures. (E) Confocal laser scanning microscopy images of HeLa cells labeled with P3 at 15 °C (top), 25 °C (middle), and 35 °C (bottom). (F) Luminescence intensity of HeLa cells recorded from the green channel (green) and the red channel (red) and the intensity ratio green/red (purple). (G) Bright images and confocal laser scanning microscopy images of living zebrafish larva after injection of P3 at 22 °C (top) and 28 °C (bottom). Ratiometric luminescence images were from the green channel to red channel. (H) Luminescence intensity of zebrafish recorded from the green channel (green) and the red channel (red) and the intensity ratio (green/red) (purple) at 22 or 28 °C. Reproduced with permission from ref. 75. Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
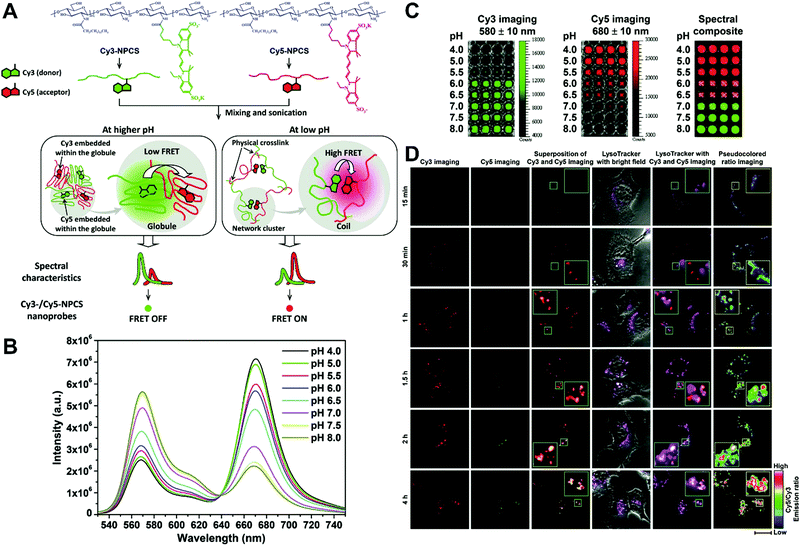 |
| | Fig. 5 (A) Schematic illustration showing a dual-emission nanoprobe that can sense changes in the environmental pH, based on the concept of pH-responsive FRET of a biocompatible polyelectrolyte, NPCS, conjugated with a donor (Cy3) or an acceptor (Cy5) moiety. (B) FRET spectra, and (C) Dual-emission pH images of Cy3-/Cy5-NPCS-15% nanoparticle suspensions. (D) Mapping spatial pH changes in living cells. Dual-emission fluorescence images (scale bar, 20 μm) of cells treated with Cy3-/Cy5-NPCS NPs for distinct durations taken by a confocal laser scanning microscope at 543 nm. The corresponding pseudocolored ratio images were obtained by analyzing the ratio of the signal intensities of Cy5 to Cy3 imaging channels. Reproduced with permission from ref. 76. Copyright 2010, American Chemical Society. | |
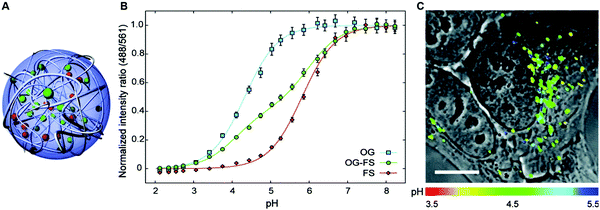 |
| | Fig. 6 (A) Schematic illustration of the cross-linked triple-labeled polyacrylamide nanoparticle. (B) In vitro calibration of the triple-labeled sensor with both OG and FS, and two dual-labeled sensors with either OG or FS. (C) Uptake of the triple-labeled sensor by a HepG2 cell after 24 h incubation and washing and imaged with confocal microscopy. Reproduced with permission from ref. 100. Copyright 2011, American Chemical Society. | |
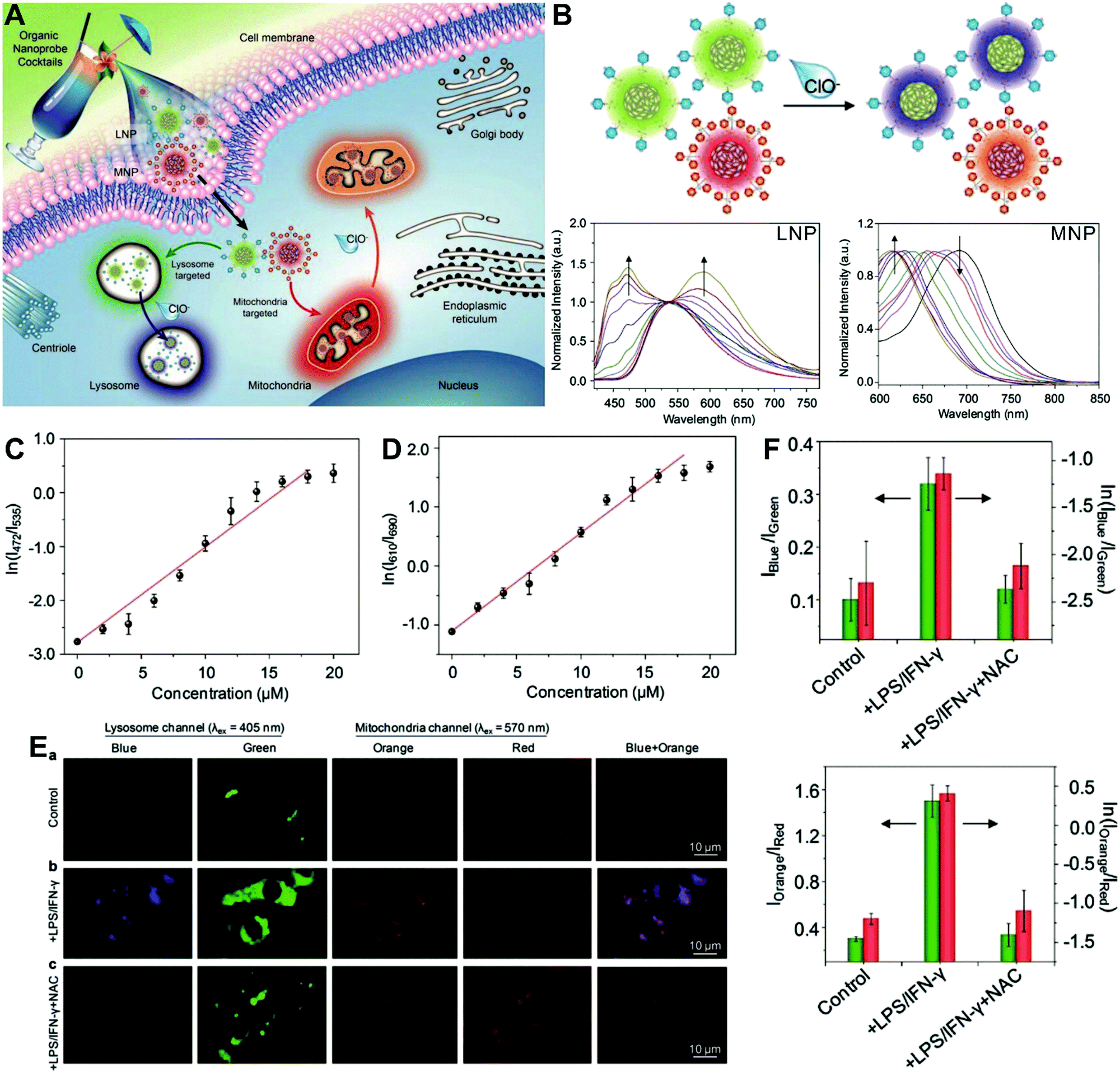
The aforementioned dual-labeled or triple-labeled nanosensors have overcome many of the problems encountered by conventional single-labeled nanosensors and have significantly improved the sensitivity and reliability of OMSI techniques. However, such approaches usually require at least two fluorophores to be integrated into the sensing system, which results in difficulties in terms of manufacturing and cost. To overcome these issues, dual-emission nanoprobes with a single fluorophore are attractive. Fraser et al. first reported single-component iodide-substituted difluoroboron dibenzoylmethane-poly(lactic acid) (BF2dbm(I)PLA) as a dual-emissive probe for ratiometric tumor hypoxia imaging in vivo through modulating fluorescence and phosphorescence.103 Single-fluorophore dual-emission nanoprobes have also been used for ratiometric sensing and imaging of ROS,104–107 enzyme activity108 and hypoxia,109 as well as for monitoring intracellular drug release.110 For instance, Pu et al. developed a cocktail design with organic semiconducting nanoparticles (OSNs) to construct target-responsive dual-emission polymer dots (Pdots) for ratiometric hypochlorite (HClO) imaging in vitro (Fig. 7A).111 By using a nanoprecipitation method, two HClO sensing units of phenothiazine were integrated into two different semiconducting oligomers to generate two organelle-targeted nanoprobes, named lysosome-targeted nanoprobe (LNP) and mitochondria-targeted nanoprobe (MNP), respectively. In vitro fluorescence analyses (Fig. 7B) showed that with the addition of HClO, the emission at 535 nm from LNP gradually reduced and shifted to 480 nm, while the emission at 690 nm from MNP gradually reduced and shifted to 610 nm. The ratiometric HClO response showed a linear relationship between the fluorescence ratio of I472/I535 or I610/I690 and the HClO concentration, with a limit of detection (LOD) of 0.17 or 0.19 μM, (Fig. 7C and D). Additionally, living cell multicolor ratiometric imaging of HClO was demonstrated by incubating the nanoprobe cocktail with RAW 264.7 cells. Two positive stimuli, including lipopolysaccharide (LPS) and interferonγ (IFNγ) were used to induce the generation of ROS, and NacetylLcysteine (NAC), a ROS scavenger, was used as a negative control. All images were acquired with the selective excitation of LNP at 405 nm and excitation of MNP at 570 nm, respectively. The results in Fig. 7E reveal the increased fluorescence signals in the blue and orange channels after the addition of LPS and IFNγ when compared with the control group. With the subsequent addition of NAC, the fluorescence signals from the two channels are remarkably reduced. The ratiometric fluorescence analysis shown in Fig. 7F reveals that the ratios of I472/I535 and I610/I690 were 0.32 and 1.5 in the cells treated with LPS/IFNγ, and the average HClO concentrations were calculated to be approximately 9.0 and 8.0 μM in lysosomes and mitochondria, respectively. These findings demonstrate the potential of the multicolor LNP/MNP nanococktail for simultaneous probing of HClO concentration changes in lysosome and mitochondria.
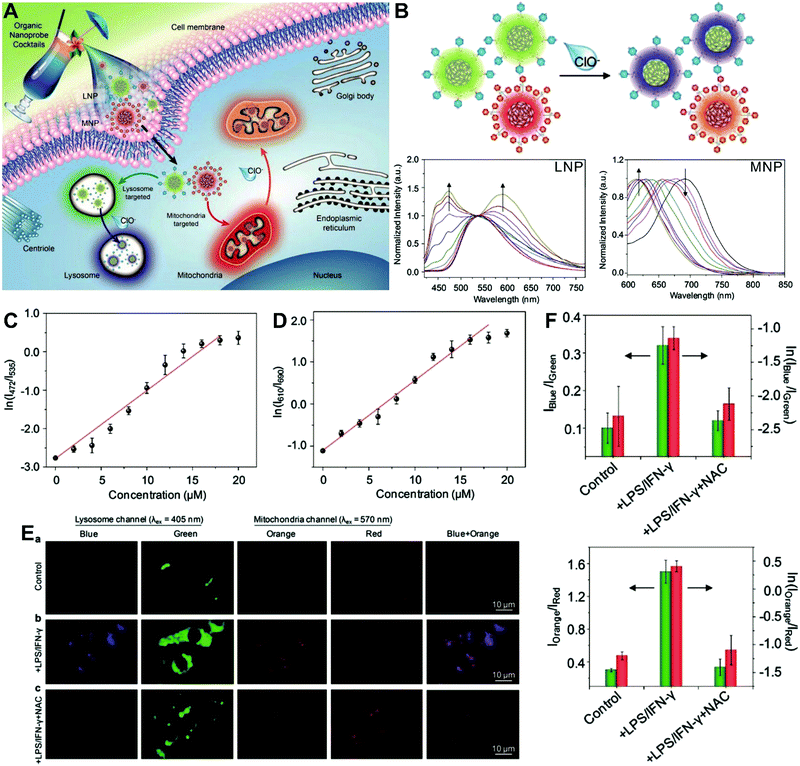 |
| | Fig. 7 (A) Schematic illustration of organelle-differentiated multilocal and multicolor fluorescence imaging of endogenous HClO in macrophage cells using the organic nanoprobe cocktails composed of lysosome-targeted nanoprobe (LNP) and mitochondria-targeted nanoprobe (MNP). (B) Normalized fluorescence spectra of the cocktail nanoprobe solution with different HClO concentrations under the excitation of 405 nm (left) and 570 nm (right) light, respectively. (C and D) The logarithmic value of ratiometric fluorescence signals (ln(I472/I535)) and (ln(I610/I690)) as a function of HClO concentration. (E) Multilocal and multicolor imaging of HClO in murine macrophage cells (RAW 264.7): (a) without any treatment, (b) with LPS/IFN-γ, (c) with LPS/IFN-γ and NAC. (F) Quantification of the ratiometric fluorescence signals of the blue (orange) channel to that of the green (red) channel (Iblue/Igreen) or (Iorange/Ired) and their logarithmic values from macrophage cells with different treatments of E. Reproduced with permission from ref. 111. Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
3.1.2 Nanoparticles with dyes located within the core and shell.
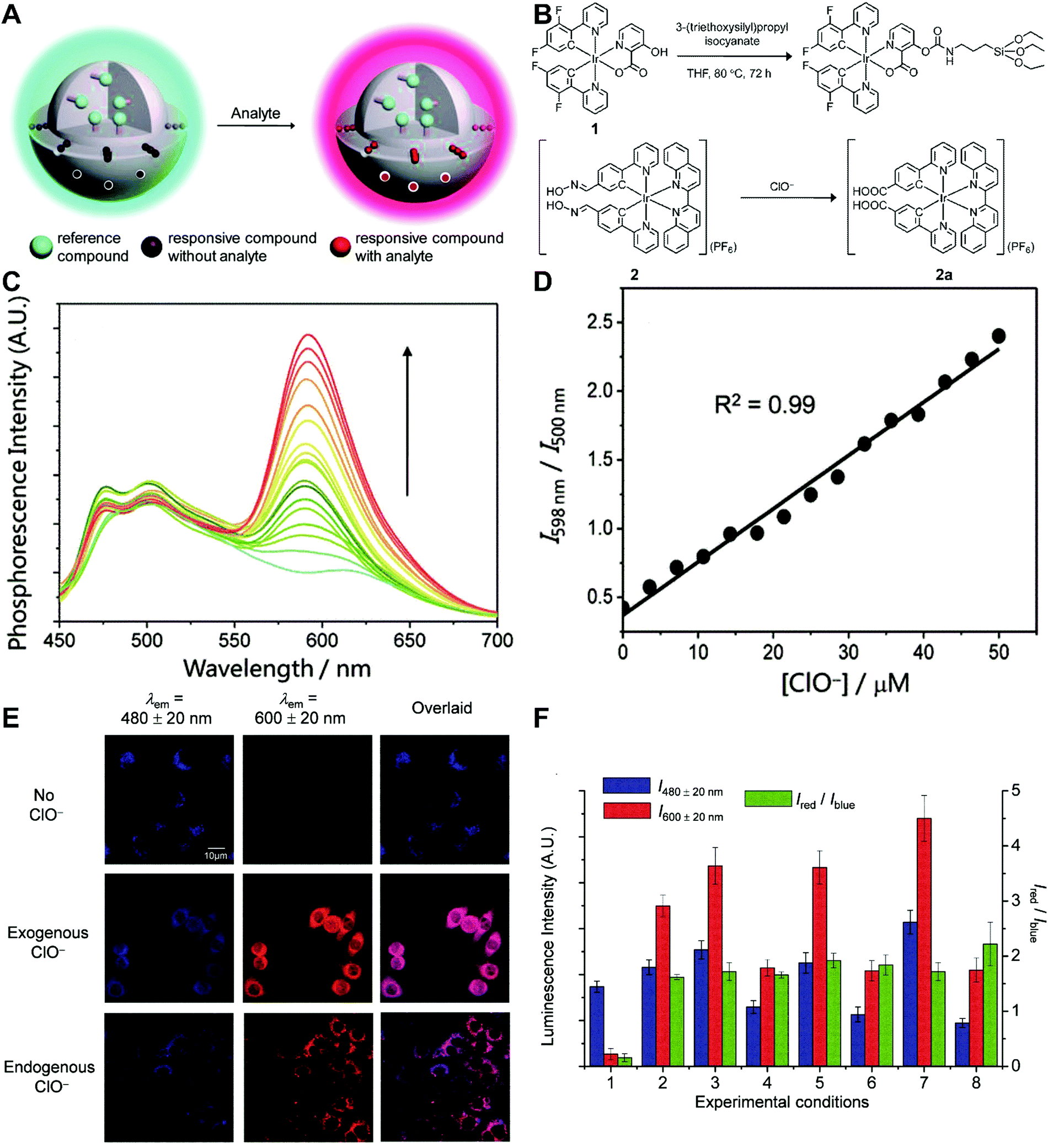
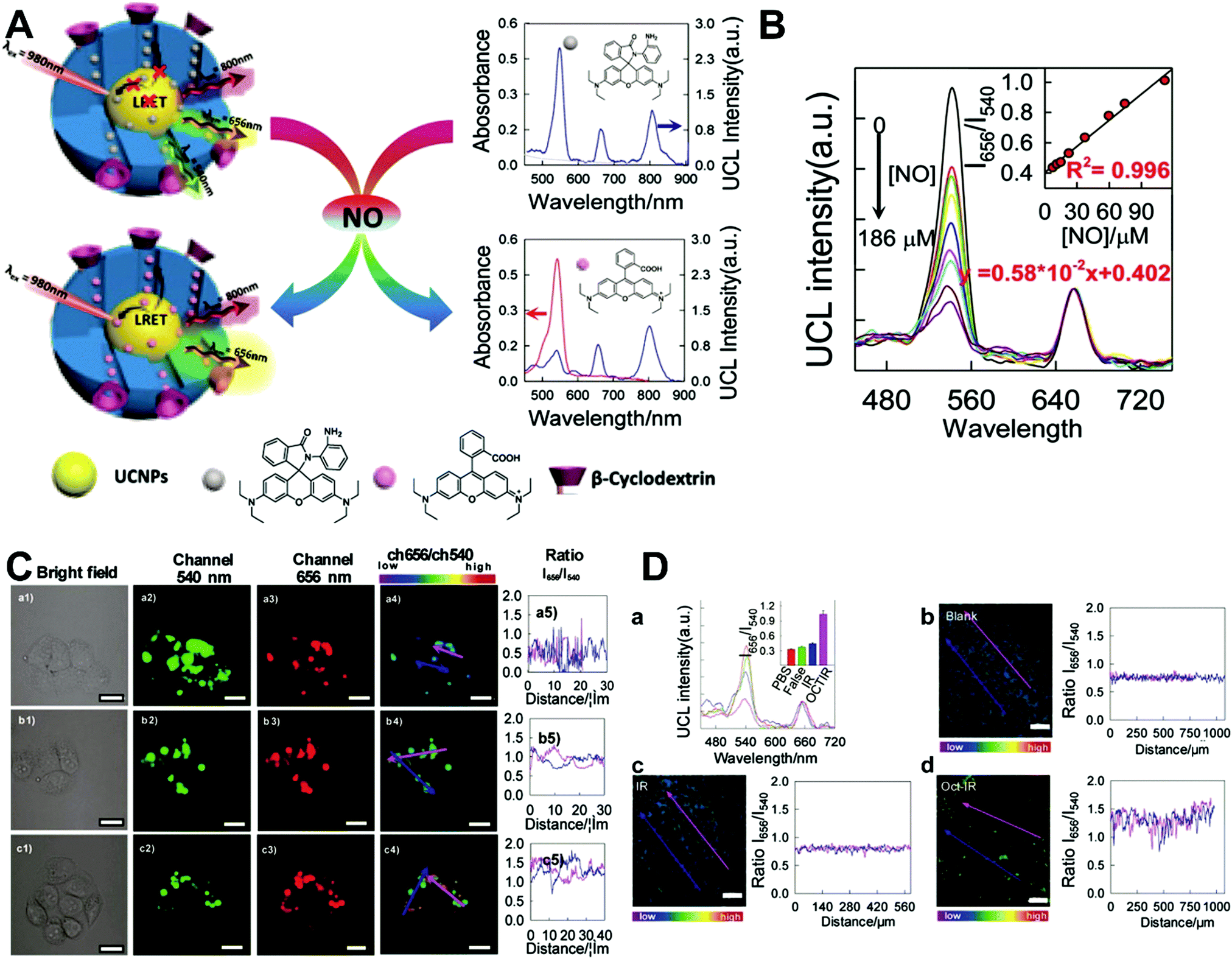
Although the technique of physically incorporating multiple dye molecules into one nanoprobe for ratiometric sensing and imaging has experienced great progress, such matrix networks provide little protection against self-quenching or bleaching owing to the bulk accumulation of dye molecules inside individual nanoprobes, which may deteriorate the sensing performance to some extent. Core–shell nanostructures are introduced as an alternative platform to construct dual-emission fluorescence nanoprobes because the unique nanostructure can effectively separate target-sensitive and reference fluorophores, thereby preventing severe self-quenching from the overcrowding of dye molecules. Additionally, the core and shell also allow for independent modifications to address individual requirements. Owing to their unique advantages, core–shell structured silica nanoparticles have been used as nanocarriers for dual-emitting ratiometric nanoprobes, where target-inert dyes are first doped into the silica core to act as a reference element, and target-active dyes are coated onto the silica shell as sensing elements. Wiesner et al. prepared a silica-based core–shell dual-emission nanoprobe for ratiometric pH sensing by layering a sensor-dye-rich silica shell onto a reference-dye-rich silica core.112 Subsequently, a series of dual-emission nanoprobes based on core–shell silica nanocomposites were designed and established for ratiometric detection of diverse targets, including pH,113 cysteine,114 Zn2+,115 hypoxia,116 and ROS.117,118 Recently, Huang's group designed a core–shell structured phosphorescent nanoparticle (SiO2-1@mSiO2-2) as a ratiometric nanoprobe for the detection of both exogenous and endogenous HClO in living cells (Fig. 8A).119 To achieve this aim, two phosphorescent iridium(III) complexes, labeled as 1 and 2 were immobilized into the inner solid silica core as an internal reference, and immobilized into the outer mesoporous silica shell as a HClO sensing element, respectively (Fig. 8B). When HClO was present, complex 2 was oxidized to complex 2a with a phosphorescence enhancement at 598 nm (Fig. 8B). Results from in vitro data showed that upon the addition of HClO, the red phosphorescence at 598 nm significantly increased, while the blue phosphorescence at 500 nm remained unchanged, thereby allowing ratiometric sensing of HClO with a ratio between I598nm/I500nm that linearly correlated with HClO concentration (Fig. 8C and D). Additionally, intracellular luminescence imaging analysis for this probe indicated a 10-fold enhancement in the I598nm/I500nm ratio with the treatment of HClO, with a further increase in the ratio obtained by LPS and phorbol myristate acetate (PMA) stimulation (Fig. 8E and F). To extend the application of these silica-based dual-emission core–shell nanostructures for ratiometric sensing and imaging, some other self-luminous fluorescent nanomaterials, such as carbon dots (Cdots),120,121 zeolite nanoparticles,122 and upconversion nanoparticles (UCNPs)123,124 have recently been proposed to replace the solid core of dye-doped silica cores as a reference for ratiometry. A typical example is described by Yang et al., who designed a Cdot-based ratiometric nanosensor for monitoring the change of intracellular GSH level and estimating the redox state in cancer cells using mesoporous SiO2 encapsulated RhB-loaded Cdot composite nanoparticles (RCDCNs) as a dual-emission core–shell type nanoprobe.121 Recently, Li et al. presented a core–shell type ratiometric nanoprobe for measuring nitric oxide (NO) in biological fluids, live cells, and tissues (Fig. 9A).125 This core–shell dual-emission nanoprobe was designed with UCNPs in the core as a donor and reference for luminescence resonance energy transfer (LRET), and RhB-derived molecules (RdMs)-encapsulated mesoporous silica in the shell as an acceptor, respectively. To avoid dye leaching, a β-cyclodextrin (βCD) layer was further modified onto the exterior of the particle to form the final product, labeled as UCNP@RdMMSN@βCD. Results from in vitro upconversion luminescence (UCL) spectrum analysis indicated that with the addition of NO, the UCL emission from UCNPs at 540 nm gradually decreased, whereas the UCL emission at 655 nm exhibited a negligible change (Fig. 9B). A possible reason for this is that NO induced the ring-opening reaction of RdMs to produce RdB with strong absorption between 500 and 600 nm, providing a good spectral overlap with the green UCL emission at 540 nm from the UCNPs for modulating LRET efficiency between UCNPs and RdMs. Using the ratio of I655/I540 as signal output, an excellent linear response between NO concentrations of 7.4 to 110 μM was obtained with a LOD of 73 nM (Fig. 9B). As shown in Fig. 9C, imaging with HeLa cells showed that the green UCL emission at 540 nm significantly decreased with increasing NO concentration, while red UCL emission had no obvious change. Thus, an increased I655/I540 ratio was observed. Ratiometric UCL sensing for monitoring NO levels in the serum and liver tissue samples was also performed. Compared with the control group (PBS), a slight increase in the ratio of I655/I540 was observed in the sham and ischemia group, while a larger increase in the octreotide (Oct)-preconditioned IR (Oct-IR) group was found (Fig. 9D). A similar result was found from ratiometric UCL imaging of liver tissue slices from sham, IR, and Oct-IR rats (Fig. 9D). These results illustrated that this developed UCNP@RdMMSN@βCD can detect the change of NO levels in the serum and liver tissue with/without Oct treatment. Based on the similar silica shell–UCNP core nanostructures, ratiometric fluorescence sensing and imaging has also been employed for the detection of other analytes, such as pH,124 cysteine,126,127 hydrogen sulfide (H2S),123 Hg2+,128 and hypoxia,129,130 and monitoring intracellular drug release.131,132
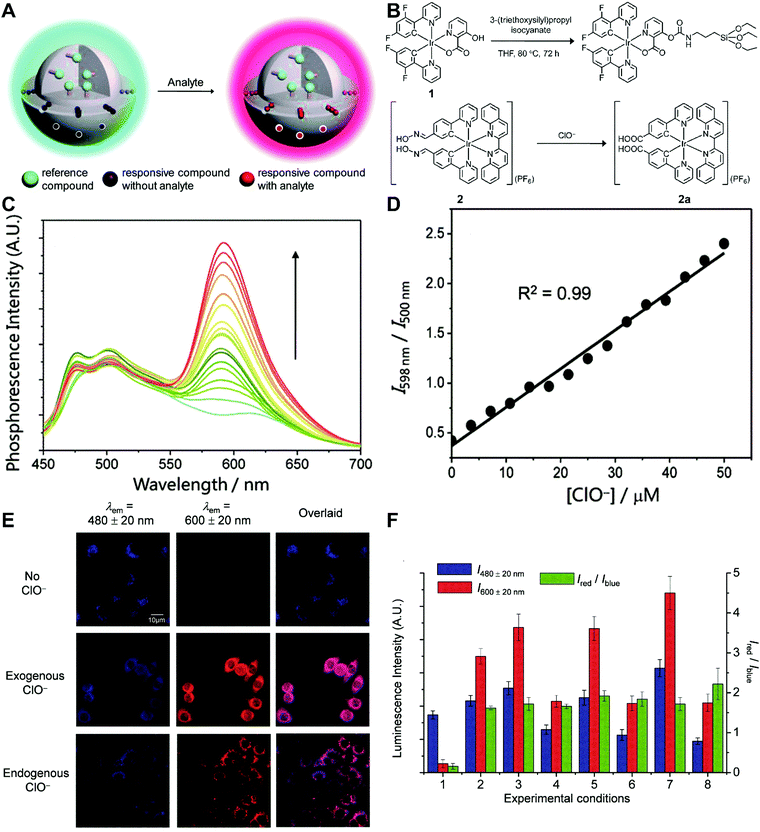 |
| | Fig. 8 (A) Design concept of core–shell typed ratiometric nanoprobes. (B) Schematic diagram of the preparation of iridium(III) silane analogue from complex 1, and recognition mechanism of complex 2 toward HClO. (C) Phosphorescence spectral traces of SiO2-1@mSiO2-2 in PBS at different HClO concentrations. (D) Plots of I598nm/I500nm as a function of HClO concentration. (E) Luminescence images of RAW264.7 cells treated with SiO2-1@mSiO2-2 (top), followed by incubation with NaClO (middle), and RAW 264.7 cells stimulated with LPS and PMA, and incubated with SiO2-1@mSiO2-2 (bottom). (F) Luminescence intensity of RAW264.7 cells recorded from the blue window (blue) and the red window (red) and the intensity ratio of Ired/Iblue (green). Reproduced with permission from ref. 119. Copyright 2015, The Royal Society of Chemistry. | |
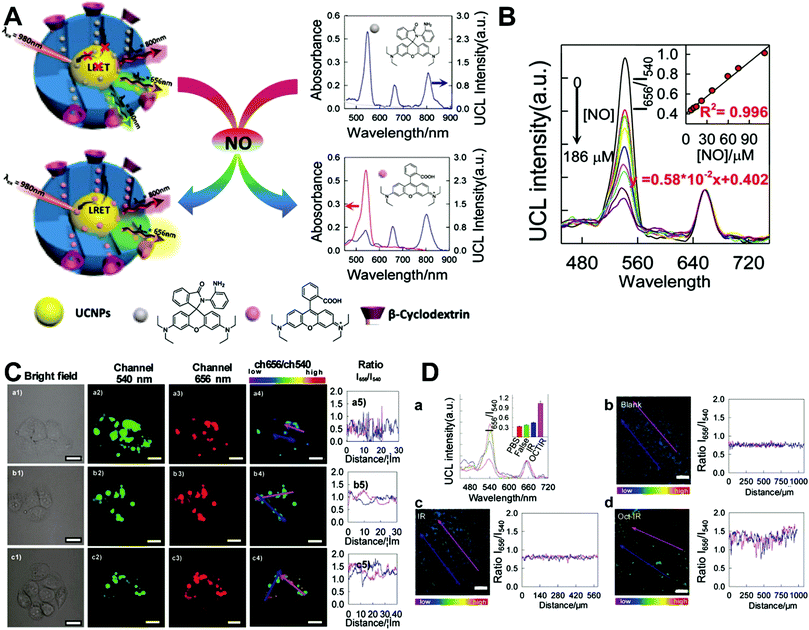 |
| | Fig. 9 (A) Schematic illustration of the sensing principle of upconversion nanoprobes for ratiometric luminescent measurement of nitric oxide. (B) In vitro response of this nanoprobe to different concentrations of nitric oxide. (C) Confocal microscopy luminescence images of HeLa cells after treatment with UCNP@RdMMSN@βCD and different concentrations of nitric oxide: (a) 0, (b) 0.2 mM, and (c) 0.4 mM, respectively. (D) UCL spectra and luminescence intensity ratios (inset) of the nanoprobes in serum (a), and luminescence ratiometric images at a depth of 300 μm of rat liver tissue slices incubated with the nanoprobes, and the corresponding intensity profile of a linear region across the liver tissue slices. Reproduced with permission from ref. 125. Copyright 2017, American Chemical Society. | |
3.2 Nanoparticle–dye nanoconjugates with dyes attached to the surface
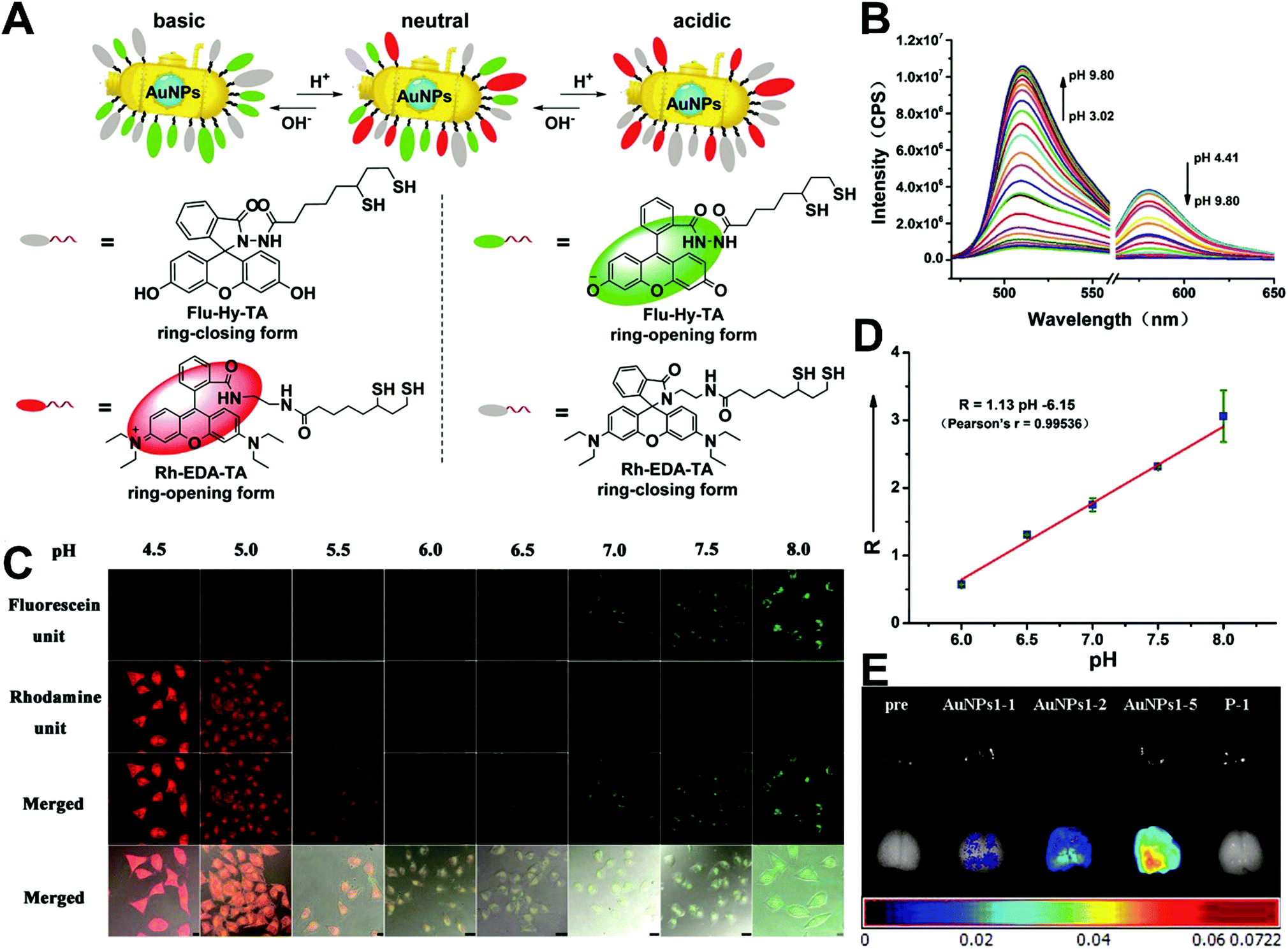
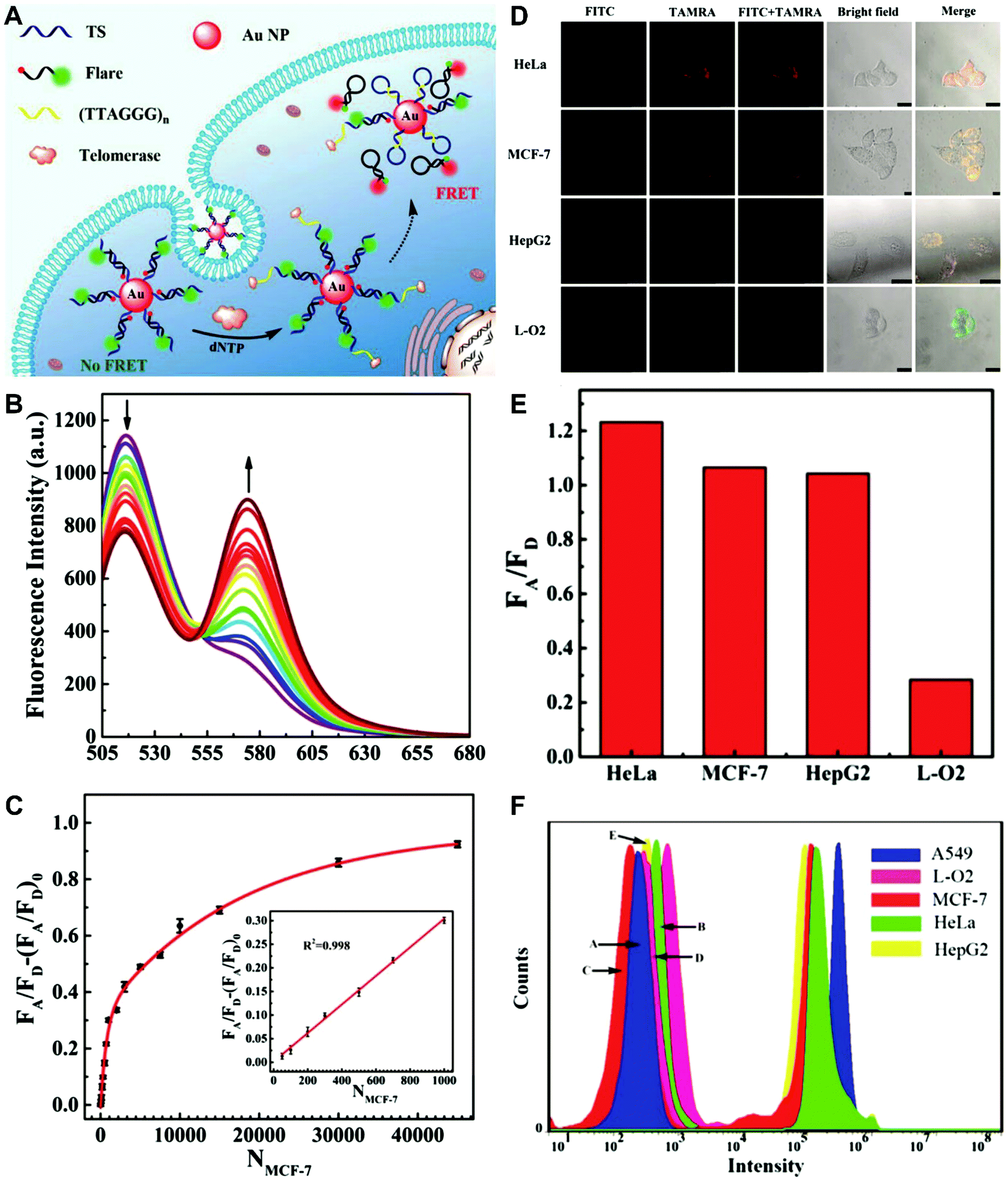
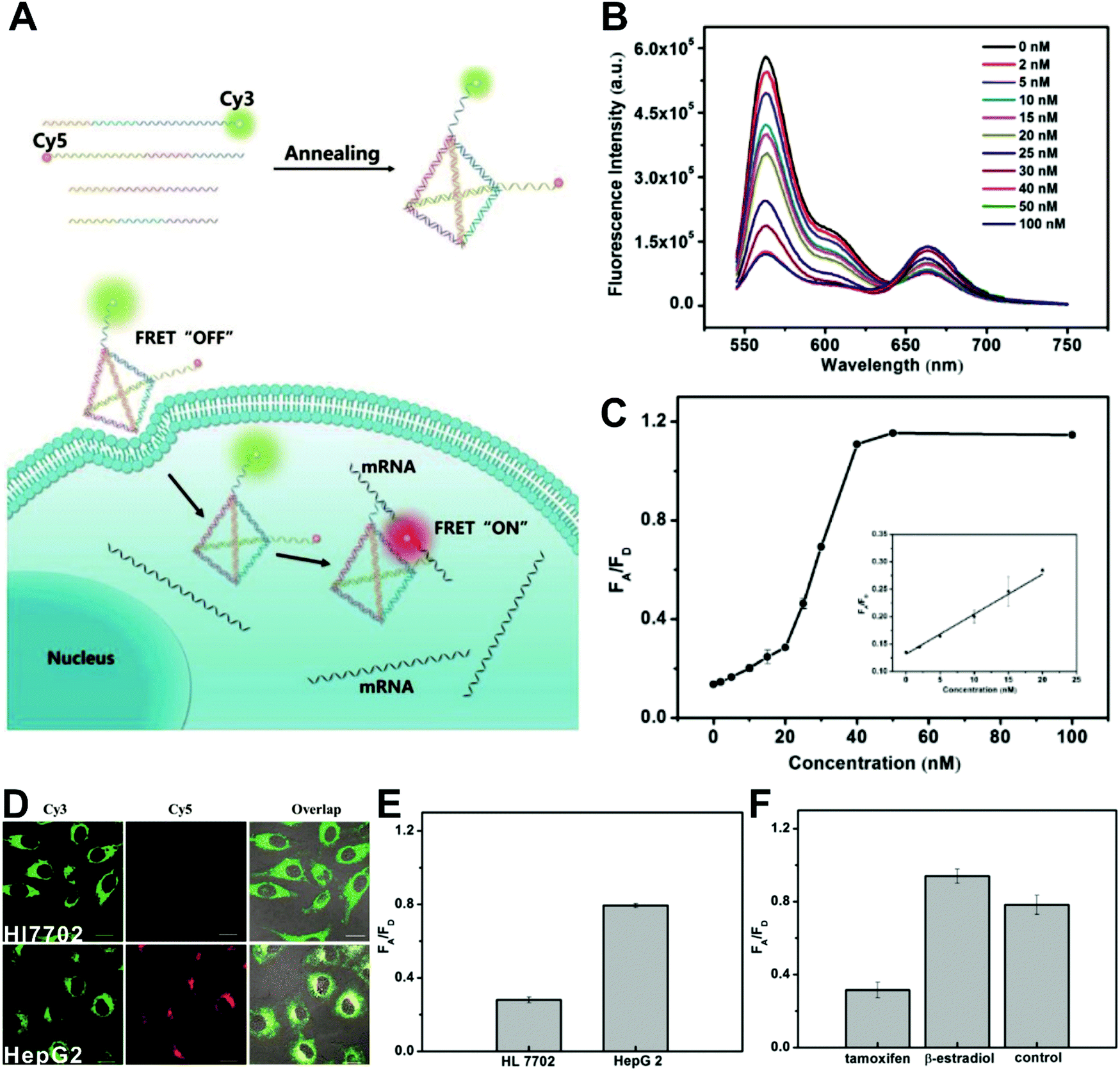
The simplest way to obtain dual-emitting nanoparticle–dye nanoconjugates for ratiometry is by conjugating two fluorescent dyes with different emission properties onto the surface of one non-luminous nanoparticle as a nanocarrier. In this case, one fluorescent dye is target-insensitive and can act as a reference for ratiometric pairing, and the other one is target-sensitive for specific response to target analytes or molecular events. Because of their remarkable advantages such as ease of synthesis and functionalization,133 excellent biocompatibility,134 and cell penetration ability,135 gold nanoparticles (AuNPs) have been introduced as non-luminous nanoparticle scaffolds for achieving dual-emitting nanoparticle–dye nanoconjugates.136,137 Yu and co-workers presented a dual-emission “gold nano-submarine” as a fluorescence nanoprobe for ratiometric imaging of pH in cells and in vivo (Fig. 10A).137 The designed “gold nano-submarine” was composed of AuNPs as nanocarriers and modified with pH-sensitive thiolated rhodamine and fluorescein derivatives through Au–S bonds, respectively. While the fluorescence at 510 nm from fluorescein significantly increased with increasing pH, the fluorescence at 580 nm declined gradually (Fig. 10B). Through use of the two emissions for ratiometry, intercellular imaging experiments displayed good linearity over the pH range from 6.0 to 8.0 (Fig. 10C and D). Encouraged by the live-cell analysis, additional in vivo imaging experiments were performed with Balb/c-nu mice, in which the results in Fig. 10E show that the “nano-submarine” could rapidly travel across the blood brain barrier (BBB) to realize accurate and sensitive imaging of pH in the brain, with the potential for early diagnosis and therapy of central nervous system diseases. Moreover, owing to their excellent fluorescence quenching ability, AuNP-based fluorescent DNA nanoprobes have gained increasing interest for intracellular sensing and imaging, especially as single-fluorophore-labeled nanobeacons and nanoflares.138–143 However, the accuracy and reliability of sensing and imaging based on these single-emission-based nanoprobes will likely be compromised by the local distribution of nanoprobes, the fluctuations of light sources or detectors, nuclease degradation, and protein absorption. Two-fluorophore-labeled AuNP-based nanoflares for ratiometric fluorescence sensing based on FRET are an effective method to overcome these limitations of existing AuNP-based nanoprobes. Recently, several AuNP-based ratiometric nanoflares have been implemented for reliable intracellular fluorescence imaging of mRNA,144 pH,145 K+,146 and telomerase.147 For example, Xu's group described a AuNP-based FRET nanoflare for ratiometric fluorescence imaging to monitor tumor-related telomerase activities in living cells (Fig. 11A).147 In this work, the designed nanoprobes consisted of telomerase-primer-sequence-modified AuNPs and nanoflares labeled with fluorescent donors (FITC) and acceptors (TAMRA) at two terminals for low FRET efficiency. With telomerase, the presence of target could trigger the displacement of nanoflares from the primer sequences to induce the formation of hairpin structures, thereby leading to the close proximity of donors and acceptors to produce high FRET efficiency. In vitro fluorescence analysis in Fig. 11B indicated the fluorescence signal from donors and acceptors exhibited a decrease at 517 nm and an increase at 576 nm with increasing telomerase concentration, and a good linear detection range from 50 to 1000 HeLa cells (Fig. 11C). Additionally, intracellular imaging analyses were carried out in three cancer lines of HeLa, MCF-7, and HepG2, as well as one normal cell line of L-O2. Results in Fig. 11D and E indicate that a higher telomerase activity was observed in all three types of cancer cells over normal L-O2 cells. Later, this result was further confirmed by flow cytometry analysis (Fig. 11F). All of these results demonstrate that AuNP-based FRET nanoflares can effectively distinguish cancer cells from normal cells, and can also quantify the changes of telomerase activity in living cells. In addition to the use of AuNPs as non-luminous nanoscaffolds, other particle nanocarriers including bacteriophage particles,148,149 dendrimers,150 poly(N-isopropylacrylamide) (PNIPAM) nanospheres,151 and virus particles,152 have also been used to construct similar dual-emitting nanoparticle–dye nanocomplexes with or without the incorporation of FRET technology, thereby allowing for ratiometric sensing and imaging of pH in vitro and even in tumor tissues in vivo.
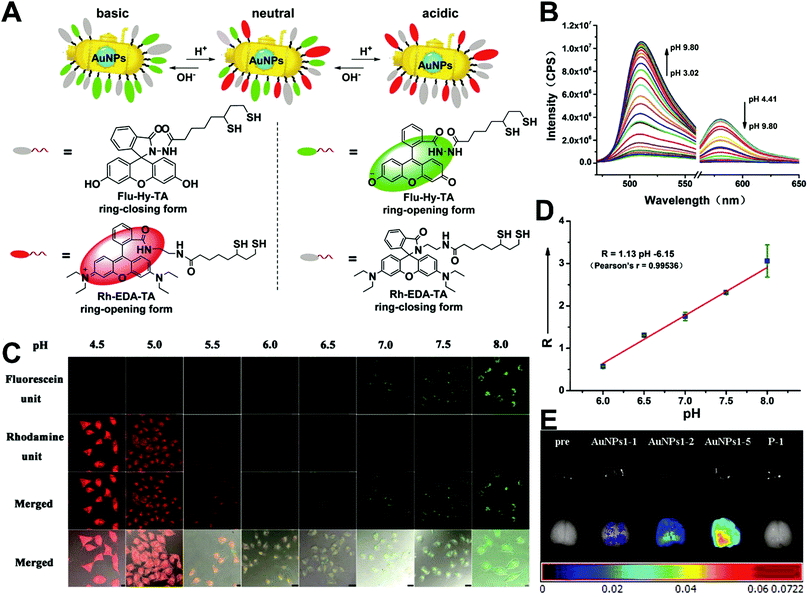 |
| | Fig. 10 (A) Structures of Rh–EDA–TA and Flu–Hy–TA at different pH. (B) Fluorescence emission spectral changes of “gold nano-submarines” at different pH values. (C) Confocal microscopy images of HeLa cells clamped at pH 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, and 8.0, respectively. (D) Intracellular pH calibration curve of “gold nano-submarines” in HeLa cells. (E) Examination of the intact BBB penetration of “gold nano-submarines” in mice. Reproduced with permission from ref. 137. Copyright 2016, American Chemical Society. | |
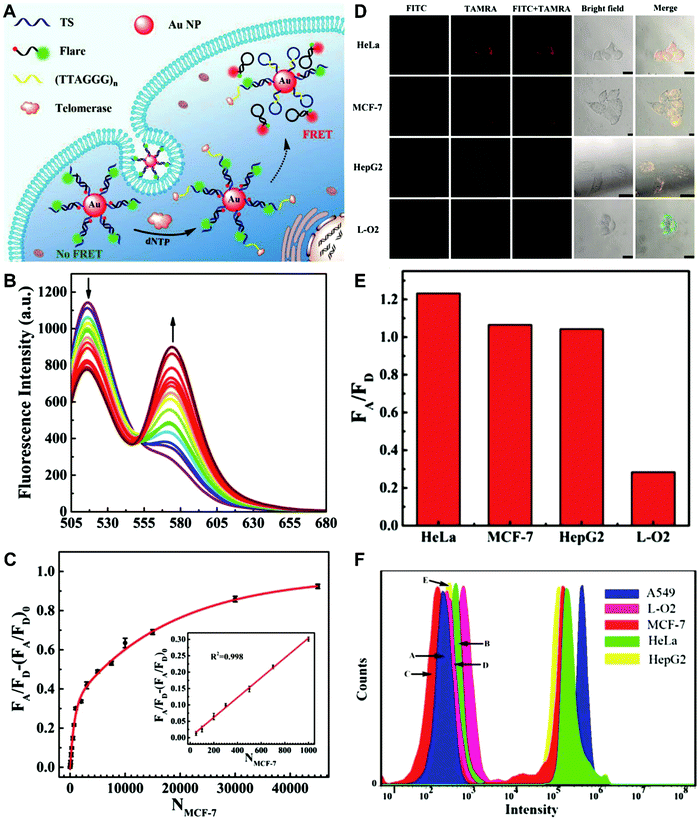 |
| | Fig. 11 (A) Schematic illustration of the FRET nanoprobe for ratiometric imaging of intracellular telomerase. (B) Fluorescence emission spectra of the designed probe in response to telomerase from different numbers of MCF-7 cells. (C) The relationship between the fluorescence ratio of acceptor to donor (FA/FD) and the number of cells. (D) Confocal images of HeLa, MCF-7, HepG2, and L-O2 cells after incubation with the FRET nanoprobe. (E) Fluorescence ratio values of different cell lines. (F) Flow cytometric analysis of various cell lines after incubation with or without the FRET nanoprobe. Reproduced with permission from ref. 147. Copyright 2017, American Chemical Society. | |
Another strategy for preparing nanoparticle–dye nanoconjugates with dual emission is through simple attachment of target-sensitive fluorescence molecules onto the surface of target-insensitive self-luminous fluorescent nanoparticles that act as a detection signal for ratiometric measurement, where the fluorescent molecules can be attached onto the nanoparticle surface through physical adsorption or chemical conjugation. After conjugation, there are two possible interactions between the attached fluorophore and self-luminous core nanomaterial.
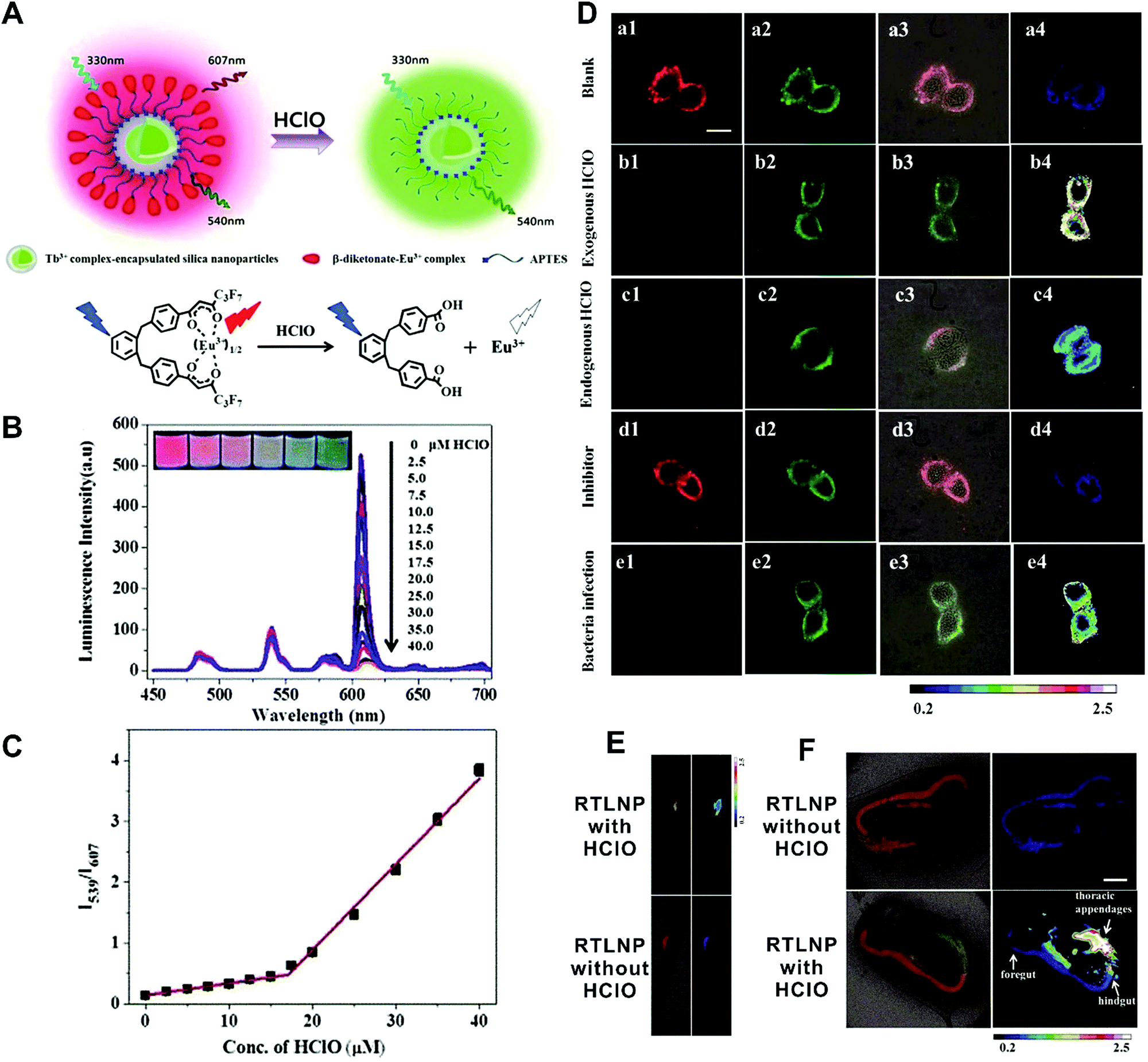
In the first situation, no obvious interaction occurs between materials, where the self-luminous nanomaterial is only used as a reference for ratiometric detection. Currently, these frequently-used self-referenced luminous nanoparticles mainly consist of dye-embedded silica,153,154 gold nanoclusters (AuNCs),155–157 fluorescent bovine serum albumin nanoparticles,158 Cdots,159,160 silicon nanodots,161 or quantum dots (QDs).162,163 Through a combination of the corresponding target-sensitive fluorescent dye units with unique responses to target analytes or molecular events, these dual-emission nanoparticle–dye nanoconjugates have been extensively applied for ratiometric sensing and imaging of diverse physical and physiological changes. For example, Tian et al. applied Cdots as a reference unit and hydroethidine (HE) as a specific response unit to develop a dual emission nanoparticle–dye nanocomplex for ratiometric sensing and imaging of superoxide anion changes upon oxidative stress in cancer cells.159 Yuan and co-workers prepared Tb3+-complex-encapsulated silica nanoparticles as self-luminous nanocarriers for a reference signal, and then β-diketonate–Eu3+ complex (1,2-bis[4′-(1′′,1′′,1′′,2′′,2′′,3′′,3′′-heptafluoro-4′′,6′′-hex-anedion-6′′-yl)-benzyl]-4-benzene–Eu3+, BHHBB–Eu3+) conjugated to the surface of silica nanoparticles to form dual-emissive nanoarchitectures for ratiometric sensing and imaging of HClO (Fig. 12A).154In vitro fluorescence measurements found that in the presence of HClO, the luminescence from Eu3+ complex at 607 nm showed a substantial decrease, whereas only a slight change was seen at 539 nm from the Tb3+ complex, thus enabling reliable ratiometric sensing for HClO (Fig. 12B). Further analysis of the luminescence quenching mechanism confirmed that the decreased luminescence of BHHBB–Eu3+ was due to the HClO-induced oxidization of the carbonyl group of β-diketonate into carboxylic acid, which caused the decomposition and luminescence quenching of BHHBB–Eu3+ (Fig. 12A). Two linear dynamic ranges were obtained between the ratio of I539/I607 and HClO concentration (Fig. 12C). Intracellular ratiometric fluorescence imaging results indicated that upon the addition of HClO, the red emission dramatically decreased, while the green emission was unchanged with a significant enhancement of I539/I607 from 0.21 to 2.25 compared with the HClO-free group (Fig. 12D). Similar findings, with enhanced ratios, were seen with the treatment of LPS, IFNγ, and PMA, which were used to induce the production of endogenous HClO (Fig. 12D). Additionally, the ratio decreased in the presence of 4-aminobenzoic hydrazide (4-ABAH, a myeloperoxidase inhibitor) (Fig. 12D). Further in vivo imaging experiments of HClO-treated zebrafish and Daphnia magna indicated enhanced I539/I607 values of over 1.7 and 6.4-fold in the zebrafish and in the thoracic appendages of Daphnia magna, respectively (Fig. 12E and F). All the results confirmed that the proposed dual-emitting nanoprobes can be employed for imaging exogenous and endogenous HClO in cells and small animals. Based on a similar design principle, a large number of dual-emissive nanoparticle–dye nanostructures have been reported for ratiometric sensing and imaging of anions,164,165 metal ions,153,166 pH,167,168 temperature,160 as well as tumor hypoxia155 at the cell, tissue, and organ levels.
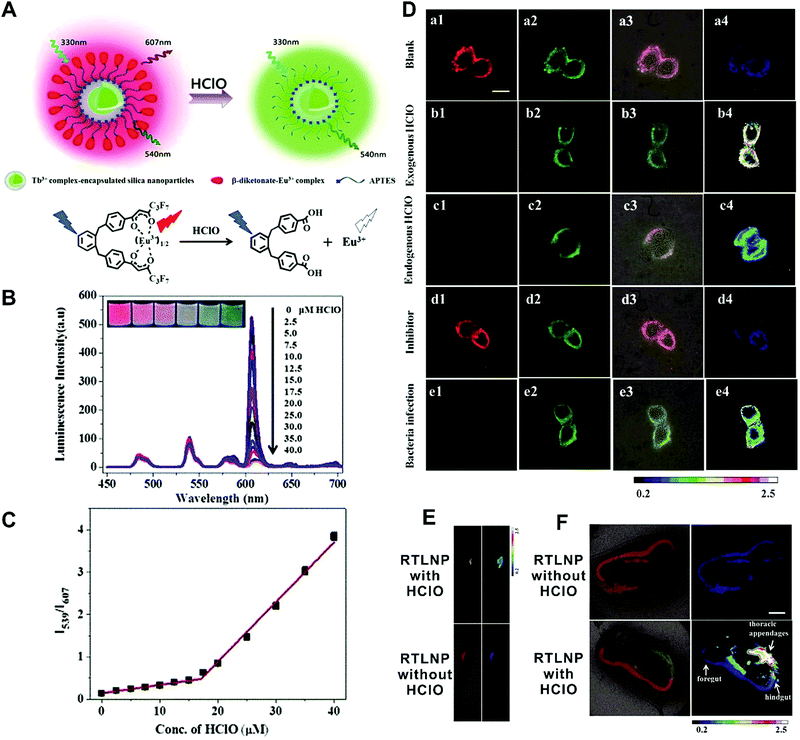 |
| | Fig. 12 (A) Design concept of a ratiometric luminescence probe based on crown-like dual-emissive silica nanoparticles modified by Tb3+ and Eu3+ complexes, and the luminescence quenching mechanism. (B) Time-gated emission spectra of the RTLNP in the presence of different concentrations of HClO. (C) The I539/I607 ratio response of the RTLNP to different concentrations of HClO. (D) Time-gated luminescence images of the RAW 264.7 cells with different treatments: (a) RTLNP without HClO, (b) RTLNP with HClO, (c) with LPS/IFN-γ/PMA and RTLNP, (d) with LPS/IFN-γ/PMA/4-ABAH and RTLNP, and (e) with Escherichia coli and RTLNP, respectively. (E and F) Time-gated luminescence images of RTLNP-loaded 5-day-old zebrafish (E) and Daphnia magna (F) with or without the treatment of HClO, respectively. Reproduced with permission from ref. 154. Copyright 2017, The Royal Society of Chemistry. | |
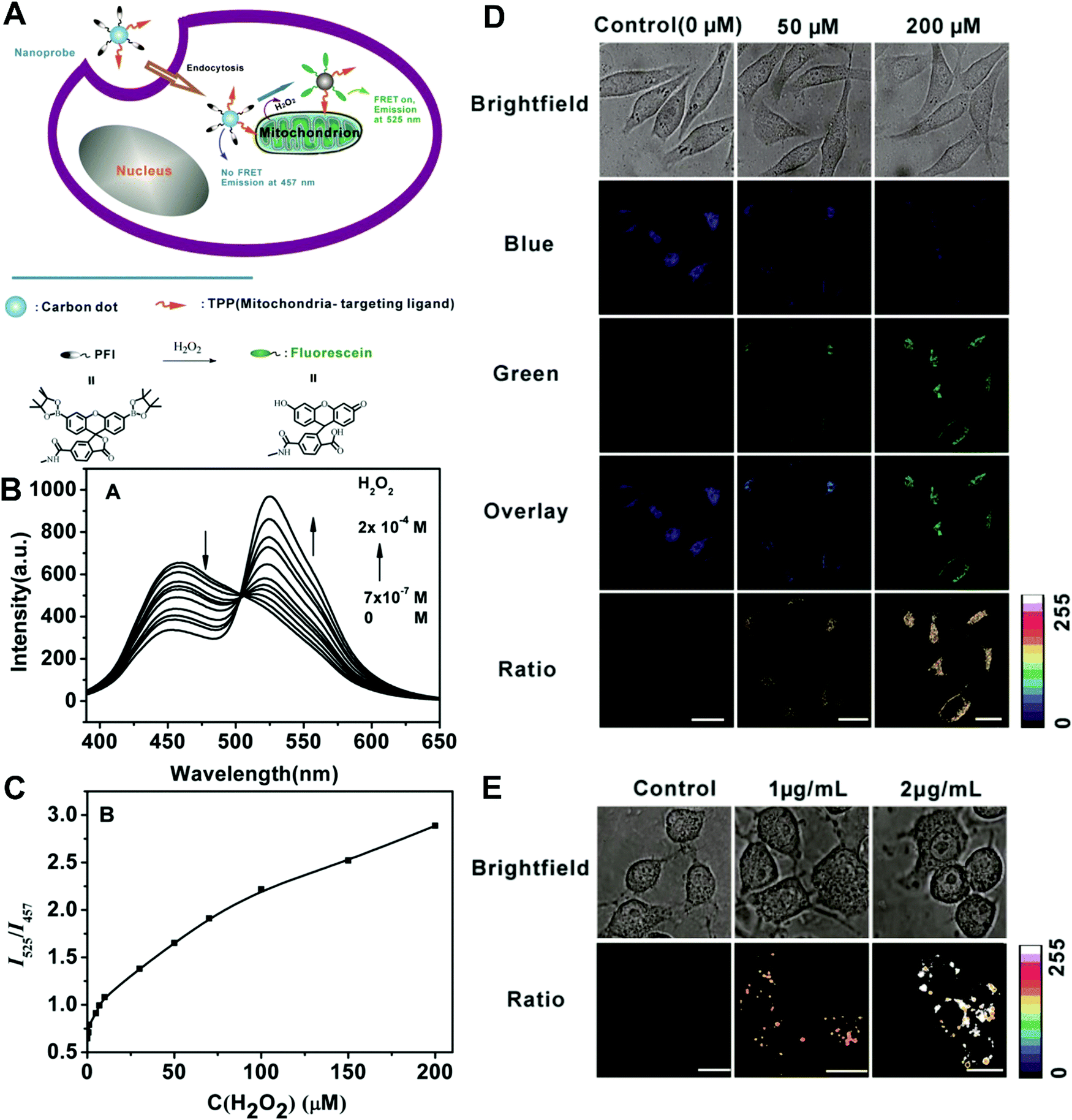
In the second situation, the self-luminous nanoparticles and the conjugated fluorescent molecules can interact with each other through FRET processes, in which the self-luminous core nanoparticles not only serve as energy donors, but also as nanocarriers for the conjugation of target recognition fluorescent acceptors. The occurrence of energy transfer between donor and acceptor triggers the production of two interconnected fluorescent signals, which can be used for ratiometry. As such, FRET systems have been widely adopted for exploring ratiometric measurement through target-induced modulation of FRET efficiency in a two-fluorophore cassette composed of donor and acceptor.169 In 2006, Nocera et al. reported on the ratiometric detection of pH based on the modulation of FRET efficiency by engineering the spectrum overlap from the absorption spectrum of pH-sensitive squaraine dye with the emission spectrum of pH-insensitive CdSe/ZnS QDs.170 Motivated by this seminal work, more FRET-based nanostructures based on nanoparticle–dye complexes have been developed for ratiometric sensing and imaging both in vitro and in vivo. Among reported dual-emitting nanoparticle–dye nanoconjugates, core nanomaterials include FRET donors such as QDs,171–176 GQDs,177 Cdots,178–183 Pdots,184–186 fluorescence MOF,187 and persistent luminescence nanoparticles (PLNPs),188 whereas the dyes for FRET acceptors have been traditional organic dyes or fluorescent proteins.189 Recently, Wu et al. reported Cdot-based fluorescent nanoprobes for ratiometric sensing and imaging of intracellular hydrogen peroxide (H2O2) based on FRET, where Cdots were used as the FRET donor and the nanocarrier for covalent attachment of a triphenylphosphonium (TPP) ligand for mitochondria targeting and a boronate-protected fluorescein (PF1) for H2O2 recognition (Fig. 13A).190 Without H2O2, the PF1 moiety is in a lactone form that is colorless and non-fluorescent, and no FRET occurs from the blue-emitting Cdots to PF1. On the contrary, the presence of H2O2 induces the conversion in the structure and spectrum of PF1 that favors FRET with decreased blue emission at 457 nm and increased green emission at 525 nm (Fig. 13B). Thus, a ratio of green/blue emission for ratiometric H2O2 detection is achieved. Fluorescence spectra analysis revealed the ratio increased steadily as the H2O2 concentration increased with a LOD of 0.75 μM (Fig. 13C). Intracellular imaging results indicated that the nanoprobe can determine exogenous H2O2 in L929 cells, and endogenously produced mitochondrial H2O2 by exposing Raw 264.7 cells to PMA (Fig. 13D and E). In addition, Singh and co-workers employed Cdot and naphthalimide-based FRET pair to build a selective ratiometric nanosensor for cancer screening by sensing thioredoxin reductase (TrxR) activity, which is overexpressed in numerous cancer cells.182
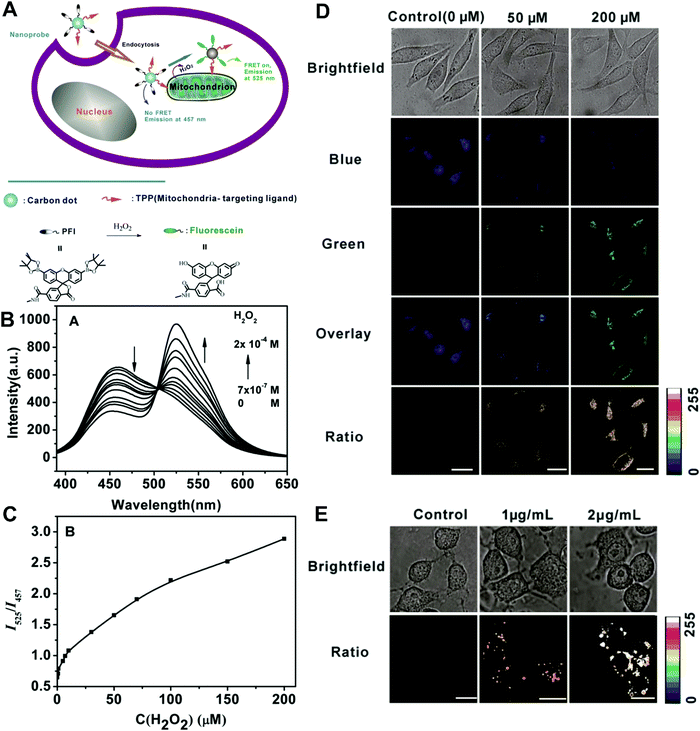 |
| | Fig. 13 (A) Schematic illustration for FRET-based ratiometric sensing of mitochondrial H2O2 in living cells by the nanoprobe. (B) Fluorescence spectra of the Mito-CD-PF1 nanoprobe in the presence of different amounts of H2O2. (C) Fluorescence intensity ratio of Mito-CD-PF1 as a function of H2O2 concentration in HEPES buffer. (D) Confocal fluorescence images of Mito-CD-PF1-stained L929 cells upon addition of 0 (control), 50 μM, and 200 μM H2O2 in the culture media. (D) Ratiometric fluorescence images of Mito-CD-PF1-stained Raw 264.7 cells with the PMA treatment at concentrations of 0, 1, and 2 μg mL−1. Reproduced with permission from ref. 190. Copyright 2013, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
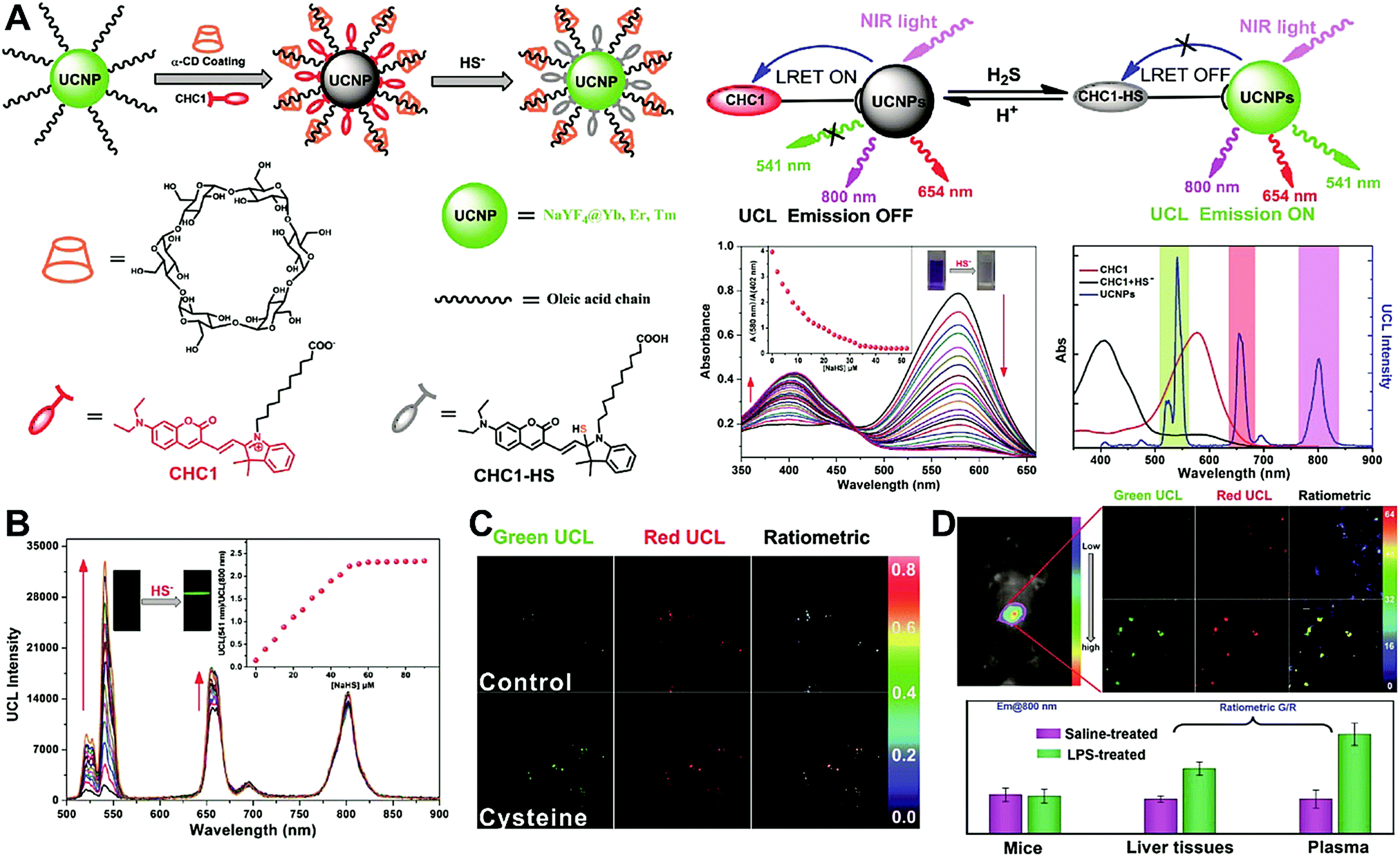
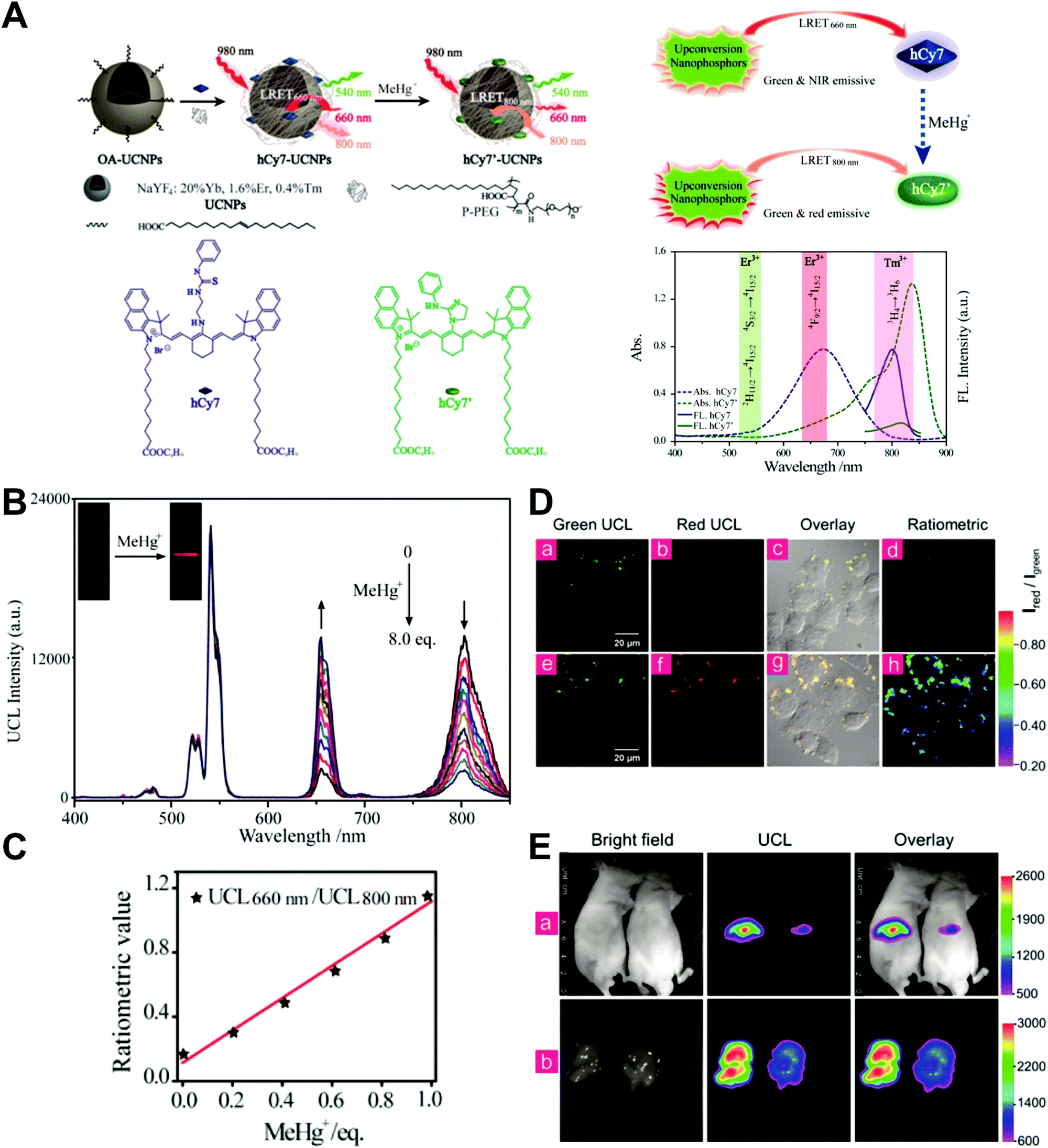
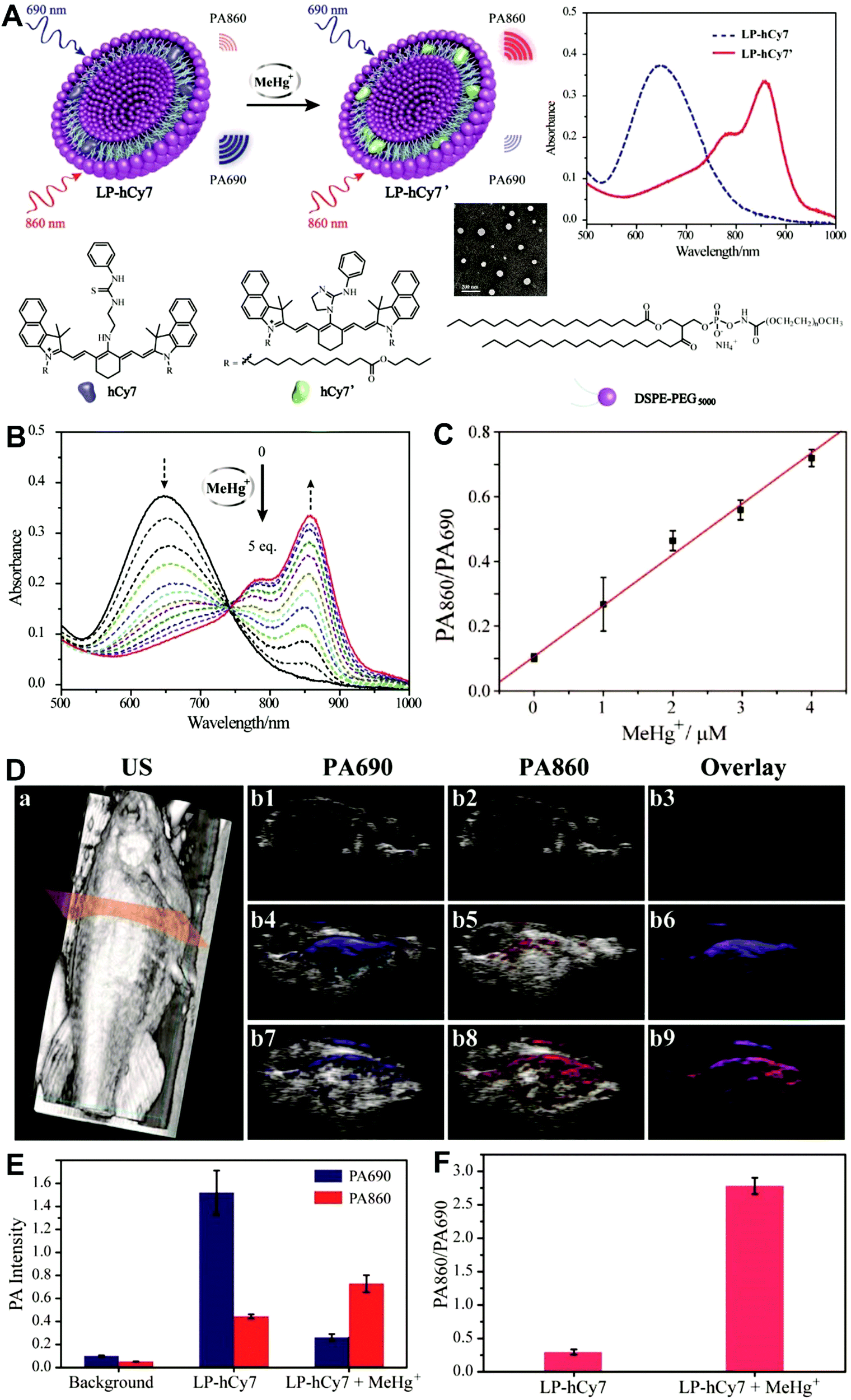
Unlike self-luminescent core materials, UCNPs exhibit many merits such as tunable multicolor emission, large anti-Stokes shift, no autofluorescence derived from biosamples, higher photostability, higher light penetration depth, and less damage to biosamples, which has spurred their development as a powerful platform for fabricating UCL imaging nanoprobes.191–195 Especially, the multiple shorter-wavelength (e.g. UV, visible, and/or NIR light) UCL emissions from different lanthanide components are observed under the single longer wavelength excitation (typically 980 nm laser excitation). Thus, the effective FRET process (also called luminescence resonance energy transfer, LRET) between the UCNP donors and energy acceptors can be realized through the modification of the absorption spectrum from the surface acceptor fluorophore to match one or more UCL emissions of UCNPs, thereby modulating LRET efficiency. The ratiometric UCL nanoprobes based on UCNPs can be used for target-induced absorption spectrum change of the acceptors on the UCNP surface, which can trigger the LRET conversion between “LRET on” and “LRET off” because of the absence or presence of the spectral overlap between the UCNP donor and the surface coated acceptor. By recording the change in LRET efficiency caused by the target, the ratiometric UCL nanosensors provide built-in self-reference correction for environmental effects and signal variations. To date, many UCNP-based ratiometric LRET nanoprobes have been developed based on surface modification with various energy acceptors for monitoring specific intracellular analytes. For example, Zhang's group presented a ratiometric UCL nanoprobe for imaging H2S (Fig. 14A).196 Here, this ratiometric nanoprobe, denoted as CHC1-UCNPs, was designed and constructed by using α-cyclodextrin (CD)-modified UCNPs as a nanocarrier for chemical coupling of coumarin–hemicyanine dye, which can be applied to detect H2S based on H2S-induced LRET from the UCL emission of UCNPs to the absorbance changes of CHC1 caused by H2S. Before adding H2S, the green UCL emission of UCNPs at 541 nm was largely quenched by the organic dye due to the strong LRET. After adding H2S, the maximum absorbance peak of CHC1 blue shifted from 588 nm to 410 nm and caused less spectral overlap with the green emission of UCNPs at 541 nm (Fig. 14A), thereby allowing for the recovery of the green UCL emission because of reduced LRET. However, no significant change was observed in the NIR UCL emission at 800 nm that could act as an internal reference for ratiometric detection of H2S (Fig. 14B). In vitro fluorescence characterization displayed an excellent linear relationship with the concentration of H2S ranging from 0 to 50 μM with a LOD of 0.13 μM (Fig. 14B). Live cell imaging experiments were completed by incubating this nanoprobe with N-ethylmaleimide (NEM)-pretreated Hela cells in the presence of a H2S donor, N-(benzoylthio)benzamide (NBB). The ratiometric UCL images were recorded with excitation at 980 nm, and results in Fig. 14C showed that a significant enhancement in the green UCL emission was observed in the cysteine-treated cells relative to the control cells, and thus a high ratio of green to red emission was obtained after the addition of cysteine for inducing the generation of H2S, illustrating that the designed CHC1-UCNPs can be applied for ratiometric monitoring of intracellular pseudo-enzymatic H2S production. Further in vivo UCL imaging for ratiometric detection of H2S level was performed by detecting H2S in a LPS-induced inflammation mouse model. Subsequently, through similar strategies, several research groups have used UCNP-based ratiometric UCL nanoprobes for determining various analytes, including important biological species,197,198 metal ions,199–202 pH,203–205 ROS,206–210 and toxins211 in living cells. An interesting work was recently presented by Li's group, who used UCNPs as FRET donors and nanocarriers for the conjugation of hydrophobic heptamethine cyanine dye (hCy7) to form dual-emission hCy7-UCNP nanoprobes by hydrophobic interactions (Fig. 15A).212 In this study, MeHg+ could induce an obvious red shift from 670 to 845 nm in the maximum absorption peaks of the conjugated Cy7. The two absorption peaks from Cy7 at 845 and 670 nm could better match the two UCL emissions at 800 and 660 nm from UCNPs, respectively, thus realizing the FRET-modulated ratiometric determination of MeHg+. In vitro study suggested that in the presence of MeHg+, the UCL emission intensity at 660 nm was significantly increased with a decrease of UCL emission at 800 nm (Fig. 15B). Using the ratio of UCL660nm/UCL800nm as a detection signal, a good linear relationship was achieved against the MeHg+ concentration with a LOD of 0.18 ppb (Fig. 15C), which is lower than those based on the use of UCL660nm/UCL540nm (0.58 ppb) or UCL800nm/UCL540nm (0.25 ppb) as signal output (the unchanged UCL emission at 540 nm was used as a reference for ratiometry). Encouraged by these results, intracellular ratiometric UCL imaging through the simple incubation of hCy7-UCNPs and HeLa cells with or without MeHg+ was recorded under the excitation of 980 nm. As shown in Fig. 15D, the addition of MeHg+ could induce a higher UCL660nm/UCL800nm ratio than that without MeHg+. Further in vivo imaging experiments for ratiometric detection were conducted with the intravenous injection of hCy7-UCNPs into mice, followed by treatment with PBS or MeHg+. Results in Fig. 15E suggested that the UCL emission at 800 nm in the liver of MeHg+-pretreated mice was obviously decreased, thus leading to a higher ratio of UCL660nm/UCL800nm. All these findings indicated that the hCy7-UCNPs could measure MeHg+ by ratiometric strategy both in vitro and in vivo.
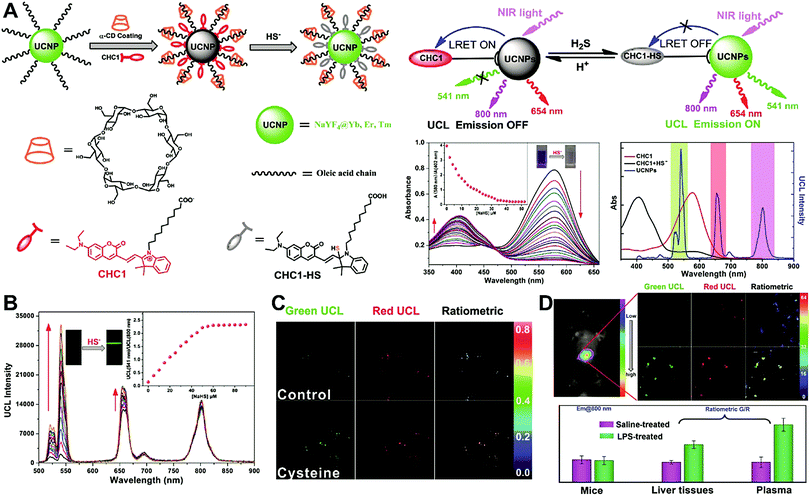 |
| | Fig. 14 (A) Schematic illustration for experimental design and proposed mechanism for the UCL detection of H2S. (B) UCL spectra of CHC1-UCNPs in HEPES buffer upon gradual addition of H2S at different concentrations, and plots of the UCL emission ratio intensity of UCL541/UCL800 as a function of H2S concentration (the inset). (C) Ratiometric UCL images of Hela cells with or without the treatment of cysteine. (D) Ratiometric UCL images of endogenous H2S levels in live mouse tissues, and the average ratiometric UCL intensities of tissues. Reproduced with permission from ref. 196. Copyright 2014, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
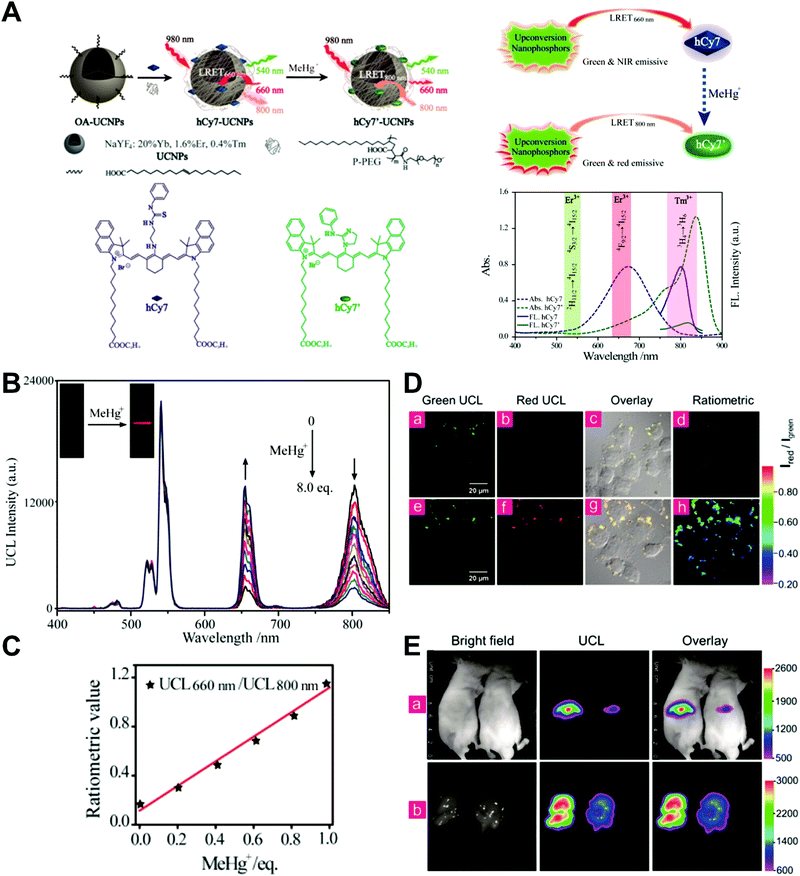 |
| | Fig. 15 (A) Schematic illustration of the synthesis of UCNPs-hCy7 and its sensing of MeHg+ with a change in UCL emission. (B) UCL spectra of hCy7-UCNPs in the aqueous solution with different concentrations of MeHg+. (C) The ratio of UCL660nm/800nm as a function of MeHg+ concentration. (D) Ratiometric UCL images in living HeLa cells (top, a–d) and MeHg+-pretreated Hela Cells (bottom, e–h) incubated with hCy7-UCNPs, with ratiometric UCL images from the ratio of red to green channels. (E) In vivo UCL images of hCy7-UCNPs-pretreated living mice injected intravenously with normal saline (left mouse) or MeHg+ solution (right mouse) (top, a), and the corresponding UCL images of the livers (bottom, b). Reproduced with permission from ref. 212. Copyright 2013, American Chemical Society. | |
3.3 Hybrid nanoparticles with dual emission
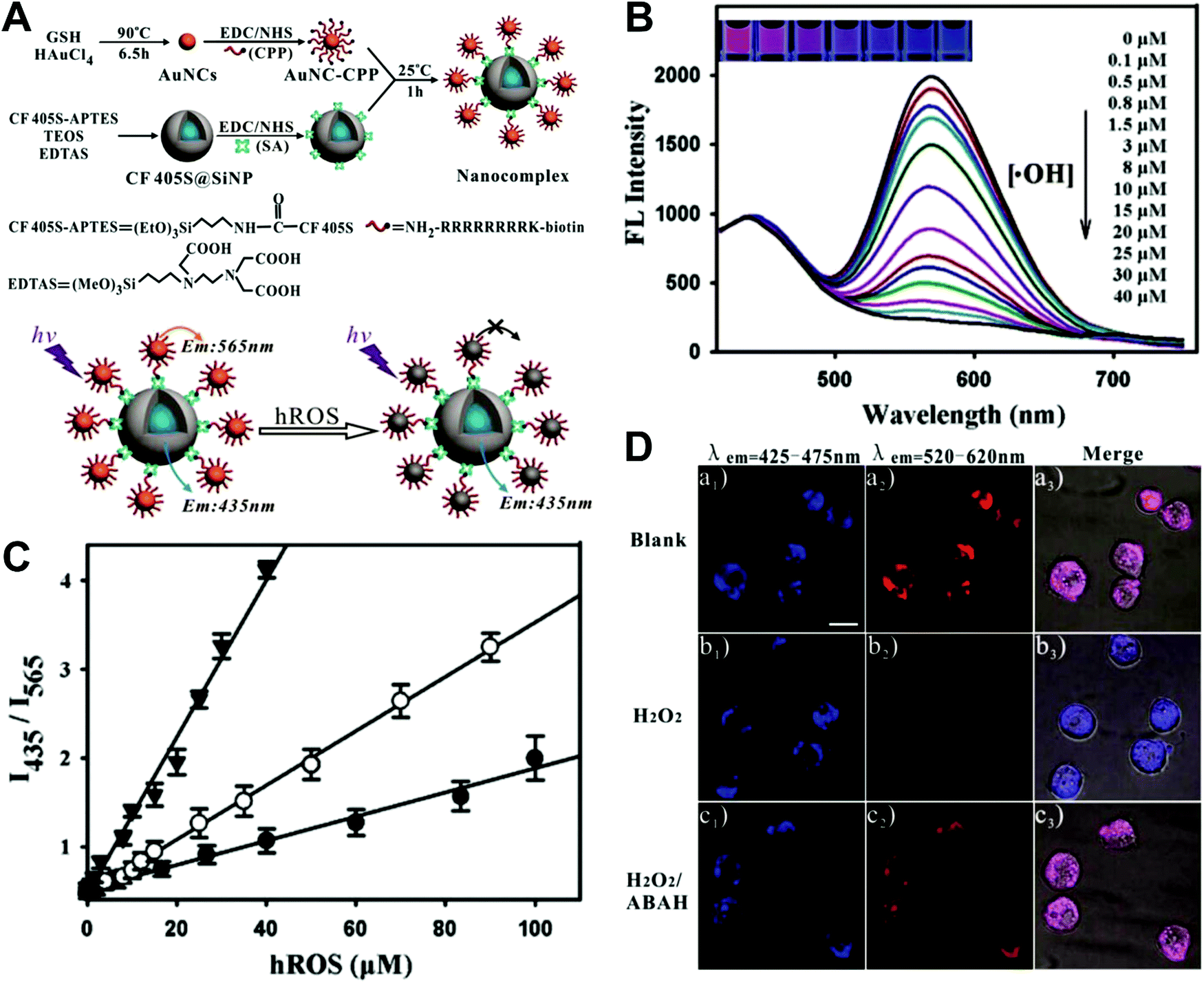
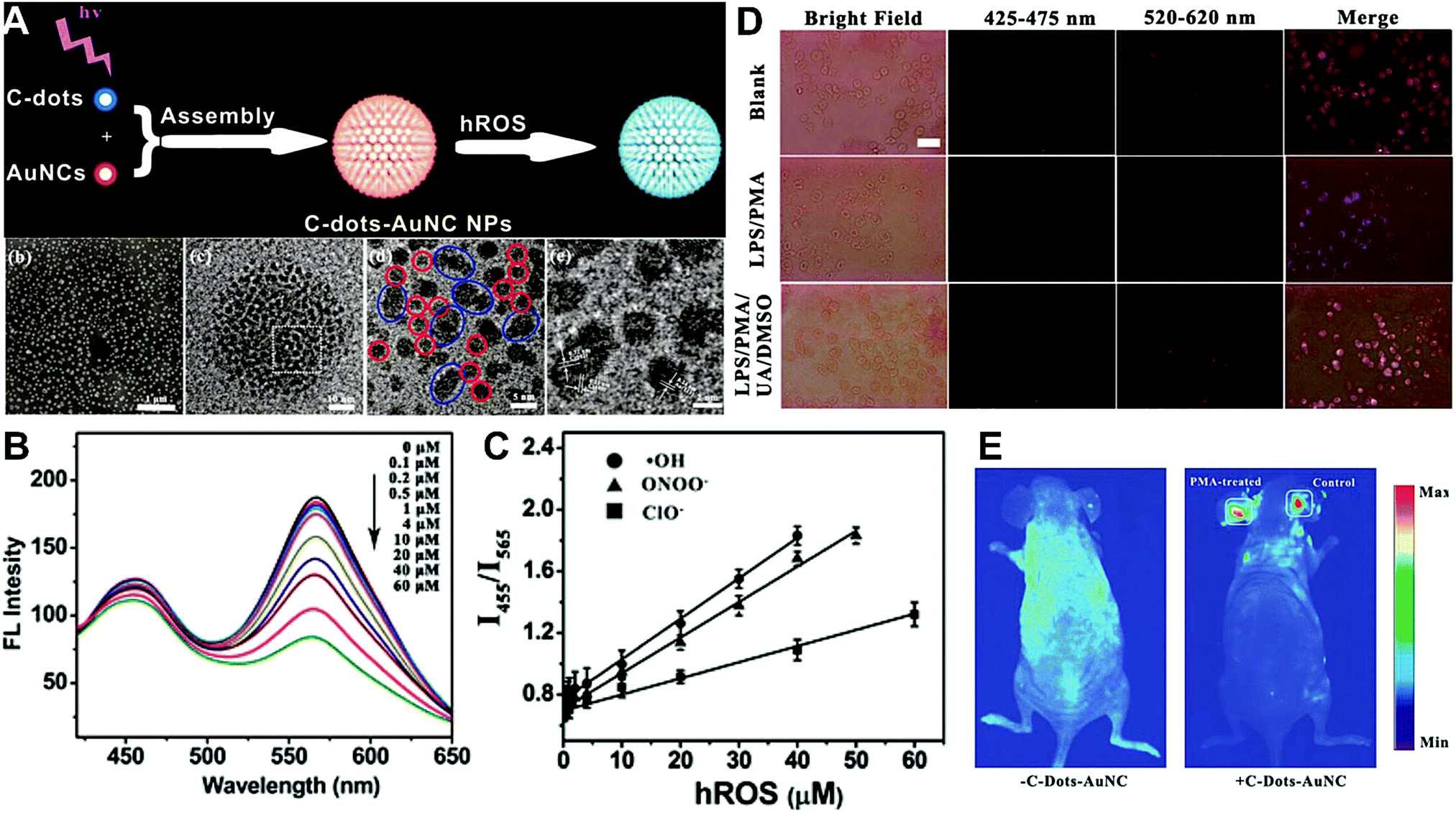
Hybrid nanoparticles are generally composed of at least two or more different kinds of nanomaterials, which have been developed as promising platforms for sensing, imaging, and even therapeutic applications.213 Hybrid nanoparticles not only retain the beneficial properties of each nanomaterial component, but also provide the opportunity for systematically tuning the features of the hybrid nanomaterials through integrating various functional components.214 Hybrid nanoparticles have become one of the most effective means for establishing dual-emission fluorescent nanoparticles, which have attracted substantial research interest. Compared with conventional organic fluorophores, dual-emission hybrid nanoparticles are obtained by the simple combination of two or more fluorescent nanomaterials with different fluorescence emission properties, which does not require elaborate molecular design and sophisticated synthesis. Moreover, dual-emission hybrid nanoparticles effectively avoid the drawback of photobleaching from traditional organic fluorescent dyes. In general, these precursor fluorescent nanomaterials mainly include dye-doped silica or polymer nanoparticles, QDs, graphene QDs (GQDs), Cdots, AuNCs, silver nanoclusters (AgNCs), and other luminescent nanomaterials. These nanomaterials can be easily conjugated or assembled together to form various hybrid nanostructures such as core–satellite nanohybrids through physical adsorption or chemical coupling. A typical example was from Lu and co-workers, who reported a dual-emission fluorescent nanohybrid (DEFN) nanoprobe for accurate sensing and imaging of high ROS (hROS) in living cells (Fig. 16A).215 In this case, the designed DEFN nanoprobe was prepared through the crown nanoparticle assembly of satellite AuNCs onto a dye-encapsulated core silica nanoparticle based on the biotin–streptavidin system and succinimide coupling chemistries. In vitro studies suggested the fluorescence at 565 nm from the AuNCs reduced remarkably with increasing hROS concentrations, whereas the fluorescence at 435 nm from the encapsulated dye remained unchanged acting as an internal reference, thus achieving reliable ratiometric sensing for hROS quantification (Fig. 16B and C). Moreover, with the decrease of fluorescence at 565 nm, an obvious change in the fluorescence color of DEFN was seen from reddish violet to blue (Fig. 16B). Live cell imaging experiments were done through simple incubation of DEFN with three different cell lines, including HeLa cells, HL-60 cells, and RAW 264.7 cells. Imaging results in Fig. 16D showed that the red fluorescence of AuNCs significantly diminished in the presence of H2O2. After the addition of a myeloperoxidase inhibitor, ABAH, to suppress the generation of hROS, the red emission was recovered, while the blue emission remained unchanged. Similar results were obtained in the presence of other hROS species, demonstrating that the DEFN nanoprobes can be used for rapid and sensitive sensing and imaging of hROS in living cells. Similar dual-emission core–satellite nanohybrids have also been applied for ratiometric sensing and imaging of metal ions,216–223 pH,224,225 ROS,226 small molecules with biological activity,227,228 temperature,229 and tumor hypoxia.230 Besides, a binary heterogeneous assembled nanohybrid with dual-emission was recently reported by Qu and colleagues for ratiometric sensing of hROS, where the nanohybrid, called Cdots–AuNC, was designed and constructed on the basis of the assembly of Cdots and AuNCs by a carbodiimide-activated coupling reaction (Fig. 17A).231 The developed Cdots–AuNC showed dual emission fluorescence at 455 and 565 nm for ratiometric sensing, one signal from the AuNCs that could specifically respond to hROS with the other signal from the Cdots serving as an internal reference. In vitro fluorescence analysis revealed that with the increase of hROS concentration, a substantially decreased fluorescence was observed at 565 nm, whereas no obvious change was observed at 455 nm (Fig. 17B). An excellent linear correlation was obtained between ratiometric fluorescence and the concentration of hROS, with a LOD of 0.5 μM (Fig. 17C). In addition, the dual-emission Cdots–AuNC allowed for rapid imaging and monitoring of hROS concentration changes caused by LPS and PMA treatment in RAW 264.7 cells, with high sensitivity and contrast, and the produced ROS could also be scavenged by dimethyl sulfoxide (DMSO) and uric acid (UA) to induce the fluorescence recovery (Fig. 17D). Further experiments in vivo showed the ability of Cdots–AuNC nanoprobes for reliable and sensitive detection of hROS in mice with local ear inflammation induced by successive exposure to PMA (Fig. 17E). Nonetheless, conventional synthetic approaches for dual-emission nanohybrids generally rely on a “three-step” approach, incorporating two separate preparation steps for two component nanomaterials with different emission bands, and a conjugation or assembly step of the two nanomaterials through various physical and chemical reactions. This strategy requires tedious multistep synthesis and sophisticated modification procedures and is not ideal for clinical implementation.
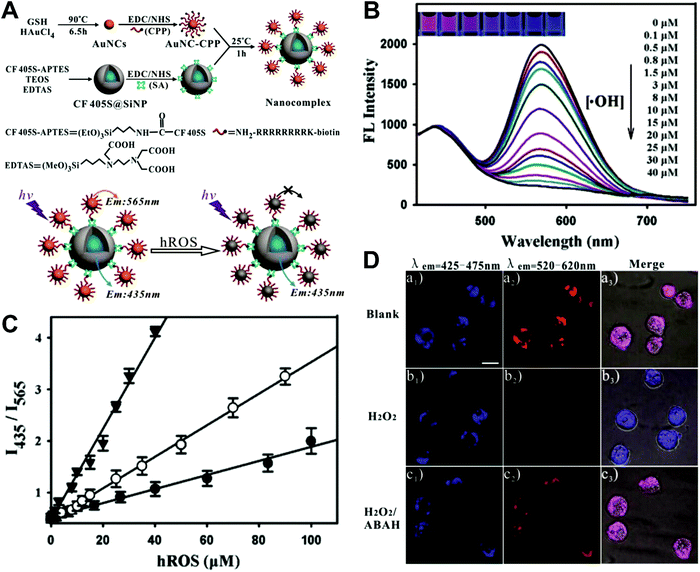 |
| | Fig. 16 (A) Schematic illustration of the DEFN synthesis, and its sensing ability for hROS detection. (B) Fluorescence spectral responses of the DEFN to hROS of varying concentrations. (C) Working curves of the DEFN-based ratiometric sensor in response to hROS, including ˙OH (triangle), ONOO− (circle), and ClO− (dot). (D) Confocal fluorescence microscopy images of HL-60 cells with different treatments of (a) no stimulation, (b) H2O2, and (c) H2O2 and ABAH, after incubating with DEFN, respectively. Reproduced with permission from ref. 215. Copyright 2013, American Chemical Society. | |
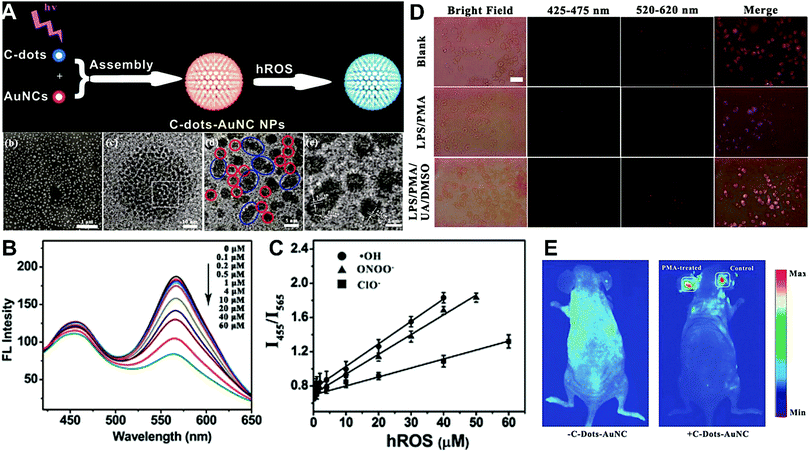 |
| | Fig. 17 (A) Schematic illustration of the construction of Cdots–AuNC and the working principle of the detection of hROS, and the corresponding SEM and TEM images of Cdots–AuNC (a–d). (B) Fluorescence spectra of Cdots–AuNC in the presence of hROS at various concentrations. (C) Ratiometric fluorescence as a function of the hROS concentration. (D) Bright field and fluorescence images of live murine macrophages (RAW 264.7) under different treatments of only Cdots–AuNC (top), LPS/PMA/Cdots–AuNC (middle), and LPS/PMA/UA/DMSO/Cdots–AuNC, respectively. (E) In vivo imaging of hROS using Cdots–AuNC in an acute local inflammation in the ear by topical application of PMA. The left ears of the mice were treated with PMA, while the right ears were set as control. Reproduced with permission from ref. 231. Copyright 2013, American Chemical Society. | |
3.4 Single nanoparticle with intrinsic dual emission
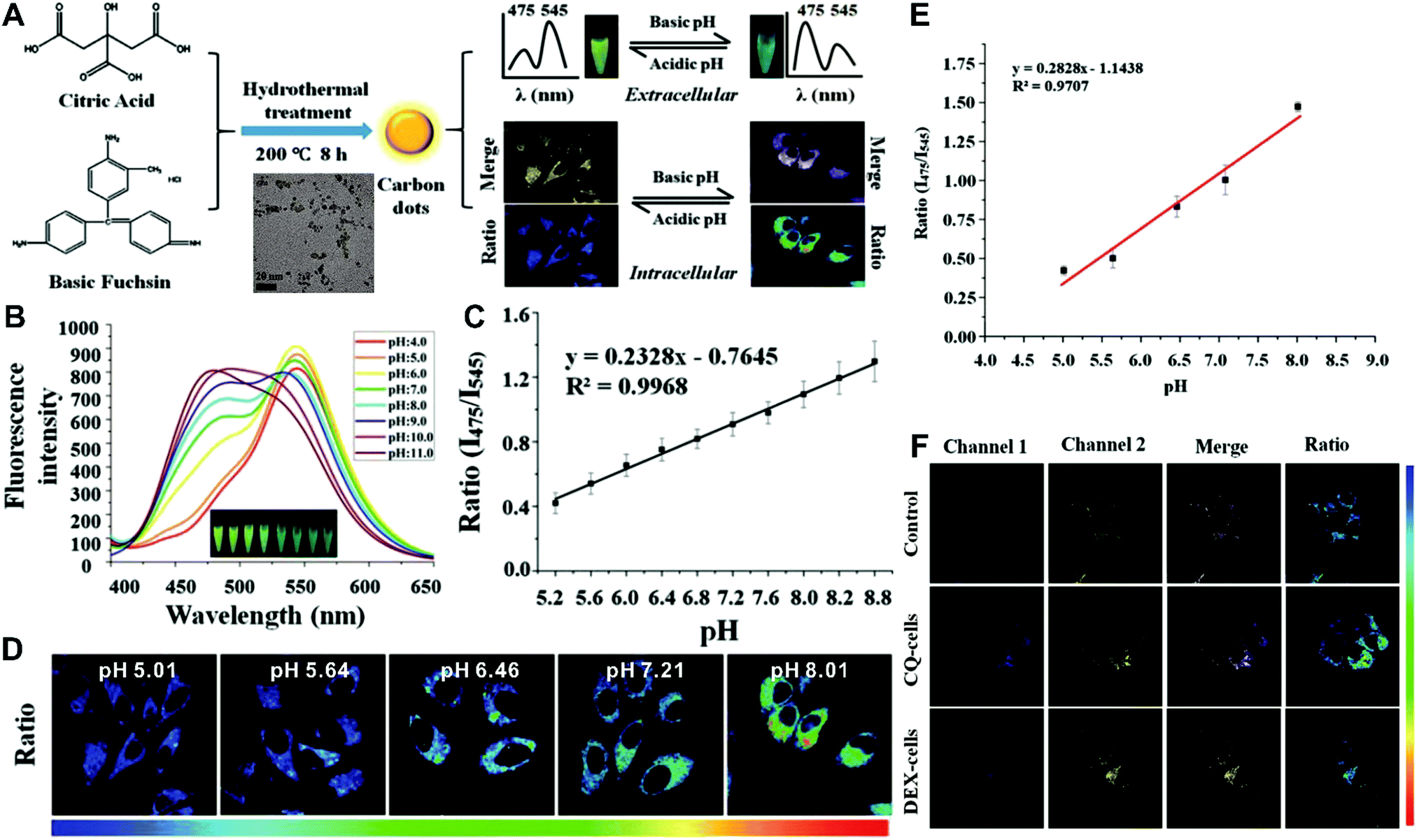
Although the above-mentioned strategies have been successfully employed to fabricate ratiometric nanoprobes for fluorescent molecular sensing and imaging, all these ratiometric designs require at least two or more kinds of fluorophores such as organic dyes, fluorescent proteins, and fluorescent nanomaterials (e.g. QDs, Cdots, Pdots, and AuNCs). To achieve ratiometric sensing, two or more fluorophores are required to be pre-conjugated or pre-assembled together through sophisticated modification of fluorophores, and cumbersome synthetic procedures. As a consequence, these design strategies for ratiometric sensing systems depend on elaborate design and ingenious fabrication, which is often time-consuming, arduous and expensive. To address these shortcomings, single fluorophores with intrinsic dual-emission properties provide enormous advantages as alternatives for developing ratiometric sensing and imaging. This is because the transition from two or more fluorophores to a sole fluorophore not only signifies a fundamental breakthrough, but also significantly simplifies ratiometric sensing design, and meanwhile accelerates the development of their corresponding applications. Toward this end, increasing attempts have been devoted to exploring various nanomaterials with intrinsic dual-emissions. These reported intrinsic dual-emission nanomaterials mainly involve QDs,232–234 Cdots,235,236 and AgNCs.237 For example, Yang and co-workers first prepared D-penicillamine-passivated Mn2+-doped (CdSSe)ZnS (core)shell nanocrystals (MnQDs) for ratiometric temperature sensing, where the intrinsic dual emission peaks that allow for ratiometry rely on thermally coupled emissive states including the excitonic state and the Mn2+-dopant state in MnQDs, respectively.238 Later, several similar Mn2+-doped dual-emitting QDs were also reported for ratiometric sensing of pH,232 H2O2,233 metabolites,234 and temperature.239,240 Nevertheless, QDs exhibit relatively high toxicity, severely limiting their further applications.241,242 As an alternative, researchers have focused on exploring nontoxic dual-emission Cdots for building ratiometric sensing. Wang et al. synthesized Cdots with intrinsic dual emissions at 475 nm and 545 nm for intracellular ratiometric fluorescence pH imaging through a one-pot hydrothermal reduction of citric acid and basic fuchsin (Fig. 18A).236 Fluorescence analysis in vitro revealed that the two emissions of the as-prepared Cdots were both pH-sensitive, with the fluorescence intensity at 475 nm increasing continuously, and the fluorescence intensity at 545 nm exhibiting a slight increase from acidic to neutral conditions, and then decreasing in alkaline solutions (Fig. 18B). Through creating a ratio of the two emissions (I475nm/I545nm) for ratiometry, a good linear correlation was observed over the pH range of 5.2 to 8.8 (Fig. 18C). Fluorescence imaging experiments conducted by incubating this nanoprobe with HeLa cells indicated that the proposed nanoprobes can be applied for the detection of intracellular pH from 5.0 to 8.0 with the ratio of I475nm/I545nm as a signal readout (Fig. 18D and E). Further live cell imaging analysis in Fig. 18F illustrated the potential of this ratiometric nanoprobe for monitoring the pH changes under the stimulation of chloroquine (CQ) for pH increase and dexamethasone (DEX) for pH decrease, respectively. Besides dual-emission QDs and Cdots, dual-emitting AgNCs were also synthesized to develop ratiometric fluorescence sensing for ROS detection in living cells. Shao et al. reported the synthesis of denatured lysozyme (dLys) coated AgNCs (dLys-AgNCs), which exhibit two emission peaks at 640 nm and 450 nm from AgNCs and aromatic amino acids in the ligand of lysozyme, respectively.237 With hydroxyl free radicals, the emission at 640 nm was quenched gradually, whereas the emission at 450 nm significantly increased, thus enabling ratiometric sensing and imaging of hydroxyl radicals.
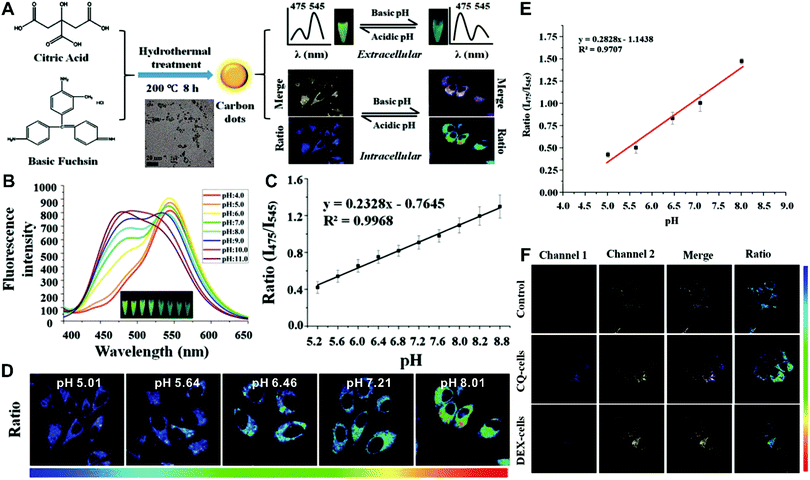 |
| | Fig. 18 (A) Schematic diagram for the preparation of label-free Cdots and their application for intracellular pH sensing. (B) Fluorescence spectrum of Cdots in PBS with different pH values ranging from 4.0 to 11.0. (C) Linear relationship of the ratiometric fluorescence intensity (I475nm/I545nm) versus pH values. (D) Ratiometric calibration of pH in living cells. (E) Calibration curve from (D). (F) Confocal microscope analysis of HeLa cells treated with CQ and DEX. Reproduced with permission from ref. 236. Copyright 2016, American Chemical Society. | |
3.5 DNA Nanostructures
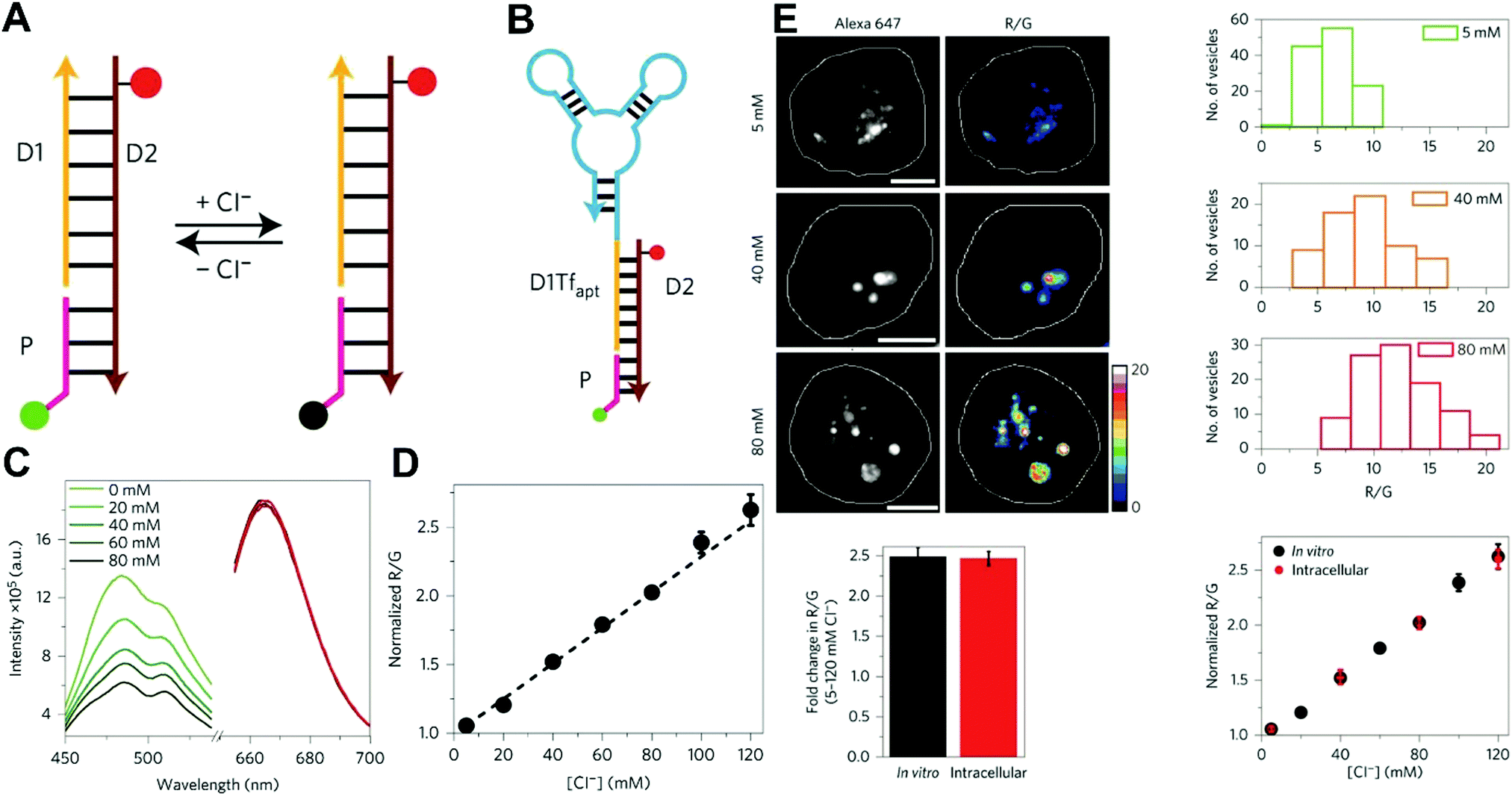
Owing to their outstanding specificity and versatility for molecular recognition, nucleic acids such as DNA, RNA, and aptamer, are well-suited as nanoscale building block materials to design and fabricate various functional nanoarchitectures.243,244 DNA-based nanostructures are artificially designed for specific response to biomolecular targets or molecular events to trigger their corresponding state changes for a measurable output, where DNA-based stimuli responses are achieved through using different mechanisms such as differential hybridization, strand displacement, enzymatic reaction, and conformation changes.245–247 DNA-based nanostructures provide several unique merits including excellent programmability and biocompatibility, high selectivity and affinity, low cytotoxicity, remarkable cell permeability, and strong resistance against enzymatic degradation, and nanoscale controllability.248–250 These excellent properties render DNA-based nanomaterials the most attractive tools for biosensing and bioimaging applications. Currently, numerous well-defined DNA-based nanostructures including 1D, 2D, and 3D nanostructures have proven that they can provide a specific biological response in vitro and in vivo.251,252 For example, several DNA tetrahedron-based nanoprobes have been reported for sensing tumor-related mRNA and microRNA in living cells.253–255 Nonetheless, these reported DNA-based nanoprobes are single intensity-dependent sensing probes that can result in false detection signals arising from nuclease digestion, nonspecific binding of protein, as well as thermodynamic fluctuations. In addition, such single intensity-dependent sensing nanoprobes are also compromised by the local distribution of nanoprobes and fluctuations from light sources and detectors. To resolve these issues, DNA-based ratiometric nanostructures have been widely proposed to eliminate the false positive signals, and meanwhile minimize the influences of system fluctuations on molecular sensing and imaging. Krishnan and coworkers described a pH-insensitive DNA nanomachine, named Clensor, for accurate sensing and imaging of chloride in organelles of living cells (Fig. 19A).256 To this end, Clensor was designed and constructed by using three modules: sensing (P), normalizing (D2), and targeting (D1). The sensing module, P, is a 12-mer peptide nucleic acid sequence labeled with a chloride-sensitive fluorescent dye, 10,10′-bis[3-carboxypropyl]-9,9′-biacridinium dinitrate (BAC). The normalizing module, D2, is a 38-mer DNA sequence labeled with a chloride-insensitive fluorescent dye, Alexa 647 fluorophore (A647). Finally, the targeting module, D1, is a 26-mer DNA sequence (Fig. 19A). To better deliver Clensor along the transferrin pathway, the targeting module (D1) of Clensor was further modified with a well-characterized RNA aptamer (Tfapt) to form a DNA-RNA chimeric oligonucleotide (D1Tfapt) (Fig. 19B). Fluorescence spectrum analysis shown in Fig. 19C indicated a decreased fluorescence at 505 nm from chloride-induced fluorescence quenching of BCA molecules with an unchanged fluorescence from A647 at 670 nm, thereby realizing ratiometric sensing of chloride ions from 5 to 200 mM (Fig. 19D). Additionally, the designed Clensor could also be used to sense chloride concentration in the endosomes of living cells (Fig. 19E). Recently, several well-designed DNA-based nanostructures employing FRET technology have also been reported for ratiometric sensing and imaging of mRNA,257 pH,258,259 and temperature260 in cells and tumor tissues. However, these reported DNA-based nanomachines are mostly 1D or 2D DNA nanostructures with limited cell permeability and relatively low stability, largely limiting their applications in biosensing and bioimaging. 3D DNA nanostructures exhibit more advantages over 1D or 2D DNA nanostructures, including higher structural rigidity, stronger cellular penetration ability, higher resistance to enzymatic degradation, as well as potential control over spatial orientation of functional ligands and multiple binding sites.248,261–266
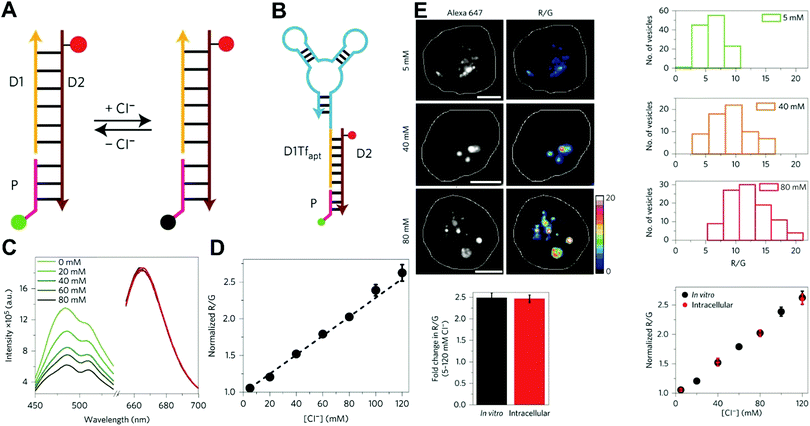 |
| | Fig. 19 (A) Structure and working principle of Clensor. P, sensing module (pink line) containing a Cl−-sensitive fluorophore, BAC (green filled circle); D2, normalizing module (brown line) containing a Cl−-insensitive fluorophore, Alexa 647 (red filled circle); D1, targeting module (orange line). In the presence of Cl−, BAC undergoes collisional quenching, whereas the fluorescence of Alexa 647 is Cl−-independent. (B) Modified sensor design for targeting to the recycling pathway (ClensorTf). D1Tfapt, targeting module modified with an RNA aptamer (Tfapt) against the human transferrin receptor (cyan line). (C) Fluorescence emission spectra of Clensor at different concentrations of Cl−. (D) In vitro Cl− calibration profile of Clensor showing normalized Alexa 647 and BAC fluorescence intensity ratio (R/G) versus chloride concentrations. (E) Quantitative performance of Clensor within subcellular organelles. Reproduced with permission from ref. 256. Copyright 2015, Macmillan Publishers Limited. | |
In recent years, various 3D DNA nanostructures including nanotweezers, tetrahedrons, triangular prisms, and cages have been designed to construct ratiometric fluorescent nanoprobes to accurately detect and image mRNA,267 DNA,268 temperature,269 and adenosine triphosphate (ATP),270,271 and to monitor the release of siRNA272 in living cells based on FRET. For instance, Tan and colleagues designed a 3D DNA tetrahedron nanotweezer (DTNT) as an intracellular DNA nanoprobe to detect and image tumor-related mRNA through FRET technology (Fig. 20A).273 In this case, the DTNT nanoprobes were synthesized by a simple self-assembly of four single-stranded DNAs, and two of them were modified with Cy3 and Cy5 to form a FRET pair for ratiometric detection. Without target mRNA, the donor fluorophore (Cy3) and receptor fluorophore (Cy5) were spatially separated, and no FRET occurred between them. Contrastingly, with target mRNA, structural changes of the DTNT nanoprobes brought the two fluorophores into close proximity, yielding high FRET signal, thereby triggering a reversible fluorescence change with a decreased emission of Cy3 at 565 nm and an increased emission of Cy5 at 662 nm (Fig. 20B). In vitro fluorescence experiments displayed a good linear correlation between the changes in FRET signal and target concentrations ranging from 0 to 20 nM with a LOD of 0.33 nM (Fig. 20C). Moreover, intracellular imaging experiments were performed using HepG2 cells that overexpressed tumor-related biomarker TK1, with HL7702 cells serving as negative control. Results in Fig. 20D and E showed that a higher FRET signal was observed in the HepG2 cells relative to the control cells of HL7702. To further monitor intracellular TK1 mRNA expression changes, tamoxifen and β-estradiol were applied to induce the downregulation and upregulation of mRNA expression, respectively. As expected, compared with the control groups, higher and lower FRET signals were found in the β-estradiol and tamoxifen groups, respectively (Fig. 20F). These above results demonstrated that the proposed DTNT nanoprobes could not only differentiate cancer cells from normal cells, but also monitor the mRNA expression level changes in living cells. Later, Jiang et al. used RNA aptamer to replace DNA as building block for fabricating light-up RNA nanostructures to ratiometrically determine and image the changes of tumor-related microRNA-21 expression level in living cells.274 To achieve the dual-emission RNA sensor for ratiometric imaging, the co-expression of green fluorescent protein (GFP) with RNA sensor can provide target-independent green emission as an internal reference, whereas the RNA sensor can offer an orange emission that can specifically respond to target microRNA, thus enabling ratiometric imaging of microRNA. Nevertheless, compared with DNA nanostructures, RNA-based nanostructures show lower stability and weaker tolerance against enzymatic degradation.
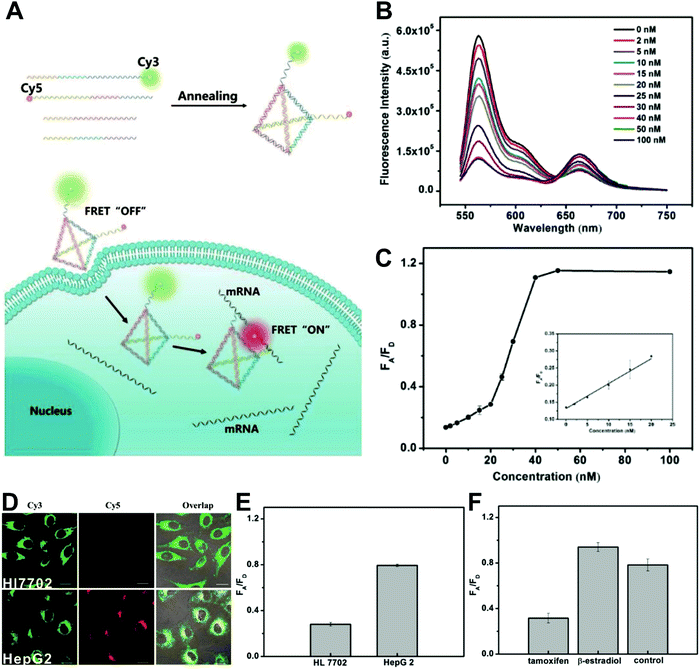 |
| | Fig. 20 (A) Schematic illustration of synthesis and mechanism of the DTNT nanoprobe for tumor-related mRNA detection in living cells. (B) Fluorescence response in the presence of different concentrations of synthetic DNA targets, ranging from 0 to 100 nM. (C) The relationship between the fluorescence emission ratio of acceptor to donor (FA/FD) and target concentration. (D) Fluorescence image of TK1 mRNA in HepG2 and HL7702 cells by DTNT nanoprobe. (E) Histogram of the relative fluorescence intensity (A/D) of the above two cell lines from D. (F) Histogram of the relative fluorescence intensity (A/D) of the following three groups, including tamoxifen-treated group, the β-estradiol-treated group, and control group. Reproduced with permission from ref. 273. Copyright 2017 American Chemical Society. | |
3.6 Challenges for designing ratiometric fluorescence nanoprobes
Although great progress has been made over the past few decades in designing ratiometric fluorescence nanoprobes, there are still quite a few challenges that impede their further development, both for basic research and clinical applications. Firstly, most of these reported dual-emission nanoprobes depend on versatile traditional organic fluorophores with fluorescence emissions less than 650 nm. Thus, in vivo imaging applications are further hampered by the high autofluorescence background from biosamples, high light scatting from tissues, and limited tissue penetration depth.275 Although this dilemma has been largely solved by introducing near-infrared (NIR) fluorescent dyes with emissions between 650 and 900 nm as alternative fluorophores, the improvement is often limited because only one NIR dye is typically used as a sensor dye or reference dye in these so-called NIR ratiometric nanoprobes, while the other still exhibits an emission below 650 nm. Improved in vivo imaging could potentially be achieved through the usage of two NIR dyes with separated emission bands in individual nanoprobes. In addition, novel NIR dyes with emissions in the NIR II window (900 to 1700 nm) have recently received widespread attention for in vivo biomedical imaging because of their diminished light scattering and deeper tissue penetration compared with shorter wavelength visible or NIR I contrast agents. Moreover, the usage of fluorophores with an ultralong fluorescence lifetime or large Stokes shift can also provide a means of filtering out autofluorescence background. Secondly, photobleaching is another factor that constrains the widespread application of ratiometric fluorescence nanoprobes, a problem that can be overcome by using fluorophores with high photostability such as QDs,276–278 introducing luminogens with aggregation induced emission (AIE) properties,279 increasing scan speed to minimize light dose,280 and exploring controlled light-exposure microscopy.281 Thirdly, relative to ratiometric fluorescent detection of single target analyte, multiplexed ratiometric nanosensors for simultaneous detection of several targets is very appealing owing to their convenience, shorter analysis time, and lower cost. However, only a few ratiometric fluorescent nanoprobes have been reported for multiplexing and simultaneous intracellular biosensing and bioimaging.282–287 As such, more efforts should be devoted to designing ratiometric nanoprobes for multiplexed sensing and imaging in vitro, and even in vivo, in which minimizing the cross-talk between different types of fluorophores should be considered. Fourthly, although various designs have been reported for developing ratiometric fluorescent nanoprobes, most of them suffer from drawbacks regarding stability and synthesis. For example, with dye-doped ratiometric nanoprobes, dye leakage hinders their practical application because leached dyes harm cells, and the uneven leaching rates of sensing and reference dyes lead to inaccurate results.288 Although an extra protective layer such as a polymer can be added onto the nanoparticle surface to minimize dye leakage, the nanoprobe's responsiveness towards its target may also be weakened, leading to decreased sensitivity. In addition, for nanoparticle–dye nanoconjugates, dye molecules attached on the surface of nanoparticles are directly in contact with the surrounding environment without any protection. As a result, changes in the environment, such as pH and ion strength, will likely degrade the nanoprobe performance. Additionally, dual-emission nanohybirds involve tedious multistep synthesis and sophisticated modification, and single nanoparticles with intrinsic dual emission also require elaborate molecular design and complicated synthesis. Finally, DNA-nanostructure-based dual-emission nanoprobes may encounter nuclease digestion and nonspecific protein binding that can cause false positive signals. Thus, careful molecular design and complex synthetic methods are also needed. Among the five design strategies reported so far, the use of single nanoparticles with intrinsic dual-emission properties appears to be an attractive fluorescence ratiometry method owing to its superior simplicity and reliability over other methods.
4. Ratiometric SERS nanoprobes
SERS has been considered as an ideal alternative to fluorescence for molecular sensing and imaging because of its outstanding ability to resist photobleaching and autofluorescence.289–292 Additionally, SERS exhibits advantages including high sensitivity, high spectral resolution for multiplexed analysis, fast response, low background signal, resistance to matrix interferences in diverse systems, as well as noninvasiveness for biological samples.293–298 These excellent properties render SERS an ideal molecular sensing and imaging platform for targeting biological molecule concentrations, or real-time monitoring of the pathological and physiological changes in cells, tissues, and even organs. For example, SERS-based sensing and imaging has been widely used for sensitive detection of biological markers overexpressed in cancer cells.299 However, the nonspecific accumulation of SERS-active nanoprobes results in a high background distribution of SERS nanoprobes regardless of the targeting moiety. Both targeted and untargeted nanoprobes can accumulate more or less in both benign and diseased tissues due to non-biomarker-related reasons such as nonspecific pooling, diffusion, uneven distribution and washout, etc., resulting in ambiguous or erroneous detection readouts. Although a wash step has been introduced to remove nonspecifically bound nanoprobes to maximize the contrast between disease and normal tissue regions, it decreases but does not eliminate the random pooling of these nonspecifically adsorbed nanoprobes. In addition, the working distance between a detector or lensing element and the interrogated surface affects the strength of the detected signal. Like all optical imaging modalities, the Raman signal intensity strictly depends on the working distance, where the absolute intensity diminishes with increasing working distance. To address these problems, ratiometric SERS nanoprobes have been proposed to describe the biomarker distribution on the basis of the specific binding of targeted SERS nanoprobes, while also minimizing the ambiguities due to nonspecific nanoprobe binding and other nonspecific contrast mechanisms (e.g. variations in working distance and detection angle, uneven delivery and washout, uneven laser illumination, etc.).300 Not only that, SERS are superior to fluorescence for ratiometric sensing and imaging since different kinds of SERS nanoprobes are identical with respect to morphology and surface properties, ensuring the similar nonspecific effects for all SERS nanoprobes. Finally, these SERS nanoprobes can be excited with a single wavelength, and emit Raman spectra with a similar wavelength range, ensuring that all nanoprobes are irradiated identically and are influenced by the same tissue optical properties, thus avoiding wavelength-dependent effects often encountered in ratiometric fluorescence detection. As such, ratiometric SERS nanoprobes have been extensively applied for quantitative detection of biomolecules of interest and monitoring environmental variables at cellular and organ levels. Currently, SERS-based ratiometric strategies can be classified into three types: (i) multiple nanoparticles with multiple Raman probes, (ii) single nanoparticles with dual Raman probes, and (iii) single nanoparticles with target-response activatable Raman probes (Fig. 21).
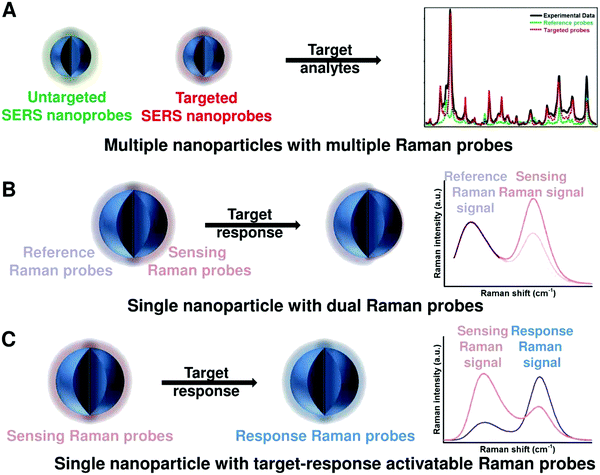 |
| | Fig. 21 Design strategies for ratiometric SERS nanoprobes. (A) Multiple nanoparticles with multiple Raman probes. (B) Single nanoparticles with dual Raman probes. (C) Single nanoparticles with target-response activatable Raman probes. | |
4.1 Multiple nanoparticles with multiple Raman probes
Generally, ratiometric SERS sensing and imaging can be achieved by physically mixing two different SERS nanoprobes with distinct Raman signatures, including one targeted nanoprobe acting as a sensing element and the other nontargeted nanoprobe functioning as a negative control, thereby yielding a ratio for reliable quantitative measurement. The ratiometric approach can effectively mitigate the ambiguity from nonspecific nanoparticle accumulation and uneven nanoparticle delivery, a universal problem in the molecular imaging field that often leads to erroneous image interpretation. A ratiometric SERS-based nanosensor for quantitative discrimination of normal and cancerous prostate cells was reported by Moskovits and colleagues,301 who investigated two sets of SERS nanoprobes, one for targeting the neuropilin-1 (NRP-1) receptors of cancer cells through the RPARPAR peptide and the other for a positive control (PC) to bind both normal and cancer cells through the HIV-derived TAT peptide (Fig. 22A and B). Here, the two types of SERS biotags were synthesized by using silver nanoparticles as SERS substrates to provide two enhanced SERS signals from two different Raman reporters of methylene blue and thionin, and two cell-targeting peptides as targeting ligands for specific recognition of normal and cancer cells, respectively. A cocktail of NRP-1-coated SERS nanoprobe and PC-coated SERS nanoprobe at a volume ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were incubated with NRP-1 overexpressing cancer PPC-1 cells and noncancerous RWPE-1 cells, respectively. Results from SERS and bright field imaging in Fig. 22C indicated that the PPC-1 cells were stained as a mosaic with a mean ratio of approximately 1.0, whereas the SERS mapping of noncancerous RWPE-1 cells displayed very little red that caused a low mean ratio of approximately 0.3, with only a few red points outside the cells from the nonspecific binding of SERS nanoprobe. This result was in accordance with that from average-cell signals (Fig. 22D). Inspired by this work, two similar ratiometric SERS nanoprobes were fabricated for the detection of microscopic residual cancerous tissue through ex vivo tissue imaging and in vivo SERS mapping based on anti-epidermal growth factor receptor (EGFR) antibody and folate as targeting ligands, respectively.302,303 Nevertheless, the sensing and imaging of a single biomarker only provides limited detection information, which is insufficient for accurate clinical diagnosis of cancer owing to its high heterogeneity. Instead, multiplexed imaging analysis of multiple biomarkers with higher signal resolution exhibits higher accuracy and reliability. In 2008, Gambhir et al.304 reported a multiplexed SERS imaging strategy for in vivo tumor targeting in mouse models, but this study was only based on a single Raman signal intensity, not for ratiometric detection. Encouraged by this study, numerous multiplexed SERS imaging nanoprobes with ratiometric readout have been recently introduced for simultaneous detection of multiple cancer biomarkers ex vivo and in vivo. Broadly speaking, ratiometric multiplexed SERS sensing and imaging is accomplished through the combination of two or more different kinds of targeted SERS nanoprobes acting as sensing elements of multiple biomolecules and one nontargeted nanoprobe serving as a reference control, thus favoring different ratios of SERS intensity based on targeted and nontargeted nanoprobes for multiplexed ratiometric determination. An excellent work was done in this field by Garai et al. in 2013,305 Gambhir and Contag labs developed an endoscopic Raman imaging system utilizing SERS nanoparticles, while further improvement of this approach was carried out by Liu's group, who presented a battery of double,26,306–308 triple,309 and quadruple24,310 SERS nanoprobes for ratiometric SERS imaging of cell surface biomarkers both in vitro and in vivo. This team recently also performed a clinical study using this strategy for intraoperative guidance of lumpectomy (Fig. 23A).23,24 In this case, a quadruple ratiometric Raman-encoded molecular imaging (REMI) technique, based on four different targeted SERS nanoprobes and one untargeted control, was applied for simultaneous quantification of the expression levels of four protein biomarkers: HER2, ER, EGFR, and CD44. REMI was carried out with 57 fresh tissue specimens from lumpectomy and mastectomy patients. Each specimen was stained with a cocktail of these five SERS nanoprobes through a convection-enhanced staining method, followed by raster-scanned imaging and spectral demultiplexing. Results indicated a high sensitivity of 89.3% and a high specificity of 92.1% for the detection of breast carcinoma. In addition, REMI analysis of these four biomarkers was in good agreement with histopathology (IHC and H&E staining). Earlier, a similar REMI-based intraoperative imaging technique was also developed to rapidly quantify the expression level of four breast cancer biomarkers: EGFR, HER2, CD44 and CD24 at the margins of freshly resected breast tissues within 15 min (Fig. 23B–E). In brief, the above results showed that the developed REMI method could be employed as an alternative to frozen-section analysis as an intraoperative guidance tool for detection of residual tumor at lumpectomy margins in order to obviate the need for re-excision.
:1 were incubated with NRP-1 overexpressing cancer PPC-1 cells and noncancerous RWPE-1 cells, respectively. Results from SERS and bright field imaging in Fig. 22C indicated that the PPC-1 cells were stained as a mosaic with a mean ratio of approximately 1.0, whereas the SERS mapping of noncancerous RWPE-1 cells displayed very little red that caused a low mean ratio of approximately 0.3, with only a few red points outside the cells from the nonspecific binding of SERS nanoprobe. This result was in accordance with that from average-cell signals (Fig. 22D). Inspired by this work, two similar ratiometric SERS nanoprobes were fabricated for the detection of microscopic residual cancerous tissue through ex vivo tissue imaging and in vivo SERS mapping based on anti-epidermal growth factor receptor (EGFR) antibody and folate as targeting ligands, respectively.302,303 Nevertheless, the sensing and imaging of a single biomarker only provides limited detection information, which is insufficient for accurate clinical diagnosis of cancer owing to its high heterogeneity. Instead, multiplexed imaging analysis of multiple biomarkers with higher signal resolution exhibits higher accuracy and reliability. In 2008, Gambhir et al.304 reported a multiplexed SERS imaging strategy for in vivo tumor targeting in mouse models, but this study was only based on a single Raman signal intensity, not for ratiometric detection. Encouraged by this study, numerous multiplexed SERS imaging nanoprobes with ratiometric readout have been recently introduced for simultaneous detection of multiple cancer biomarkers ex vivo and in vivo. Broadly speaking, ratiometric multiplexed SERS sensing and imaging is accomplished through the combination of two or more different kinds of targeted SERS nanoprobes acting as sensing elements of multiple biomolecules and one nontargeted nanoprobe serving as a reference control, thus favoring different ratios of SERS intensity based on targeted and nontargeted nanoprobes for multiplexed ratiometric determination. An excellent work was done in this field by Garai et al. in 2013,305 Gambhir and Contag labs developed an endoscopic Raman imaging system utilizing SERS nanoparticles, while further improvement of this approach was carried out by Liu's group, who presented a battery of double,26,306–308 triple,309 and quadruple24,310 SERS nanoprobes for ratiometric SERS imaging of cell surface biomarkers both in vitro and in vivo. This team recently also performed a clinical study using this strategy for intraoperative guidance of lumpectomy (Fig. 23A).23,24 In this case, a quadruple ratiometric Raman-encoded molecular imaging (REMI) technique, based on four different targeted SERS nanoprobes and one untargeted control, was applied for simultaneous quantification of the expression levels of four protein biomarkers: HER2, ER, EGFR, and CD44. REMI was carried out with 57 fresh tissue specimens from lumpectomy and mastectomy patients. Each specimen was stained with a cocktail of these five SERS nanoprobes through a convection-enhanced staining method, followed by raster-scanned imaging and spectral demultiplexing. Results indicated a high sensitivity of 89.3% and a high specificity of 92.1% for the detection of breast carcinoma. In addition, REMI analysis of these four biomarkers was in good agreement with histopathology (IHC and H&E staining). Earlier, a similar REMI-based intraoperative imaging technique was also developed to rapidly quantify the expression level of four breast cancer biomarkers: EGFR, HER2, CD44 and CD24 at the margins of freshly resected breast tissues within 15 min (Fig. 23B–E). In brief, the above results showed that the developed REMI method could be employed as an alternative to frozen-section analysis as an intraoperative guidance tool for detection of residual tumor at lumpectomy margins in order to obviate the need for re-excision.
 |
| | Fig. 22 (A) Structural description of SBT system and spectra. (B) Schematic of the SERS cell mapping experiment. NRP- and PC-SBTs are synthesized, combined, and added to either noncancerous or cancer cells suspended in DMEM supplemented with 10% FBS. (C) Two-dimensional mappings for cancer (PPC-1) and normal cells (RWPE-1). (D) Box plots of the NRP/PC ratio for both PPC-1 and RWPE-1 populations after application of the logarithmic transformation to render both distributions normal. Reproduced with permission from ref. 301. Copyright 2011, National Academy of Sciences. | |
 |
| | Fig. 23 (A) Schematic of a REMI-based intraoperative imaging guidance of lumpectomy to rapidly identify residual tumors at the margins of freshly resected tissues for guiding breast-conserving surgeries. A ratiometric strategy (right inset) quantifies biomarker expression by comparing the signal from targeted NPs and nontargeted NPs. Reproduced with permission from ref. 308. Copyright 2016, Nature Publishing Group. (B) Photograph of a human breast tumor and a normal tissue specimen from one patient. (C) Ratiometric images of EGFR-NPs versus isotype-NPs, HER2-NPs versus isotype-NPs, CD44-NPs versus isotype-NPs and CD24-NPs versus isotype-NPs. (D) Validation data: H&E and IHC for EGFR, HER2, CD44, and CD24. Unlabeled scale bars represent 200 μm. (E) Cumulative results from multiple regions of interest from a total of 5 patient specimens: measured NP ratios on IHC-validated biomarker-negative and biomarker-positive tissue regions. Each data point in the plots is the average ratio from one region of interest. Reproduced with permission from ref. 24. Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
4.2 Single nanoparticle with dual Raman probes
Although the addition of one nontargeted SERS nanoprobe is an effective method for ratiometric SERS sensing, their wide applications are largely compromised by the obvious disadvantage of the requirement for at least two or more types of SERS-active nanoprobes, which rests on complicated multistep synthetic methods for each nanoprobe. An elegant design for ratiometric SERS sensing can be achieved by incorporating two different Raman reporters onto a single nanoparticle to generate ratiometric Raman signals. Recently, several related studies reported on ATP and microRNA imaging. For instance, Tang et al. reported dual-signal switchable (DSS) nanoprobes for intracellular imaging of two cancer-related microRNAs based on a fluorescence and Raman signal switching (Fig. 24A).311 To this end, two Raman dyes of Cy3 and Rox-modified DNA probes were applied to facilitate the SERS enhancement of the AuNP surface through target-induced nucleic acid hybridization. In vitro SERS analysis showed that the typical Raman peaks of Cy3 (1193, 1391, 1465, and 1586 cm−1) and Rox (1344, 1499, and 1644 cm−1) increased with increasing target microRNA concentrations, whereas the peak at 783 cm−1 from the ring breathing and asymmetric O−P−O stretching of DNA molecules remained constant, thus serving as an internal reference peak for ratiometry (Fig. 24B). A good linear relationship between SERS intensity ratio and target concentration was gained (Fig. 24C and D). In addition, intracellular SERS imaging in MCF-7 cells revealed a time-dependent increase in SERS signal (Fig. 24E and F), and further SERS intensity analysis from various cells showed relatively homogeneous distributions based on ratiometric readout (Fig. 24G). A few other studies using a single nanoparticle with dual Raman dyes for ratiometric SERS detection were also reported.312–314
 |
| | Fig. 24 (A) Schematic illustration of detection of microRNA-21 (miR-21) and microRNA-203 (miR-203) in living cells using the developed nanoprobes, and mechanism for sensing the asymmetric signal amplification of the nanoprobes for miR-21 and miR-203. (B) SERS spectra for increasing concentrations of miR-21 and miR-203. (C and D) Variances of the I1586/I783 and I1499/I783 with the concentration of miR-21 and miR-203, respectively. (E) SERS images for miR-21 and miR-203. (F) Average SERS intensity ratios for one peak in a single MCF-7 cell incubated with the proposed probes for 1, 2, 4, and 6 h. (G) Comparison of the SERS intensity ratios in each living cell versus the average intensity ratio from 100 cells, miR-21 (left) and miR-203 (right), respectively. Reproduced with permission from ref. 311. Copyright 2017 American Chemical Society. | |
4.3 Single nanoparticle with target-response activatable Raman probes
By using the above two strategies, many ratiometric SERS sensors have been demonstrated for reliable measurement of specific targets in cells, tissues, and even animals. However, in these ratiometric SERS systems, multiple nanoparticles or multiple Raman reporters are needed, making these methods complex and expensive. In contrast, ratiometric SERS nanosensors based on single nanoparticles with target activatable Raman reporter can effectively simplify the synthesis process, and largely reduce costs. Accordingly, through the integration of SERS-active nanostructures with Raman reporters that can specifically respond to targets and cause changes in SERS spectra, ratiometric SERS nanoprobes can be obtained. In this case, the SERS signals of the Raman reporters vary with changes in target concentration, while a relatively stable Raman peak that remains unchanged at different target concentrations is selected as an internal reference for ratiometric strategy. This ratiometric method has become an ideal criterion for direct sensing and imaging in living cells due to their small Raman scattering cross-sections. Till now, these targets have mainly included intracellular pH,315–318 metal ions,319 H2O2,320–322 CO,323 NO,324–326 and H2S,327etc. For example, Long et al. described a ratiometric SERS nanosensor for sensing H2S in living cells via 4-acetamidobenzenesulfonyl azide (4-AA)-modified AuNPs (AuNPs/4-AA) as activatable SERS nanoprobes (Fig. 25A).327 In this work, the 4-AA was chosen as a target-responsive Raman reporter molecule because H2S molecules can reduce the azide groups of 4-AA molecules into amino groups, leading to a SERS spectrum change of AuNPs/4-AA. Results from SERS spectrum analysis demonstrated that the presence of H2S can induce a significant SERS spectrum change of AuNPs/4-AA with an increased peak at 709 cm−1 from amino groups and a decreased peak at 1626 cm−1 from azide groups (Fig. 25B). Besides, a target concentration-independent peak was observed at 1161 cm−1 that could be used as a reference. By creating a ratio of I709/I1161 or I1626/I1161, a good linear correlation with the concentration of H2S was observed (Fig. 25C). Later, SERS spectrum detection of H2S in living cells was performed with S-adenosyl methionine (SAM) stimulation, and results found that I709/I1161 increased with extended stimulation time, whereas I1626/I1161 correspondingly reduced (Fig. 25D). These findings illustrated the feasibility of the designed AuNPs/4-AA-based SERS nanosensors for ratiometric determination of intracellular H2S. An interesting ratiometric SERS nanosensor was recently presented for simultaneous sensing and imaging of two inorganic molecules including HClO and glutathione (GSH) in living cells through applying 4-mercaptophenol (4-MP)-functionalized gold nanoflowers as target-response Raman probes, where 4-MP exhibits a reversible response to HClO and glutathione GSH.328
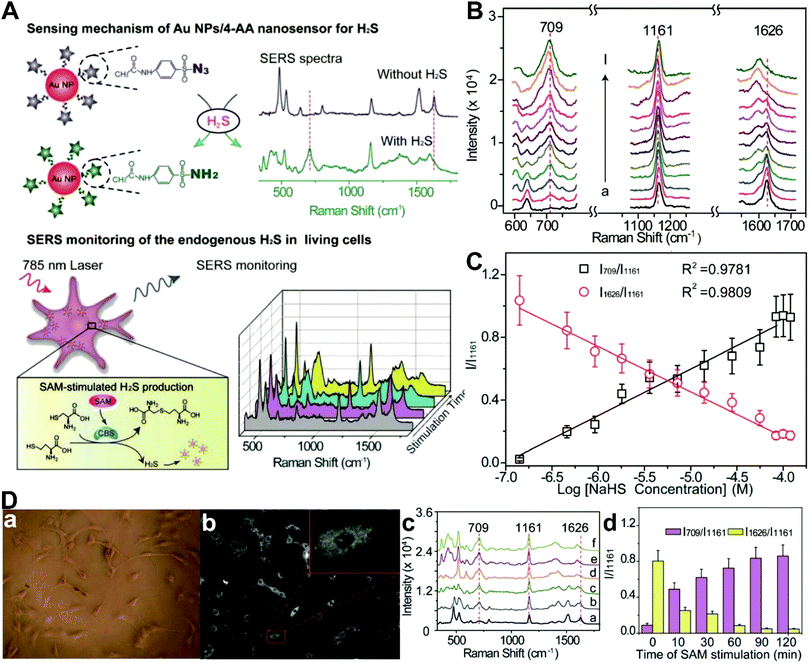 |
| | Fig. 25 (A) SERS nanosensors for monitoring endogenous H2S in living cells. (B) SERS spectra of AuNPs/4-AA in PBS in the presence of NaHS at various concentrations. (C) Plots of ratiometric peak intensities versus logarithmic NaHS concentration based on I709/I1161 and I1626/I1161. (D) SERS monitoring of endogenous H2S in living cells (rat C6 glioma cells) under SAM stimulation: bright-field images (a), DFM images (b), SERS monitoring of rat-mediated H2S with AuNPs/4-AA nanosensors under SAM stimulation with different times of 0, 10, 30, 60, 90, and 120 min (c), and the corresponding ratiometric peak intensities from (c) of I709/I1161 and I1626/I1161versus time of SAM stimulation (d). Reproduced with permission from ref. 327. Copyright 2015, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
4.4 Challenges for designing ratiometric SERS nanoprobes
Compared with fluorescence methods, SERS possesses the outstanding ability of resistance to photobleaching and autofluorescence, which makes the SERS-based method an ideal tool for molecular sensing and imaging, especially for in vivo imaging applications such as intraoperative guidance. Preclinical studies that take advantage of SERS-based imaging methods for in vivo diagnosis of cancer will promote in vivo detection without the requirement of resection and biopsy preparation, which can largely shorten the overall detection time. Although great progress has been made in building SERS nanoprobes for in vivo application, the irregular distribution of SERS nanoprobes hinders the further development of conventional single-intensity SERS nanosensors. In recent years, these problems have been effectively overcome through the introduction of ratiometric strategies. Many previous in vivo studies have demonstrated that the ratiometric SERS nanosensors can provide higher sensitivity, specificity, and reliability. However, most of these studies are performed in rodent models, and very few have advanced in humans, such as topical applications of ratiometric nanoprobes limited to accessible tissues through endoscopic probes. Nevertheless, the success of these applications in humans could help translate ratiometric SERS detection into clinical practice.
Despite the clinical translational potential of ratiometric SERS nanoprobes, there are a number of issues that need to be addressed. First, the design of ratiometric SERS nanoprobes should be further optimized. The existing ratiometric SERS nanosensors for in vivo cancer detection require at least two different types of nanoprobes for sensing and reference elements. Here, multiple-step synthesis is necessary for obtaining these nanoprobes, which is relatively complicated, time-consuming, and costly. Thus, further research should focus on simplifying the design of ratiometry to expand applications. Second, some developed ratiometric nanoprobes have been demonstrated to work well for in vivo SERS applications, but the biggest hurdle is the lack of a FDA-approved SERS nanoprobe for use in humans. Thus, it is very important to accelerate the approval process of these promising SERS nanoprobes, and with more effort dedicated to designing biocompatible and more stable nanoparticles that are well-suited for human use. Third, traditional instruments for SERS analysis mainly involve point-and-shoot devices, plate readers, as well as advanced mapping instruments. However, none of these is appropriate for in vivo SERS detection. Therefore, more advanced and portable Raman imaging instruments, such as fiberoptic-based Raman devices that can penetrate deep tissue are required to facilitate in vivo SERS applications, like Raman-guided cancer resection. For example, the first clinical endoscopic optoelectromechanical Raman device was recently designed and developed for SERS imaging of tissues during gastrointestinal endoscopy through ratiometric readout.309 Fourth, the biotoxicity of nanoprobes can obstruct the clinical application of SERS, and extensive research should be devoted to evaluating and improving the biocompatibility of nanoprobes.
5. Ratiometric photoacoustic imaging nanoprobes
Owing to their increased tissue penetration depth, photoacoustic imaging has become one of the best methods for clinically relevant sensing and imaging in vivo.329–332 Nonetheless, the photoacoustic signal intensity mainly depends on the photothermal conversion efficiency of contrast agents, which generally leads to a lower sensitivity relative to other optical measurements.331,333–335 Another problem is the limited degree of multiplexing that is achievable with PA probes due to the physics of how the signal is generated (absorption contrast), which does not allow for as many degrees of freedom for multiplexing. Recently, increasing interest has been devoted to designing and exploiting various enhanced contrast agents with higher photothermal effect and photoacoustic signals.336 Among these developed contrast agents, nanoparticle-based contrast agents have been explored for improving photoacoustic sensing and imaging to detect target analyte concentrations and monitor the physiological and pathological changes.337 However, the low SBR for these photoacoustic nanoprobes are generally determined via two targeting mechanisms: passive targeting that is based on EPR effect in tumor sites, and active targeting that is based on the recognition of target molecules by a receptor.42,329,338 For traditional photoacoustic sensing and imaging, the most frequently used signal readout methods rely on the changes from analyte-induced absolute photoacoustic signal intensity, such as conventional “always on” probes.339–341 Unfortunately, this signal acquisition of absolute signal intensity is susceptible to common artifacts that are analyte-insensitive, but hard to correct for, including the variations in nanoprobe concentration, photobleaching, local light deposition, the dimension of target analytes, as well as laser irradiation.342,343 These target-independent factors lead to inaccurate analysis results. Besides, they also suffer from low analytical sensitivity and specificity. Recently, researchers have demonstrated the usage of ratiometric photoacoustic measurements to replace the absolute photoacoustic signal intensity measurements, which can elegantly tackle the above-mentioned issues. Thus, designing and developing novel ratiometric photoacoustic nanoprobes with improved performances are highly desired and critical for further exploring photoacoustic techniques for sensitive and reliable molecular sensing and imaging. Up to now, there have been two types of photoacoustic ratiometric strategies reported: (i) nanoparticle–dye nanocomplexes with dual photoacoustic absorption, and (ii) target-response activatable photoacoustic absorption switches (Fig. 26).
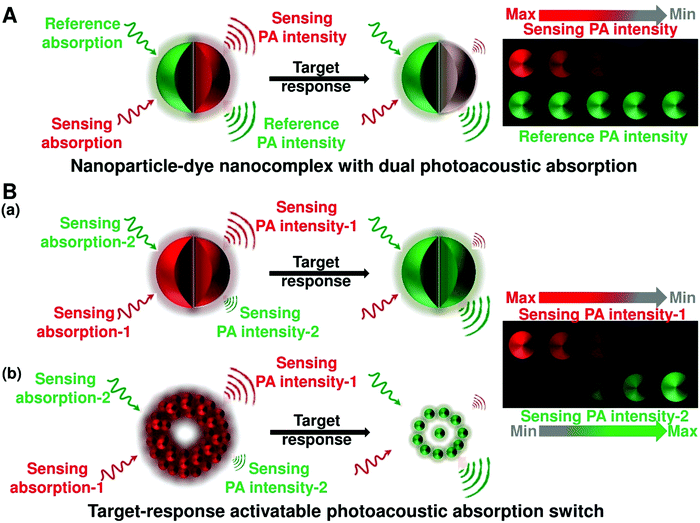 |
| | Fig. 26 Design strategies for ratiometric photoacoustic nanoprobes. (A) Nanoparticle–dye nanocomplex with dual photoacoustic absorption. (B) Target-response activatable photoacoustic absorption switch. | |
5.1 Nanoparticle–dye nanocomplex with dual photoacoustic absorption
Analogous to dye-embedded dual-emission fluorescence nanoprobes, dye-doped photoacoustic nanoprobes with dual absorption can achieve ratiometric photoacoustic detection. Two dyes with two clearly segregated absorption peaks are spontaneously encapsulated into one nanoparticle, in which one dye responds to the target molecule to yield the absorption change that can act as a sensing unit, and the other target-inert dye can serve as the reference absorption for ratiometric detection. Based on this strategy, Pu et al. designed a ratiometric photoacoustic nanoprobe (SON) for imaging pH in vivo (Fig. 27A).344 To this goal, the ratiometric nanoprobe was prepared with a target-insensitive semiconducting oligomer (SO) as an indicator and a pH-sensitive boron-dipyrromethene (BODIPY) dye for pH sensing. Under acidic environments, the hydroxyl group of BODIPY can undergo protonation, causing the absorption changes of SON. As shown in Fig. 27B, the absorption peak at 750 nm reduced obviously with decreasing pH, while no obvious absorption peak change at 680 nm was observed. Consequently, the decreased photoacoustic intensity at 750 nm was obtained with a decrease in pH (Fig. 27C and D). Fig. 27E indicated the correlation of ratiometric photoacoustic signal (PA680/PA750) with pH, showing a good linear relationship with pH ranging from 7.4 to 5.5. It suggested that this nanoprobe could be applied to measure normal physiological pH (7.4), and the pathological pH shift caused by diseases like cancer (6.4 or 5.5). In vivo photoacoustic imaging of pH was performed by local injection of nanoprobes into normal muscle and tumor tissue in a HeLa xenograft model, and then the corresponding photoacoustic images were acquired at 680 and 750 nm, respectively. Results in Fig. 27F demonstrated the relatively high photoacoustic intensities at both 680 and 750 nm in muscle tissue; on the contrary, lower photoacoustic intensity at 750 nm was observed compared with that at 680 nm in the tumor. Additionally, to eliminate the background interference from tissue itself, the increments of the photoacoustic intensities at 680 and 750 nm (ΔPA680 and ΔPA750) were employed to build a ratio of ΔPA680/ΔPA750 for the detection of pH in vivo, where the photoacoustic intensity increments (ΔPA) are based on the subtraction of photoacoustic intensity after and before the sample treatment. Fig. 27G showed a higher ΔPA680/ΔPA750 in the tumor tissue than that in the normal muscle tissue, illustrating a decreased pH at the tumor site. In a similar way, other research groups have also demonstrated that the design concept is well-suited for the fabrication of ratiometric photoacoustic sensing and imaging probes to monitor the changes in pH value,345 ROS level,346–348 as well as cancer-related enzyme activity349,350 both ex vivo and in vivo. These reported ratiometric photoacoustic nanoprobes were designed and constructed by altering the target-responsive sensing absorption dye and the reference absorption dye.
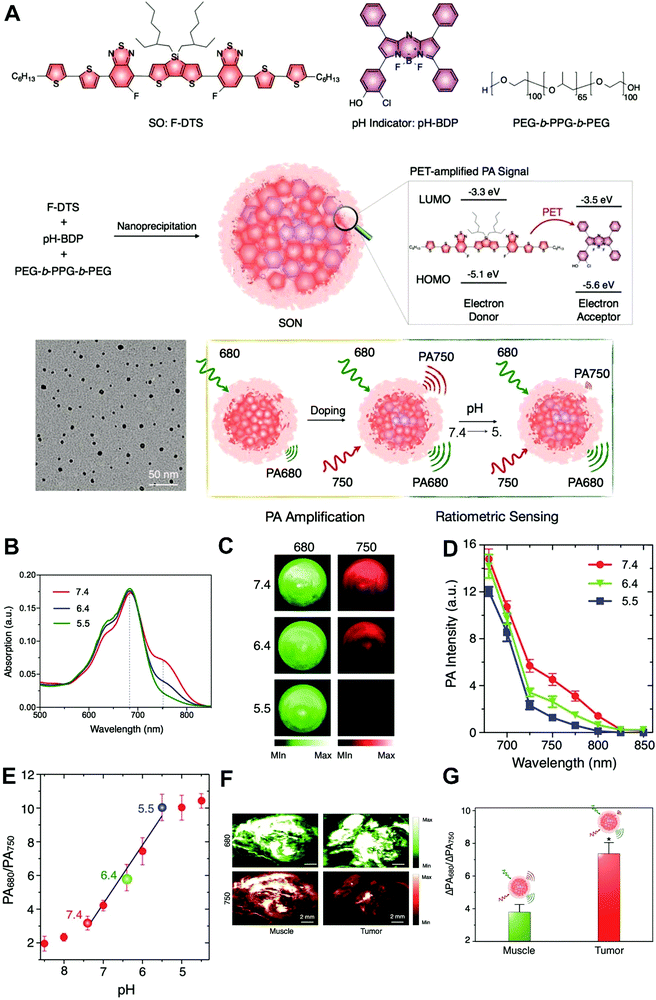 |
| | Fig. 27 (A) Schematic illustration for nanoprobe design of PET-amplified PA imaging of pH, doping-induced PA amplification, and pH-sensing mechanism. (B) UV-vis absorption spectra of SON50 at different pH. (C) PA images of the SON50 solution at pH = 7.4, 6.4, or 5.5. A pulsed laser was tuned to 680 or 750 nm for ratiometric imaging. (D) PA spectra of SON50 at different pH. (E) Quantification of the ratiometric PA signals (PA680/PA750) of SON50 at different pH. The blue line represents linear fitting from pH = 7.4 to 5.5 (R2 = 0.991). (F and G) PA images and ratiometric signals (ΔPA680/ΔPA750) of muscle and tumor with local administration of SON50. Reproduced with permission from ref. 344. Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
5.2 Target-response activatable photoacoustic absorption switch
Compared with dye-doped ratiometric photoacoustic nanoprobes, target-responsive activatable photoacoustic nanoprobes for ratiometric strategy have attracted increasing attention because of their convenience and simplicity, in which only one target-responsive absorption dye is needed to produce an absorption shift induced by targets, and then the shifted absorption results in a reversible change of the two absorption peaks with a corresponding increase and decrease. As a consequence, the ratio from these two absorptions can be utilized to allow for ratiometric measurement. Ashkenazi's groups used a well-known pH-sensitive SNARF-5F, seminaphthorhodafluors-5F, as a sensing unit for ratiometric pH detection with a reversible absorption change at 532 and 564 nm.351 Several improved ratiometric nanosensors for pH detection were reported recently by Wang and co-workers with the same pH indicator of SNARF-5F.352,353 Very recently, we reported a ratiometric photoacoustic nanoprobe (LP-hCy7) for detecting methylmercury (MeHg+), a major neurotoxin (Fig. 28A).354 The nanoprobe was synthesized based on MeHg+-responsive near-infrared (NIR) cyanine dye (hCy7), which can be easily encapsulated in the lipid bilayer of nanoliposomes via hydrophobic interactions. In the presence of MeHg+, hCy7 in the nanoliposome can be converted to hCy7’ based on mercury-triggered cyclization reaction, thus leading to an absorption shift from 690 to 860 nm (Fig. 28A). As a result, the absorbance at 690 nm reduced significantly with increasing MeHg+ concentration, whereas the absorbance at 860 nm increased remarkably. Thus, the photoacoustic intensities at 690 and 860 nm exhibited obvious decrease and increase, respectively, which caused an increase in the ratio of the two photoacoustic signals (PA860/PA690) with increased concentration of MeHg+ (Fig. 28B and C). A linear correlation was obtained between PA860/PA690 and MeHg+ concentrations, with a LOD of 2.0 ppb (Fig. 28C). In vivo PA imaging for real-time monitoring of MeHg+ level was conducted in zebrafish with a treatment of LP-hCy7 and MeHg+, and meanwhile two control groups received either no treatment or only LP-hCy7. Results in Fig. 28D and E displayed no obvious photoacoustic signals at 690 and 860 nm in the control group without any treatment, and a relatively high PA signal at 690 nm was observed with a relatively low PA signal at 860 nm in the control group with the treatment of only LP-hCy7. In contrast, with the incubation of LP-hCy7 and MeHg+, the PA signal at 690 nm significantly decreased along with PA signal increase at 860 nm. Consequently, the ratio of PA860/PA690 reached up to 2.82 when the zebrafish was treated with LP-hCy7 and MeHg+, which is approximately 10-fold higher than that (0.27) obtained without MeHg+ (Fig. 28F). In addition, similar results were observed from PA imaging in mice. In recent years, more examples have demonstrated the feasibility of this design strategy for ratiometric PA sensing and imaging of pH values,355,356 and metal ions.342,343,357
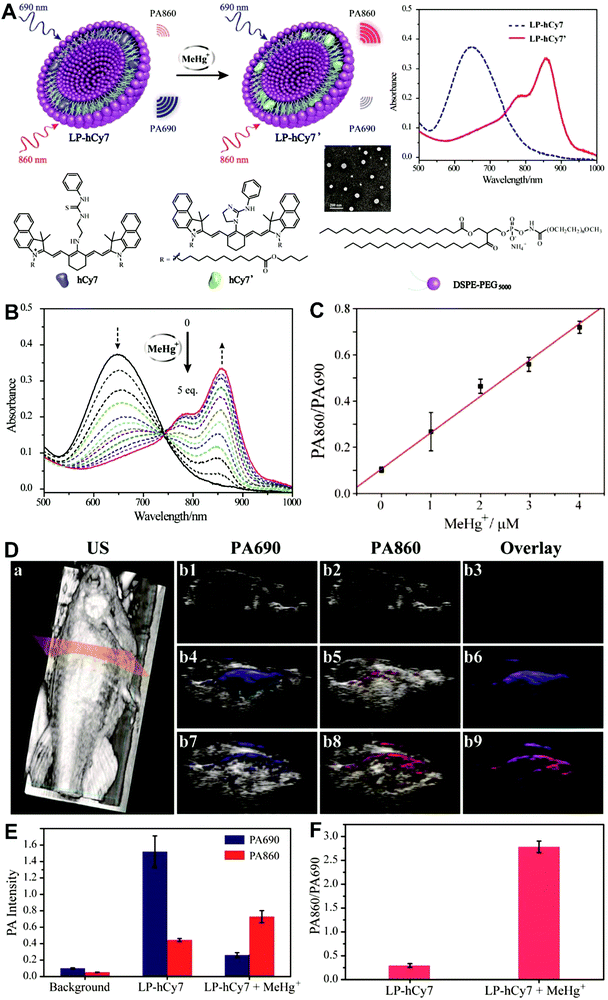 |
| | Fig. 28 (A) Schematic illustration of the proposed strategy for ratiometric photoacoustic imaging of MeHg+. (B) Absorbance response of LP-hCy7 as a function of MeHg+ concentration in an aqueous solution. (C) Plot of PA860/PA690 of LP-hCy7 against the concentration of MeHg+ ions. (D) 3D ultrasonic (US) image of zebrafish for illustration of photoacoustic imaging in transection of abdomen (a). (b) Merged US and PA images of untreated zebrafish (b1–b3), LP-hCy7 incubated zebrafish (b4–b6), and MeHg+/LP-hCy7 treated zebrafish (b7–b9) at 690 (left) and 860 nm (right), respectively. (E) Corresponding quantified PA intensity at 690 nm (blue) and 860 nm (red) for (D-b). (F) Ratios of PA860/PA690 obtained from (D-b). Reproduced with permission from ref. 354. Copyright 2017, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | |
Stimuli-responsive nanoswitches are another design strategy for constructing ratiometric PA nanosensors, where the presence of target can induce a reversible nanoswitch from monomeric to aggregated states, which can induce a conformation-dependent absorption spectral change. As a result, the stimuli-responsive PA nanoswitch can be achieved from the absorption spectral shifts and changes. Thus, ratiometric detection was performed on the basis of the absorption ratio before/after the shift. Using this strategy, Zheng's group described a stimuli-responsive PA nanoswitch from ordered J-aggregates to dispersed monomers (Fig. 29).358 Here, this nanoswitch was achieved through the conjugation of light-absorbing bacteriopheophorbide α (Bchl) and 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine to prepare bacteriopheophorbide α-lipid (Bchl-lipid) with an ordered J-aggregated structure (Fig. 29A and B). Later, the J-aggregating nanoparticles (JNPs) were formed by the simple mixing of Bchl-lipid, dipalmitoylphosphatidylcholine (DPPC), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (mPEG2000-DPPE) based on the freeze–thaw extrusion method (Fig. 29B). The formed JNP nanoprobes exhibited a strong NIR absorption at 824 nm with a red-shift of 74 nm relative to the porphyrin monomer absorption at 750 nm (Fig. 29C). As shown in Fig. 29D, upon increasing temperature, the J-aggregate absorption from the JNP at 824 nm reduced obviously, whereas the monomeric absorption of porphyrin at 750 nm increased. The reversible absorption spectral change provided a potential method for fabricating ratiometric PA nanosensors for real-time detection of temperature. Similarly, the design concept of stimuli-responsive PA nanoswitches has also been employed for ratiometric imaging and monitoring of intracellular pH359 and caspase-1 activity.360
 |
| | Fig. 29 (A) Structure of bacteriopheophorbide-lipid with axes showing the Qy and Qx transition dipoles for the chromophore. (B) Schematic of the J-aggregating nanoparticle (JNP) prepared with 15% Bchl-lipid, 80% host phospholipid, and 5% mPEG2000-DPPE. Insets: (left) Representative transmission electron micrograph of JNP prepared with dipalmitoylphosphatidylcholine (DPPC) (JNP16) and (right) color photographs of JNP16 sample below and above phase transition temperature. (C) Absorption spectra of JNP16 in the intact (blue) and detergent disrupted (red) state. (D) Representative near-infrared absorption spectrum of JNP prepared with dipalmitoylphosphotidylcholine (JNP16) upon heating from 25 to 50 °C. Reproduced with permission from ref. 358. Copyright 2014 American Chemical Society. | |
5.3 Challenges for designing ratiometric photoacoustic nanoprobes
In recent years, PA imaging technology, a newly-developed imaging modality, has been developed as one of the most popular imaging approaches, especially for in vivo imaging. Numerous unmet needs for biological and medical applications have greatly incentivized the technical progress for improved imaging performances in this field. For example, various excellent PA contrast agents, like nanoparticle-based contrast agents, have been designed and explored to enhance the sensitivity. Nonetheless, these developed PA contrast agents are mostly at the proof-of-concept level for preclinical studies without approval for clinical use. Besides, conventional PA contrast agents are built on single-intensity-based signal acquisition that is easily influenced by various artificial factors and background interferences, largely limiting further development of traditional PA imaging. PA imaging with ratiometric readout has been proven as a feasible strategy to overcome this problem, and enhances the sensitivity and reliability of conventional PA imaging. However, unlike other ratiometric imaging modalities, ratiometric PA imaging for in vivo applications has been far less developed. The feasibility of ratiometric PA imaging in vivo should be further evaluated in preclinical and clinical applications, such as imaging tumor microenvironments and for in vivo disease imaging. Besides, other challenges also need to be addressed to promote their clinical applications. First, from a technical point of view, the PA signal mainly relies on absorption-induced heat production, indicating that PA imaging exhibits a lower sensitivity than fluorescence. Again, this absorption-based signal-generation mechanism also may limit the degree of multiplexing that can be achieved. But, an improved sensitivity for PA imaging can be obtained by designing contrast agents with higher photothermal conversion efficiency, which can contribute to reducing the dosage of contrast agents necessary for minimization of toxicity. Second, the PA imaging depth is determined by the light attenuation rate in the optically opaque tissues. Although PA imaging can provide relatively high tissue penetration, the depth is limited to several centimeters. A simple method for gaining higher penetration depth is through increasing the laser energy, but exorbitant laser exposure does harm to the living subjects. Third, compared with traditional short-wavelength absorbing materials, the design and fabrication of NIR-absorbing nanoprobes as ratiometric PA contrast agents can further improve the imaging performances including higher sensitivity and tissue depth. This is because the use of NIR light for NIR-absorbing nanoprobes can effectively reduce light scattering and avoid the background absorption from tissue samples such as blood and other biological compounds, thus leading to a higher SBR. For instance, NIR dyes with optical absorption at the second NIR window have been introduced as exogenous contrast agents for deeper PA imaging with high sensitivity.361,362 Fourth, many preclinical PA detection systems have been commercialized, however, these systems for clinical use still require authentication and approval. Additionally, more advanced imaging devices with improved sensitivity, portability, durability, low cost, and easy operation for non-specialists can help accelerate development as a well-accepted clinical bioimaging modality.
6. Ratiometric BRET nanoprobes
Over the past decades, fluorescence-based molecular sensing and imaging technologies have aroused extensive interest, and great breakthroughs have been made. However, they are limited by the necessity of external excitation light, which can lead to the strong autofluorescence from background, light scattering, and photobleaching.363 Chemiluminescence or bioluminescence-based bioimaging does not require external light excitation and thereby dramatically reduces the background autofluorescence signal from biosamples to enable higher SBR than fluorescence-based imaging, allowing more sensitive signal detection for in vivo deep tissue imaging.364–366 In addition, the lack of external excitation light can effectively avoid the phototoxicity-induced tissue damage and the possible photoactivation of azide functional groups.367 Such properties make the chemiluminescence or bioluminescence potentially superior to fluorescence as a bioimaging tool. Nonetheless, the major challenge of chemiluminescence or bioluminescence-based imaging is the inherent weak luminescence signal from the frequently-used luminescent agents such as luciferase and luminol. Recently, some improved luminescent agents, including genetically encoded luciferase enzyme or QDs have been introduced to obtain optimized chemiluminescence or bioluminescence imaging through utilizing chemiluminescence or bioluminescence resonance energy transfer (CRET or BRET).368 Similar to FRET, the CRET and BRET provide a great opportunity for the development of ratiometric chemiluminescence or bioluminescence sensing and imaging, which is favorable for removing the influences from various possible interferences, such as the changes in optical path length, the fluctuations of detectors, the expression level of indicators, as well as tissue movement and growth artifacts.369 Owing to such advantages, CRET or BRET-based ratiometric nanoprobes have been widely developed in recent years for investigating some important biological species and functions. The related progress in this field has been reviewed by Kim et al. and Mezzanotte et al.368,370 Thus, in this section, we will select several representative examples to provide a brief illustration for the proof of concept. Imamura and colleagues reported a BRET-based nanosensor (BTeam) for imaging ATP level in living cells.371 As shown in Fig. 30A, BTeam consists of three parts, including a variant of yellow fluorescent protein (YFP), an ATP binding domain of the ε subunit from the bacterial ATP synthase, and an ATP-nonconsuming NanoLuciferase (NLuc). Upon the addition of NLuc substrate (furimazine), two separated emission peaks at 455 and 527 nm were observed from NLuc and YFP, respectively. With increasing ATP concentration, the emission peak at 527 nm obviously increased along with a decrease in the emission peak at 455 nm (Fig. 30B). Accordingly, the emission ratio of YFP/NLuc, termed BRET ratio, was recorded for ratiometric detection of ATP (Fig. 30C). For intracellular basal ATP level detection, live cell imaging experiments with BTeam were conducted in several cell lines, including HeLa, Cos7, HepG2, HEK293A, PC12, and B16F10. Fig. 30D indicated a standard curve for ATP quantitation based on the BRET ratios versus ATP concentrations. According to this standard curve, the corresponding ATP concentrations for these six kinds of cell lines were calculated to be 3.8 ± 0.1, 3.7 ± 0.1, 4.1 ± 0.2, 3.7 ± 0.04, 3.9 ± 0.3, and 3.7 ± 0.2 mM, respectively, and meanwhile these results were confirmed by conventional firefly luciferase assay (Fig. 30E). Further luminescent imaging of individual cells with BTeam was recorded for ratiometric ATP detection, and the results indicated that the cytosol showed a higher BRET ratio than that of the mitochondria (Fig. 30F and G). All these findings demonstrated that the developed BRET-based ratiometric nanosensor is very suitable for reliable quantitative analysis and real-time tracing of intracellular ATP level, even at the single-cell level. With a similar design principle, BRET-based nanosensors have also been developed for ratiometric sensing and imaging of other intracellular events, including pH,372 calcium ions,367,369,373 cAMP,374 caspase activity,375 drug screening,376 and protein–protein interactions,377 with applications that span from high-throughput assays to live-cell and live-animal imaging.
 |
| | Fig. 30 (A) Schematic illustration of the proposed BTeam biosensor. (B) ATP-dependent luminescence spectral changes of purified BTeam. (C) ATP-dependent BRET ratio changes of purified BTeam. (D) Standard curve for calculation of ATP concentration based on the BRET ratio values (mean ± SD) of purified BTeam. (E) Comparison of intracellular ATP concentrations determined by BTeam (closed bar) and firefly luciferase (open bar). (F) Luminescence images of NLuc (left) and YFP (middle), and BRET ratio (right, pseudocolored) of HeLa cells stably expressing cyt-BTeam (upper) or transiently expressing mit-BTeam (bottom). (G) Comparison of BRET ratio values between cytosol and mitochondria at the single HeLa cell level. Reproduced with permission from ref. 371. Copyright 2016, Nature Publishing Group. | |
7. Summary and outlook
Ratiometric optical nanoprobes effectively eliminate many of the fluctuations and ambiguities in image contrast due to nonspecific sources of signal and background, providing an increased SBR and more reliable detection. Research for designing and fabricating ratiometric optical nanoprobes has attracted great interest, and major advances have been made in recent years. In this review, we provide a comprehensive and systematic overview on the design of ratiometric optical nanoprobes and their applications in molecular sensing and imaging. These developed ratiometric optical nanoprobes mainly involve three different signal generation mechanisms including fluorescence, SERS, and PA. Table 1 summarizes the design strategies for fabricating these three classes of nanoprobes, and provides a brief comparison. Such optical nanoprobes share the same concept of ratiometry, but each of them has its own unique characteristics. Ratiometric fluorescence nanoprobes can provide an intuitive visualization of physical or pathological changes, making them more suitable for real-time detection and tracking of dynamic processes involving target analytes or molecular events in vitro. Advanced ratiometric fluorescence imaging approaches for intracellular and even in vivo application are performed by introducing longer-wavelength NIR dyes (over 900 nm) or nanocrystals with an increased fluorescence lifetime for time-resolved detection. In particular for intraoperative guidance with ratiometric fluorescence imaging, real-time visual visualization of residual tumors at the surgical margins is essential for reliable judgment of complete surgical excision. Ratiometric SERS nanoparticles can offer rich spectral fingerprints generated by exogenous Raman reporter molecules that do not exist in organisms, thus resulting in lower background levels for imaging with a higher SBR. Moreover, different Raman reporters show distinct and unique Raman fingerprint spectra, and thus through the combination of SERS-active substrates, multiple Raman spectra can be obtained for multiplexed detection. Compared with fluorescence-based nanoprobes, ratiometric SERS nanoprobes are well-suited for denser multiplexed in vivo imaging with advanced, portable, or handheld Raman devices. For intraoperative guidance, ratiometric SERS imaging has some advantages for real-time and dynamic monitoring of the surgical margins to ensure complete surgical resection. However, spectral detection is often slow, and makes real-time microscopic imaging challenging with SERS, which could be a barrier for clinic adoption. Compared with fluorescence and Raman imaging, ratiometric PA imaging provides deeper tissue penetration, rendering it more appropriate for in vivo deep tissue imaging. Moreover, ratiometric PA imaging is appropriate for many applications of image-mediated intraoperative guidance due to the generation of three-dimensional depth-resolved images. However, the relatively low imaging sensitivity and limited capacity for multiplexing necessitates the further development of improved PA contrast agents, such as by utilizing long-wavelength absorbing materials that can minimize the inherent background absorption from biosamples.
Table 1 Summary and comparison of the design strategies for various ratiometric optical nanoprobes
| Ratiometric nanoprobes |
Design strategy |
Advantages |
Disadvantages |
| Fluorescence |
Two dye-embedded nanoparticles with dual emission |
Nanoparticles with dyes randomly distributed in the interior |
Simplicity, without complicated synthesis and elaborate molecular design |
Dye leakage, self-quenching, photobleaching, phototoxicity |
| Nanoparticles with dyes located within the core and shell |
Reduced self-quenching, simplicity, without complicated synthesis and elaborate molecular design |
Complicated synthesis and additional modification, dye leakage, photobleaching, phototoxicity |
| Nanoparticle–dye nanoconjugates with dyes attached to the surface |
Simplicity, without complicated synthesis and elaborate molecular design |
Additional dye surface modification, dyes without any protection that are sensitive to the environmental changes, such as pH, ion strength, photobleaching, phototoxicity |
| Hybrid nanoparticles with dual emission |
No organic fluorophore, reduced photobleaching |
The requirement of multiple-step synthesis and surface modification, phototoxicity, structural stability |
| Single nanoparticles with intrinsic dual emission |
Avoids multiple fluorophores resulting in a simplified ratiometric design |
Complicated synthesis and elaborate molecular design, phototoxicity |
| DNA nanostructures with dual emission |
High selectivity and affinity, high programmability, excellent biocompatibility, remarkable cell permeability, and nanoscale controllability |
Nuclease digestion, nonspecific protein binding that will lead to a false positive signal, elaborate molecular design and complex synthesis, photobleaching, phototoxicity |
| Surface enhanced Raman scattering |
Multiple nanoparticles with multiple Raman probes |
Avoids elaborate molecular design for each SERS-active nanoprobe |
The requirement of multiple-step synthesis and surface modification, complex spectral decoupling, expensive, hard to operate, with additional recognition molecules (e.g. antibody, peptide), heterogeneous and unequal distributions of probes |
| Single nanoparticles with dual Raman probes |
Simplified ratiometric sensing design, without complicated synthesis |
The requirement of two different Raman reporter molecules, with additional dye surface modification, with addition recognition molecules |
| Single nanoparticles with target-response activatable Raman probes |
Low background noise, high signal to background ratio, real-time correlation between probe states, without complicated synthesis, without additional recognition molecules, the most promising design |
With elaborate molecular design |
| Photoacoustic |
Nanoparticle–dye nanocomplex with dual photoacoustic absorption |
Simplicity, without complicated synthesis and elaborate molecular design |
The requirement of two different absorption dyes |
| Target-response activatable photoacoustic absorption switch |
Low background noise, high signal to background ratio, real-time correlation between probe states, without additional recognition molecules, the most promising design |
Complicated synthesis and elaborate molecular design |
| Bioluminescence |
Luciferase-based bioluminescence resonance energy transfer |
Without external light excitation, free from phototoxicity, avoiding photo-induced physiological reaction, no autofluorescence from the biosamples, high signal to background ratio |
Weak bioluminescence signal, strict distance-dependent FRET, the stability and reproducibility of BRET pairs |
Although the sensing and imaging applications using these three classes of ratiometric optical nanoprobes have been demonstrated at the cellular scale and in small animal models, there are still several limitations. First, ratiometric strategies efficiently mitigate the problems of nonspecific and misleading sources of signal and background fluctuations, but face many of the same limitations of any single imaging modality, which can make it hard to provide an accurate disease diagnosis. Thus, the integration of whole-body imaging methods for multi-scale information, including computed tomography (CT), magnetic resonance imaging (MRI), and positron emission tomography (PET), are potentially valuable in certain clinical applications as a complementary tool for accurate early disease diagnosis. Second, multimodal ratiometric nanoprobes that include both optical and non-optical signaling molecules are an appealing strategy for obtaining multi-scale information at several spatial and temporal scales, potentially allowing for a reliable disease diagnosis. However, the design and construction of multimodality ratiometric nanoprobes for sensing and imaging is compromised by the huge differences in the fundamental contrast mechanisms of different imaging modalities. Thus, integrating multimodality ratiometric imaging into a single nanoprobe is a very interesting and challenging task. Third, while the usage of ratiometry enables higher accuracy and reliability compared with traditional detection strategies, to date only a few studies have investigated this. This is because nanoprobes for multiplexed ratiometric analysis depend on the optimal combination of at least one reference unit and two or more target-responsive sensing units, which require elaborate molecular design and complex synthesis. Fourth, “all-in-one” nanotheranostic platforms for disease diagnosis and therapy have received increasing attention in recent years. Further studies should focus on the utilization of ratiometric nanoprobes as a theranostic nanocarrier to allow real-time and dynamic imaging of disease and drug delivery, as well as image-guided targeted therapy. Fifth, the EPR effect and micropinocytosis of nanoprobes are highly relevant to the processes of nanoparticle accumulation and clearance in vivo, but there exist substantial differences between mouse models used in early preclinical investigations and humans. Therefore, more preclinical and clinical trials, including magnifying the imaging to the cellular level and increasing the field of view should be encouraged to ascertain how well these ratiometric nanoprobes may be translated to the clinic. Sixth, nanoparticles with high purity and excellent monodispersity are crucial since these variables are closely related to their functions. Thus, post-production purification and characterization can contribute to obtaining high-quality nanoparticles for application. However, in practical industrial production processes, key core technologies for large-scale purification are still lacking. Thus, there is a need to develop more advanced technologies for nanoprobe purification and characterization to promote their further commercialization and clinical application. For instance, the presence of nanoparticle tracking analysis (NTA) and inductively coupled plasma mass spectrometry (ICP-MS) provides possibilities for accurately quantifying nanoprobes. Seventh, the colloid stability of nanoprobes is very important for their wide application. Currently, the most widely-used post-production modification approach to improve the nanoparticle stability over a wide range of buffer conditions is through simply attaching PEG moieties (“pegylation”) onto the nanoparticle surface. However, to enable higher long-term stability, some key considerations including geometry shape, loading density, molecular weight, charge as well as purification method, need to be optimized before application. Moreover, in addition to pegylation, new methods to ensure high stability in a wide range of buffer solutions are needed, such as incorporating ligand molecules like DNAs, proteins, and cytomembranes. Eighth, for in vivo imaging applications, the biocompatibility of nanoprobes should be improved by careful selection of the substrate and application of protective coatings. Extensive research will be necessary to determine the most biocompatible and stable nanoprobes for use in humans, in order to promote the clinical translation and regulatory approval of these nanoprobes. Ninth, the imaging performance is determined not only by the nanoprobe itself, but also by the physics of the instrumentation. As such, the development of advanced imaging equipment with increased portability, low cost, and ease of use, as well as higher tissue penetration depth would be helpful, especially for in vivo applications. Finally, while this review mainly focuses on ratiometry, more-advanced compartmental models, such as advanced kinetic modeling and paired-agent methods,30 should receive widespread interest for quantifying molecular images and removing the effects of nonspecific sources of image contrast with the ultimate goal of achieving more accurate and reliable quantification of biological and pathological changes. In brief, we anticipate that these improvements will bring about the broadened clinical application of ratiometric optical nanoprobes for diverse purposes ranging from routine detection of physiological changes such as pH, to monitoring of pathological changes like the expression of disease biomarkers, to improving image-based intraoperative guidance, as well as noninvasive imaging of deeper tissues.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (31760485), Major Projects of the Natural Science Foundation of Jiangxi province (20161ACB20002), the Innovation Fund Designated for Graduate Students of Nanchang University (cx2015107), and the Intramural Research Program, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health. Mr Xiaolin Huang is partially supported by the China Scholarship Council (201606820007).
References
- M. Chen, X. He, K. Wang, D. He, X. Yang and H. Shi, TrAC, Trends Anal. Chem., 2014, 58, 120–129 CrossRef CAS.
- A. Schulz and C. McDonagh, Soft Matter, 2012, 8, 2579–2585 RSC.
- T. Ueno and T. Nagano, Nat. Methods, 2011, 8, 642–645 CrossRef CAS PubMed.
- M. I. Stich, L. H. Fischer and O. S. Wolfbeis, Chem. Soc. Rev., 2010, 39, 3102–3114 RSC.
- G. M. Lanza, P. M. Winter, S. D. Caruthers, A. M. Morawski, A. H. Schmieder, K. C. Crowder and S. A. Wickline, J. Nucl. Cardiol., 2004, 11, 733–743 CrossRef PubMed.
- C. Sun, J. S. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS PubMed.
- Z. Zhou, R. Bai, J. Munasinghe, Z. Shen, L. Nie and X. Chen, ACS Nano, 2017, 11, 5227–5232 CrossRef CAS PubMed.
- X. Chai, X. Zhou, A. Zhu, L. Zhang, Y. Qin, G. Shi and Y. Tian, Angew. Chem., Int. Ed., 2013, 52, 8129–8133 CrossRef CAS PubMed.
- F. Zhao, L. Zhang, A. Zhu, G. Shi and Y. Tian, Chem. Commun., 2016, 52, 3717–3720 RSC.
- C. Bremer, V. Ntziachristos and R. Weissleder, Eur. Radiol., 2003, 13, 231–243 Search PubMed.
- G. D. Luker and K. E. Luker, J. Nucl. Med., 2008, 49, 1–4 CrossRef PubMed.
- R. Weissleder, Science, 2006, 312, 1168–1171 CrossRef CAS PubMed.
- M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 121–131 CrossRef PubMed.
- Y. E. K. Lee and R. Kopelman, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 98–110 CrossRef CAS PubMed.
- H. Koo, M. S. Huh, J. H. Ryu, D.-E. Lee, I.-C. Sun, K. Choi, K. Kim and I. C. Kwon, Nano Today, 2011, 6, 204–220 CrossRef CAS.
- O. S. Wolfbeis, Chem. Soc. Rev., 2015, 44, 4743–4768 RSC.
- Q. Li, L. Liu, J.-W. Liu, J.-H. Jiang, R.-Q. Yu and X. Chu, TrAC, Trends Anal. Chem., 2014, 58, 130–144 CrossRef CAS.
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901–986 CrossRef CAS PubMed.
- J.-W. Yoo, E. Chambers and S. Mitragotri, Curr. Pharm. Des., 2010, 16, 2298–2307 CrossRef CAS PubMed.
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 24–36 CrossRef.
- V. Mohanraj and Y. Chen, Trop. J. Pharm. Res., 2006, 5, 561–573 Search PubMed.
- S. Y. Leigh, M. Som and J. T. Liu, PLoS One, 2013, 8, e62084 CAS.
- Y. Wang, N. P. Reder, S. Kang, A. K. Glaser, Q. Yang, M. A. Wall, S. H. Javid, S. M. Dintzis and J. T. Liu, Cancer Res., 2017, 77, 4506–4516 CrossRef CAS PubMed.
- Y. W. Wang, J. D. Doerksen, S. Kang, D. Walsh, Q. Yang, D. Hong and J. T. Liu, Small, 2016, 12, 5612–5621 CrossRef PubMed.
- S. Kang, Y. W. Wang, X. Xu, E. Navarro, K. M. Tichauer and J. T. Liu, J. Biophotonics, 2018, e201700246 CrossRef PubMed.
- Y. W. Wang, S. Kang, A. Khan, P. Q. Bao and J. T. Liu, Biomed. Opt. Express, 2015, 6, 3714–3723 CrossRef CAS PubMed.
- N. Hamzei, K. S. Samkoe, J. T. Elliott, R. W. Holt, J. R. Gunn, T. Hasan, B. W. Pogue and K. M. Tichauer, Austin J. Biomed. Eng., 2014, 1, 1002 Search PubMed.
-
L. Sinha, Y. Wang, C. S. Yang, A. Khan, J. T. Liu and K. M. Tichauer, Ex vivo Tissue Classification of Cell Surface Receptor Concentrations Using Kinetic Modeling, International Society for Optics and Photonics, Molecular-Guided Surgery: Molecules, Devices, and Applications, 2015, p. 9311 Search PubMed.
- K. M. Tichauer, R. W. Holt, F. El-Ghussein, S. C. Davis, K. S. Samkoe, J. R. Gunn, F. Leblond and B. W. Pogue, J. Biomed. Opt., 2013, 18, 016003 CrossRef PubMed.
- K. M. Tichauer, Y. Wang, B. W. Pogue and J. T. Liu, Phys. Med. Biol., 2015, 60, R239 CrossRef PubMed.
- M. A. Haidekker and E. A. Theodorakis, J. Mater. Chem. C, 2016, 4, 2707–2718 RSC.
- S. Kumar, T. Verma, R. Mukherjee, F. Ariese, K. Somasundaram and S. Umapathy, Chem. Soc. Rev., 2016, 45, 1879–1900 RSC.
- J. T. Liu, M. W. Helms, M. J. Mandella, J. M. Crawford, G. S. Kino and C. H. Contag, Biophys. J., 2009, 96, 2405–2414 CrossRef CAS PubMed.
- D. Srikun, E. W. Miller, D. W. Domaille and C. J. Chang, J. Am. Chem. Soc., 2008, 130, 4596–4597 CrossRef CAS PubMed.
- A. P. Demchenko, J. Fluoresc., 2010, 20, 1099–1128 CrossRef PubMed.
- J. Fan, M. Hu, P. Zhan and X. Peng, Chem. Soc. Rev., 2013, 42, 29–43 RSC.
- L. Yuan, W. Lin, K. Zheng and S. Zhu, Acc. Chem. Res., 2013, 46, 1462–1473 CrossRef CAS PubMed.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- D. Andina, J.-C. Leroux and P. Luciani, Chem. – Eur. J., 2017, 23, 13549–13573 CrossRef CAS PubMed.
- C. Wang, Z. Wang, T. Zhao, Y. Li, G. Huang, B. D. Sumer and J. Gao, Biomaterials, 2018, 157, 62–75 CrossRef CAS PubMed.
- P. Zhang, A. G. Cheetham, L. L. Lock, Y. Li and H. Cui, Curr. Opin. Biotechnol., 2015, 34, 171–179 CrossRef CAS PubMed.
- Q. Miao and K. Pu, Bioconjugate Chem., 2016, 27, 2808–2823 CrossRef CAS PubMed.
- M. J. Ruedas-Rama, J. D. Walters, A. Orte and E. A. Hall, Anal. Chim. Acta, 2012, 751, 1–23 CrossRef CAS PubMed.
- H.-S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699–4722 RSC.
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- J. Rao, A. Dragulescu-Andrasi and H. Yao, Curr. Opin. Biotechnol., 2007, 18, 17–25 CrossRef CAS PubMed.
- P. Wu, X. Hou, J.-J. Xu and H.-Y. Chen, Nanoscale, 2016, 8, 8427–8442 RSC.
- H. A. Clark, R. Kopelman, R. Tjalkens and M. A. Philbert, Anal. Chem., 1999, 71, 4837–4843 CrossRef CAS PubMed.
- Y.-E. L. Koo, Y. Cao, R. Kopelman, S. M. Koo, M. Brasuel and M. A. Philbert, Anal. Chem., 2004, 76, 2498–2505 CrossRef CAS PubMed.
- J. Peng, X. He, K. Wang, W. Tan, Y. Wang and Y. Liu, Anal. Bioanal. Chem., 2007, 388, 645–654 CrossRef CAS PubMed.
- Q. Yu, K. Y. Zhang, H. Liang, Q. Zhao, T. Yang, S. Liu, C. Zhang, Z. Shi, W. Xu and W. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 5462–5470 CAS.
- W. Pan, H. Wang, L. Yang, Z. Yu, N. Li and B. Tang, Anal. Chem., 2016, 88, 6743–6748 CrossRef CAS PubMed.
- Y.-P. Chen, H.-A. Chen, Y. Hung, F.-C. Chien, P. Chen and C.-Y. Mou, RSC Adv., 2012, 2, 968–973 RSC.
- Y. Zhang, S. Li and Z. Zhao, Anal. Chem., 2016, 88, 12380–12385 CrossRef CAS PubMed.
- Y. Takei, S. Arai, A. Murata, M. Takabayashi, K. Oyama, S. I. Ishiwata, S. Takeoka and M. Suzuki, ACS Nano, 2013, 8, 198–206 CrossRef PubMed.
- L. He, X. Yang, F. Zhao, K. Wang, Q. Wang, J. Liu, J. Huang, W. Li and M. Yang, Anal. Chem., 2015, 87, 2459–2465 CrossRef CAS PubMed.
- Y. Bao, H. De Keersmaecker, S. Corneillie, F. Yu, H. Mizuno, G. Zhang, J. Hofkens, B. Mendrek, A. Kowalczuk and M. Smet, Chem. Mater., 2015, 27, 3450–3455 CrossRef CAS.
- M. J. Patrick, J. M. Janjic, H. Teng, M. R. O’Hear, C. W. Brown, J. A. Stokum, B. F. Schmidt, E. T. Ahrens and A. S. Waggoner, J. Am. Chem. Soc., 2013, 135, 18445–18457 CrossRef CAS PubMed.
- J. Tian, H. Chen, L. Zhuo, Y. Xie, N. Li and B. Tang, Chem. – Eur. J., 2011, 17, 6626–6634 CrossRef CAS PubMed.
- S. Lu, W. Xu, J. Zhang, Y. Chen, L. Xie, Q. Yao, Y. Jiang, Y. Wang and X. Chen, Biosens. Bioelectron., 2016, 86, 176–184 CrossRef CAS PubMed.
- R. F. Wang, H. Q. Peng, P. Z. Chen, L. Y. Niu, J. F. Gao, L. Z. Wu, C. H. Tung, Y. Z. Chen and Q. Z. Yang, Adv. Funct. Mater., 2016, 26, 5419–5425 CrossRef CAS.
- Y. H. Seo, M. J. Cho, O. J. Cheong, W.-D. Jang, T. Y. Ohulchanskyy, S. Lee, D. H. Choi, P. N. Prasad and S. Kim, Biomaterials, 2015, 39, 225–233 CrossRef CAS PubMed.
- J. Park, J. Lee, J. Kwag, Y. Baek, B. Kim, C. J. Yoon, S. Bok, S.-H. Cho, K. H. Kim and G.-O. Ahn, ACS Nano, 2015, 9, 6511–6521 CrossRef CAS PubMed.
- X.-d. Wang, H. H. Gorris, J. A. Stolwijk, R. J. Meier, D. B. Groegel, J. Wegener and O. S. Wolfbeis, Chem. Sci., 2011, 2, 901–906 RSC.
- J. Napp, T. Behnke, L. Fischer, C. Würth, M. Wottawa, D. R. M. Katschinski, F. Alves, U. Resch-Genger and M. Schäferling, Anal. Chem., 2011, 83, 9039–9046 CrossRef CAS PubMed.
- Z. Feng, P. Tao, L. Zou, P. Gao, Y. Liu, X. Liu, H. Wang, S. Liu, Q. Dong and J. Li, ACS Appl. Mater. Interfaces, 2017, 9, 28319–28330 CAS.
- L. Lu, C. Feng, J. Xu, F. Wang, H. Yu, Z. Xu and W. Zhang, Biosens. Bioelectron., 2017, 92, 101–108 CrossRef CAS PubMed.
- Y. Wu, J. Liu, J. Ma, Y. Liu, Y. Wang and D. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 14396–14405 Search PubMed.
- R. Xu, Y. Wang, X. Duan, K. Lu, D. Micheroni, A. Hu and W. Lin, J. Am. Chem. Soc., 2016, 138, 2158–2161 CrossRef CAS PubMed.
- X.-Y. Xu and B. Yan, J. Mater. Chem. C, 2016, 4, 8514–8521 RSC.
- P. Wu, J. Wang, C. He, X. Zhang, Y. Wang, T. Liu and C. Duan, Adv. Funct. Mater., 2012, 22, 1698–1703 CrossRef CAS.
- T. Xia, F. Zhu, K. Jiang, Y. Cui, Y. Yu and G. Qian, Dalton Trans., 2017, 46, 7549–7555 RSC.
- T. Xia, Y. Cui, Y. Yang and G. Qian, ChemNanoMat, 2017, 3, 51–57 CrossRef CAS.
- J.-T. Yi, T.-T. Chen, J. Huo and X. Chu, Anal. Chem., 2017, 89, 12351–12359 CrossRef CAS PubMed.
- Z. Chen, K. Y. Zhang, X. Tong, Y. Liu, C. Hu, S. Liu, Q. Yu, Q. Zhao and W. Huang, Adv. Funct. Mater., 2016, 26, 4386–4396 CrossRef CAS.
- Y.-L. Chiu, S.-A. Chen, J.-H. Chen, K.-J. Chen, H.-L. Chen and H.-W. Sung, ACS Nano, 2010, 4, 7467–7474 CrossRef CAS PubMed.
- L. Chen, L. Wu, J. Yu, C.-T. Kuo, T. Jian, I.-C. Wu, Y. Rong and D. Chiu, Chem. Sci., 2017, 8, 7236–7245 RSC.
- H. s. Peng, J. A. Stolwijk, L. N. Sun, J. Wegener and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2010, 122, 4342–4345 CrossRef.
- T. Liu, J. Hu, Z. Jin, F. Jin and S. Liu, Adv. Healthcare Mater., 2013, 2, 1576–1581 CrossRef CAS PubMed.
- J. Wang, S. R. MacEwan and A. Chilkoti, Nano Lett., 2017, 17, 1226–1232 CrossRef CAS PubMed.
- F. Ye, C. Wu, Y. Jin, Y.-H. Chan, X. Zhang and D. T. Chiu, J. Am. Chem. Soc., 2011, 133, 8146–8149 CrossRef CAS PubMed.
- Y. Cui, R. Song, J. Yu, M. Liu, Z. Wang, C. Wu, Y. Yang, Z. Wang, B. Chen and G. Qian, Adv. Mater., 2015, 27, 1420–1425 CrossRef CAS PubMed.
- X. Zhou, H. Liang, P. Jiang, K. Y. Zhang, S. Liu, T. Yang, Q. Zhao, L. Yang, W. Lv and Q. Yu, Adv. Sci., 2016, 3, 1500155 CrossRef PubMed.
- Q. Zhao, X. Zhou, T. Cao, K. Y. Zhang, L. Yang, S. Liu, H. Liang, H. Yang, F. Li and W. Huang, Chem. Sci., 2015, 6, 1825–1831 RSC.
- H. Shi, X. Ma, Q. Zhao, B. Liu, Q. Qu, Z. An, Y. Zhao and W. Huang, Adv. Funct. Mater., 2014, 24, 4823–4830 CrossRef CAS.
- R. I. Dmitriev, S. M. Borisov, H. Düssmann, S. Sun, B. J. Müller, J. Prehn, V. P. Baklaushev, I. Klimant and D. B. Papkovsky, ACS Nano, 2015, 9, 5275–5288 CrossRef CAS PubMed.
- A. V. Kondrashina, R. I. Dmitriev, S. M. Borisov, I. Klimant, I. O'Brien, Y. M. Nolan, A. V. Zhdanov and D. B. Papkovsky, Adv. Funct. Mater., 2012, 22, 4931–4939 CrossRef CAS.
- C. M. Lemon, E. Karnas, X. Han, O. T. Bruns, T. J. Kempa, D. Fukumura, M. G. Bawendi, R. K. Jain, D. G. Duda and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 9832–9842 CrossRef CAS PubMed.
- X. Wu, Z. Li, L. Yang, J. Han and S. Han, Chem. Sci., 2013, 4, 460–467 RSC.
- H. Wang, P. Zhang, Y. Hong, B. Zhao, P. Yi and J. Chen, Polym. Chem., 2017, 8, 5795–5802 RSC.
- J. Qiao, Z. Liu, Y. Tian, M. Wu and Z. Niu, Chem. Commun., 2015, 51, 3641–3644 RSC.
- K. Han, S.-B. Wang, Q. Lei, J.-Y. Zhu and X.-Z. Zhang, ACS Nano, 2015, 9, 10268–10277 CrossRef CAS PubMed.
- K. Sun, Y. Tang, Q. Li, S. Yin, W. Qin, J. Yu, D. T. Chiu, Y. Liu, Z. Yuan and X. Zhang, ACS Nano, 2016, 10, 6769–6781 CrossRef CAS PubMed.
- C. Zhao, X. Zhang, K. Li, S. Zhu, Z. Guo, L. Zhang, F. Wang, Q. Fei, S. Luo and P. Shi, J. Am. Chem. Soc., 2015, 137, 8490–8498 CrossRef CAS PubMed.
- X. Zhang, Q. Hu, T. Xia, J. Zhang, Y. Yang, Y. Cui, B. Chen and G. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 32259–32265 CAS.
- H. Sun, T. L. Andresen, R. V. Benjaminsen and K. Almdal, J. Biomed. Nanotechnol., 2009, 5, 676–682 CrossRef CAS PubMed.
- H. Sun, K. Almdal and T. L. Andresen, Chem. Commun., 2011, 47, 5268–5270 RSC.
- R. V. Søndergaard, J. R. Henriksen and T. L. Andresen, Nat. Protoc., 2014, 9, 2841–2858 CrossRef PubMed.
- M. M. Elsutohy, V. M. Chauhan, R. Markus, M. A. Kyyaly, S. J. Tendler and J. W. Aylott, Nanoscale, 2017, 9, 5904–5911 RSC.
- R. V. Benjaminsen, H. Sun, J. R. Henriksen, N. M. Christensen, K. Almdal and T. L. Andresen, ACS Nano, 2011, 5, 5864–5873 CrossRef CAS PubMed.
- X.-H. Wang, H.-S. Peng, H. Ding, F.-T. You, S.-H. Huang, F. Teng, B. Dong and H.-W. Song, J. Mater. Chem., 2012, 22, 16066–16071 RSC.
- X. Hu, Y. Li, T. Liu, G. Zhang and S. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 15551–15560 CAS.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- A. J. Shuhendler, K. Pu, L. Cui, J. P. Uetrecht and J. Rao, Nat. Biotechnol., 2014, 32, 373–380 CrossRef CAS PubMed.
- Y.-H. Fu, C.-Y. Chen and C.-T. Chen, Polym. Chem., 2015, 6, 8132–8143 RSC.
- Y. Huang, P. Zhang, M. Gao, F. Zeng, A. Qin, S. Wu and B. Z. Tang, Chem. Commun., 2016, 52, 7288–7291 RSC.
- J. Peng, X. Hou, F. Zeng and S. Wu, Biosens. Bioelectron., 2017, 94, 278–285 CrossRef CAS PubMed.
- Q. Fei, L. Zhou, F. Wang, B. Shi, C. Li, R. Wang and C. Zhao, Dyes Pigm., 2017, 136, 846–851 CrossRef CAS.
- F. R. Kersey, G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, ACS Nano, 2010, 4, 4989–4996 CrossRef CAS PubMed.
- W. Chen, Q. Huang, W. Ou, Y. Hao, L. Wang, K. Zeng, H. Guo, J. Li and Y. N. Liu, Small, 2014, 10, 1261–1265 CrossRef CAS.
- C. Yin, H. Zhu, C. Xie, L. Zhang, P. Chen, Q. Fan, W. Huang and K. Pu, Adv. Funct. Mater., 2017, 27, 1700493 CrossRef.
- A. Burns, P. Sengupta, T. Zedayko, B. Baird and U. Wiesner, Small, 2006, 2, 723–726 CrossRef CAS PubMed.
- L. Yang, Y. Chen, Z. Yu, W. Pan, H. Wang, N. Li and B. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 27512–27521 CAS.
- W. Xu, X. Zhao, W. Lv, H. Yang, S. Liu, H. Liang, Z. Tu, H. Xu, W. Qiao and Q. Zhao, Adv. Healthcare Mater., 2014, 3, 658–669 CrossRef CAS PubMed.
- C. He, W. Zhu, Y. Xu, Y. Zhong, J. Zhou and X. Qian, J. Mater. Chem., 2010, 20, 10755–10764 RSC.
- H. Sun, J. Zhang, K. Y. Zhang, S. Liu, H. Liang, W. Lv, W. Qiao, X. Liu, T. Yang and Q. Zhao, Part. Part. Syst. Charact., 2015, 32, 48–53 CrossRef CAS.
- S. Liu, J. Zhao, K. Zhang, L. Yang, M. Sun, H. Yu, Y. Yan, Y. Zhang, L. Wu and S. Wang, Analyst, 2016, 141, 2296–2302 RSC.
- G. Chen, F. Song, J. Wang, Z. Yang, S. Sun, J. Fan, X. Qiang, X. Wang, B. Dou and X. Peng, Chem. Commun., 2012, 48, 2949–2951 RSC.
- K. Y. Zhang, J. Zhang, Y. Liu, S. Liu, P. Zhang, Q. Zhao, Y. Tang and W. Huang, Chem. Sci., 2015, 6, 301–307 RSC.
- G. Hidalgo, A. Burns, E. Herz, A. G. Hay, P. L. Houston, U. Wiesner and L. W. Lion, Appl. Environ. Microbiol., 2009, 75, 7426–7435 CrossRef CAS PubMed.
- Y. Liu, Y. Tian, Y. Tian, Y. Wang and W. Yang, Adv. Mater., 2015, 27, 7156–7160 CrossRef CAS PubMed.
- T. Doussineau, M. Smaihi and G. J. Mohr, Adv. Funct. Mater., 2009, 19, 117–122 CrossRef CAS.
- S. Liu, L. Zhang, T. Yang, H. Yang, K. Y. Zhang, X. Zhao, W. Lv, Q. Yu, X. Zhang and Q. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 11013–11017 CAS.
-
T. Näreoja, T. Deguchi, S. Christ, R. Peltomaa, N. Prabhakar, E. Fazeli, N. Perälä, J. M. Rosenholm, R. Arppe and T. Soukka, Anal. Chem., 2017, 89, 1501–1508 Search PubMed.
- N. Wang, X. Yu, K. Zhang, C. A. Mirkin and J. Li, J. Am. Chem. Soc., 2017, 139, 12354–12357 CrossRef CAS PubMed.
- L. Zhao, J. Peng, M. Chen, Y. Liu, L. Yao, W. Feng and F. Li, ACS Appl. Mater. Interfaces, 2014, 6, 11190–11197 CAS.
- Y. Guan, S. Qu, B. Li, L. Zhang, H. Ma and L. Zhang, Biosens. Bioelectron., 2016, 77, 124–130 CrossRef CAS PubMed.
- X. Ge, L. Sun, B. Ma, D. Jin, L. Dong, L. Shi, N. Li, H. Chen and W. Huang, Nanoscale, 2015, 7, 13877–13887 RSC.
- J. Liu, Y. Liu, W. Bu, J. Bu, Y. Sun, J. Du and J. Shi, J. Am. Chem. Soc., 2014, 136, 9701–9709 CrossRef CAS PubMed.
- W. Lv, T. Yang, Q. Yu, Q. Zhao, K. Y. Zhang, H. Liang, S. Liu, F. Li and W. Huang, Adv. Sci., 2015, 2, 1500107 CrossRef PubMed.
- J. Lai, B. P. Shah, Y. Zhang, L. Yang and K.-B. Lee, ACS Nano, 2015, 9, 5234–5245 CrossRef CAS PubMed.
- K. Li, Q. Su, W. Yuan, B. Tian, B. Shen, Y. Li, W. Feng and F. Li, ACS Appl. Mater. Interfaces, 2015, 7, 12278–12286 CAS.
- X. Huang, Z. Xu, Y. Mao, Y. Ji, H. Xu, Y. Xiong and Y. Li, Biosens. Bioelectron., 2015, 66, 184–190 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- D. A. Giljohann, D. S. Seferos, P. C. Patel, J. E. Millstone, N. L. Rosi and C. A. Mirkin, Nano Lett., 2007, 7, 3818–3821 CrossRef CAS PubMed.
- M. J. Marín, F. Galindo, P. Thomas and D. A. Russell, Angew. Chem., Int. Ed., 2012, 51, 9657–9661 CrossRef PubMed.
-
K.-K. Yu, K. Li, H.-H. Qin, Q. Zhou, C.-H. Qian, Y.-H. Liu and X.-Q. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 22839–22848 Search PubMed.
- S. Song, Z. Liang, J. Zhang, L. Wang, G. Li and C. Fan, Angew. Chem., Int. Ed., 2009, 121, 8826–8830 CrossRef.
- A. Jayagopal, K. C. Halfpenny, J. W. Perez and D. W. Wright, J. Am. Chem. Soc., 2010, 132, 9789–9796 CrossRef CAS PubMed.
- W. Pan, T. Zhang, H. Yang, W. Diao, N. Li and B. Tang, Anal. Chem., 2013, 85, 10581–10588 CrossRef CAS PubMed.
- A. E. Prigodich, D. S. Seferos, M. D. Massich, D. A. Giljohann, B. C. Lane and C. A. Mirkin, ACS Nano, 2009, 3, 2147–2152 CrossRef CAS PubMed.
- A. E. Prigodich, P. S. Randeria, W. Briley, N. Kim, W. L. Daniel, D. A. Giljohann and C. A. Mirkin, Anal. Chem., 2012, 84, 2062–2066 CrossRef CAS PubMed.
- N. Li, C. Chang, W. Pan and B. Tang, Angew. Chem., Int. Ed., 2012, 51, 7426–7430 CrossRef CAS PubMed.
- Y. Yang, J. Huang, X. Yang, K. Quan, H. Wang, L. Ying, N. Xie, M. Ou and K. Wang, J. Am. Chem. Soc., 2015, 137, 8340–8343 CrossRef CAS PubMed.
- J. Huang, L. Ying, X. Yang, Y. Yang, K. Quan, H. Wang, N. Xie, M. Ou, Q. Zhou and K. Wang, Anal. Chem., 2015, 87, 8724–8731 CrossRef CAS PubMed.
- Y. Yang, J. Huang, X. Yang, K. Quan, N. Xie, M. Ou, J. Tang and K. Wang, Chem. Commun., 2016, 52, 11386–11389 RSC.
- X.-J. Yang, K. Zhang, T.-T. Zhang, J.-J. Xu and H.-Y. Chen, Anal. Chem., 2017, 89, 4216–4222 CrossRef CAS PubMed.
- S. A. Hilderbrand, K. A. Kelly, M. Niedre and R. Weissleder, Bioconjugate Chem., 2008, 19, 1635–1639 CrossRef CAS PubMed.
- Y. Tian, M. Wu, X. Liu, Z. Liu, Q. Zhou, Z. Niu and Y. Huang, Adv. Healthcare Mater., 2015, 4, 413–419 CrossRef CAS PubMed.
- L. Albertazzi, B. Storti, L. Marchetti and F. Beltram, J. Am. Chem. Soc., 2010, 132, 18158–18167 CrossRef CAS PubMed.
- Y. T. Tsai, J. Zhou, H. Weng, J. Shen, L. Tang and W. J. Hu, Adv. Healthcare Mater., 2014, 3, 221–229 CrossRef CAS PubMed.
- L. Chen, Y. Wu, Y. Lin and Q. Wang, Chem. Commun., 2015, 51, 10190–10193 RSC.
- C. Zong, K. Ai, G. Zhang, H. Li and L. Lu, Anal. Chem., 2011, 83, 3126–3132 CrossRef CAS PubMed.
- H. Ma, B. Song, Y. Wang, D. Cong, Y. Jiang and J. Yuan, Chem. Sci., 2017, 8, 150–159 RSC.
- Q. Yu, T. Huang, Y. Li, H. Wei, S. Liu, W. Huang, J. Du and Q. Zhao, Chem. Commun., 2017, 53, 4144–4147 RSC.
- M. Zhuang, C. Ding, A. Zhu and Y. Tian, Anal. Chem., 2014, 86, 1829–1836 CrossRef CAS PubMed.
- S. Sun, X. Ning, G. Zhang, Y. C. Wang, C. Peng and J. Zheng, Angew. Chem., Int. Ed., 2016, 128, 2467–2470 CrossRef.
-
Q. Yang, Z. Ye, M. Zhong, B. Chen, J. Chen, R. Zeng, L. Wei, H.-W. Li and L. Xiao, ACS Appl. Mater. Interfaces, 2016, 8, 9629–9634 Search PubMed.
- X. Gao, C. Ding, A. Zhu and Y. Tian, Anal. Chem., 2014, 86, 7071–7078 CrossRef CAS PubMed.
- L. Wei, Y. Ma, X. Shi, Y. Wang, X. Su, C. Yu, S. Xiang, L. Xiao and B. Chen, J. Mater. Chem. B, 2017, 5, 3383–3390 RSC.
- Y. Zhang, S. Guo, S. Cheng, X. Ji and Z. He, Biosens. Bioelectron., 2017, 94, 478–484 CrossRef CAS PubMed.
- M. Amelia, A. Lavie-Cambot, N. D. McClenaghan and A. Credi, Chem. Commun., 2011, 47, 325–327 RSC.
- A. Shamirian, H. Samareh Afsari, A. Hassan, L. W. Miller and P. T. Snee, ACS Sens., 2016, 1, 1244–1250 CrossRef CAS PubMed.
- L. Bau, F. Selvestrel, M. Arduini, I. Zamparo, C. Lodovichi and F. Mancin, Org. Lett., 2012, 14, 2984–2987 CrossRef CAS PubMed.
- S. Liu, J. Zhang, D. Shen, H. Liang, X. Liu, Q. Zhao and W. Huang, Chem. Commun., 2015, 51, 12839–12842 RSC.
- Y. Shi, Z. Chen, X. Cheng, Y. Pan, H. Zhang, Z. Zhang, C.-W. Li and C. Yi, Biosens. Bioelectron., 2014, 61, 397–403 CrossRef CAS PubMed.
- J. Fu, C. Ding, A. Zhu and Y. Tian, Analyst, 2016, 141, 4766–4771 RSC.
- B. Chu, H. Wang, B. Song, F. Peng, Y. Su and Y. He, Anal. Chem., 2016, 88, 9235–9242 CrossRef CAS PubMed.
- W. Xu, Y. Xiong, W. Lai, Y. Xu, C. Li and M. Xie, Biosens. Bioelectron., 2014, 56, 144–150 CrossRef CAS PubMed.
- P. T. Snee, R. C. Somers, G. Nair, J. P. Zimmer, M. G. Bawendi and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 13320–13321 CrossRef CAS PubMed.
- A. Moquin, E. Hutter, A. O. Choi, A. Khatchadourian, A. Castonguay, F. O. M. Winnik and D. Maysinger, ACS Nano, 2013, 7, 9585–9598 CrossRef CAS PubMed.
- Y. Fu, C. Ding, A. Zhu, Z. Deng, Y. Tian and M. Jin, Anal. Chem., 2013, 85, 11936–11943 CrossRef CAS PubMed.
- A. Shamirian, H. S. Afsari, D. Wu, L. W. Miller and P. T. Snee, Anal. Chem., 2016, 88, 6050–6056 CrossRef CAS PubMed.
- B. Hu, L.-L. Hu, M.-L. Chen and J.-H. Wang, Biosens. Bioelectron., 2013, 49, 499–505 CrossRef CAS PubMed.
- K. Susumu, L. D. Field, E. Oh, M. Hunt, J. B. Delehanty, V. Palomo, P. E. Dawson, A. L. Huston and I. L. Medintz, Chem. Mater., 2017, 29, 7330–7344 CrossRef CAS.
- Y. Liu, X. Qu, Q. Guo, Q. Sun and X. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 4725–4732 CAS.
- W. Zhao, Y. Li, S. Yang, Y. Chen, J. Zheng, C. Liu, Z. Qing, J. Li and R. Yang, Anal. Chem., 2016, 88, 4833–4840 CrossRef CAS PubMed.
- C. Yu, X. Li, F. Zeng, F. Zheng and S. Wu, Chem. Commun., 2013, 49, 403–405 RSC.
- C. Yu, Y. Wu, F. Zeng and S. Wu, J. Mater. Chem. B, 2013, 1, 4152–4159 RSC.
- G. Wu, F. Zeng, C. Yu, S. Wu and W. Li, J. Mater. Chem. B, 2014, 2, 8528–8537 RSC.
- F. Du, Y. Ming, F. Zeng, C. Yu and S. Wu, Nanotechnology, 2013, 24, 365101 CrossRef PubMed.
- J. S. Sidhu, A. Singh, N. Garg and N. Singh, ACS Appl. Mater. Interfaces, 2017, 9, 25847–25856 CAS.
- W. Yang, J. Ni, F. Luo, W. Weng, Q. Wei, Z. Lin and G. Chen, Anal. Chem., 2017, 89, 8384–8390 CrossRef CAS PubMed.
- J. Sun, H. Mei and F. Gao, Biosens. Bioelectron., 2017, 91, 70–75 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler and J. Rao, Angew. Chem., Int. Ed., 2013, 52, 10325–10329 CrossRef CAS PubMed.
- J. Sun, P. Ling and F. Gao, Anal. Chem., 2017, 89, 11703–11710 CrossRef CAS PubMed.
- Z. Ding, J. Tan, G. Feng, Z. Yuan, C. Wu and X. Zhang, Chem. Sci., 2017, 8, 5101–5106 RSC.
- L. Zhang, J. Lei, J. Liu, F. Ma and H. Ju, Biomaterials, 2015, 67, 323–334 CrossRef CAS PubMed.
- A. M. Dennis, W. J. Rhee, D. Sotto, S. N. Dublin and G. Bao, ACS Nano, 2012, 6, 2917–2924 CrossRef CAS PubMed.
- F. Du, Y. Min, F. Zeng, C. Yu and S. Wu, Small, 2014, 10, 964–972 CrossRef CAS PubMed.
- Y. I. Park, H. M. Kim, J. H. Kim, K. C. Moon, B. Yoo, K. T. Lee, N. Lee, Y. Choi, W. Park and D. Ling, Adv. Mater., 2012, 24, 5755–5761 CrossRef CAS PubMed.
- F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010, 135, 1839–1854 RSC.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- X. Huang, Z. P. Aguilar, H. Xu, W. Lai and Y. Xiong, Biosens. Bioelectron., 2016, 75, 166–180 CrossRef CAS PubMed.
- X. Huang, Y. Liu, B. Yung, Y. Xiong and X. Chen, ACS Nano, 2017, 11, 5238–5292 CrossRef CAS PubMed.
- Y. Zhou, W. Chen, J. Zhu, W. Pei, C. Wang, L. Huang, C. Yao, Q. Yan, W. Huang and J. S. C. Loo, Small, 2014, 10, 4874–4885 CrossRef CAS PubMed.
- Y. Cen, Y.-M. Wu, X.-J. Kong, S. Wu, R.-Q. Yu and X. Chu, Anal. Chem., 2014, 86, 7119–7127 CrossRef CAS PubMed.
- J. Peng, C. L. Teoh, X. Zeng, A. Samanta, L. Wang, W. Xu, D. Su, L. Yuan, X. Liu and Y. T. Chang, Adv. Funct. Mater., 2016, 26, 191–199 CrossRef CAS.
- Q. Liu, J. Peng, L. Sun and F. Li, ACS Nano, 2011, 5, 8040–8048 CrossRef CAS PubMed.
- B. Gu, Y. Zhou, X. Zhang, X. Liu, Y. Zhang, R. Marks, H. Zhang, X. Liu and Q. Zhang, Nanoscale, 2016, 8, 276–282 RSC.
- J. Peng, W. Xu, C. L. Teoh, S. Han, B. Kim, A. Samanta, J. C. Er, L. Wang, L. Yuan and X. Liu, J. Am. Chem. Soc., 2015, 137, 2336–2342 CrossRef CAS PubMed.
- L. Tian, Z. Dai, X. Liu, B. Song, Z. Ye and J. Yuan, Anal. Chem., 2015, 87, 10878–10885 CrossRef CAS PubMed.
- R. Arppe, T. Näreoja, S. Nylund, L. Mattsson, S. Koho, J. M. Rosenholm, T. Soukka and M. Schäferling, Nanoscale, 2014, 6, 6837–6843 RSC.
- C. Li, J. Zuo, L. Zhang, Y. Chang, Y. Zhang, L. Tu, X. Liu, B. Xue, Q. Li and H. Zhao, Sci. Rep., 2016, 6, 38617 CrossRef CAS PubMed.
- Y.-X. Wu, X.-B. Zhang, D.-L. Zhang, C.-C. Zhang, J.-B. Li, Y. Wu, Z.-L. Song, R.-Q. Yu and W. Tan, Anal. Chem., 2016, 88, 1639–1646 CrossRef CAS PubMed.
- X. Zou, Y. Liu, X. Zhu, M. Chen, L. Yao, W. Feng and F. Li, Nanoscale, 2015, 7, 4105–4113 RSC.
- Y. Zhou, W. Pei, X. Zhang, W. Chen, J. Wu, C. Yao, L. Huang, H. Zhang, W. Huang and J. S. C. Loo, Biomaterials, 2015, 54, 34–43 CrossRef CAS PubMed.
- Y. Zhou, W. Pei, C. Wang, J. Zhu, J. Wu, Q. Yan, L. Huang, W. Huang, C. Yao and J. S. C. Loo, Small, 2014, 10, 3560–3567 CrossRef CAS PubMed.
- Q. Mei, W. Deng, W. Yisibashaer, H. Jing, G. Du, M. Wu, B. N. Li and Y. Zhang, Small, 2015, 11, 4568–4575 CrossRef CAS PubMed.
- Z. Chen, Z. Liu, Z. Li, E. Ju, N. Gao, L. Zhou, J. Ren and X. Qu, Biomaterials, 2015, 39, 15–22 CrossRef CAS PubMed.
- J. Liu, Y. Liu, Q. Liu, C. Li, L. Sun and F. Li, J. Am. Chem. Soc., 2011, 133, 15276–15279 CrossRef CAS PubMed.
- Y. Liu, M. Chen, T. Cao, Y. Sun, C. Li, Q. Liu, T. Yang, L. Yao, W. Feng and F. Li, J. Am. Chem. Soc., 2013, 135, 9869–9876 CrossRef CAS PubMed.
- K. M. Taylor-Pashow, J. Della Rocca, R. C. Huxford and W. Lin, Chem. Commun., 2010, 46, 5832–5849 RSC.
- C. Sanchez, K. J. Shea and S. Kitagawa, Chem. Soc. Rev., 2011, 40, 471–472 RSC.
- T. Chen, Y. Hu, Y. Cen, X. Chu and Y. Lu, J. Am. Chem. Soc., 2013, 135, 11595–11602 CrossRef CAS PubMed.
- L. Zhang, D. Wang, H. Huang, L. Liu, Y. Zhou, X. Xia, K. Deng and X. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 6646–6655 CAS.
- C. Niu, Q. Liu, Z. Shang, L. Zhao and J. Ouyang, Nanoscale, 2015, 7, 8457–8465 RSC.
- A. Zhu, Q. Qu, X. Shao, B. Kong and Y. Tian, Angew. Chem., Int. Ed., 2012, 124, 7297–7301 CrossRef.
- C. Zou, M. F. Foda, X. Tan, K. Shao, L. Wu, Z. Lu, H. S. Bahlol and H. Han, Anal. Chem., 2016, 88, 7395–7403 CrossRef CAS PubMed.
- X. Liu, N. Zhang, T. Bing and D. Shangguan, Anal. Chem., 2014, 86, 2289–2296 CrossRef CAS PubMed.
- X. Sun, P. Liu, L. Wu and B. Liu, Analyst, 2015, 140, 6742–6747 RSC.
- L. Wu, Q.-S. Guo, Y.-Q. Liu and Q.-J. Sun, Anal. Chem., 2015, 87, 5318–5323 CrossRef CAS PubMed.
- T. T. Ruckh, C. G. Skipwith, W. Chang, V. Bulovic, P. Anikeeva and H. A. Clark, ACS Nano, 2016, 10, 4020–4030 CrossRef CAS PubMed.
- K. Paek, H. Yang, J. Lee, J. Park and B. J. Kim, ACS Nano, 2014, 8, 2848–2856 CrossRef CAS PubMed.
- I. Acquah, J. Roh and D. J. Ahn, Chem. – Asian J., 2017, 12, 1724–1729 CrossRef CAS PubMed.
- Z. Li, S. Guo, Z. Yuan and C. Lu, Sens. Actuators, B, 2017, 241, 821–827 CrossRef CAS.
- H. Jin, R. Gui, J. Sun and Y. Wang, Anal. Chim. Acta, 2016, 922, 48–54 CrossRef CAS PubMed.
- P. Zhao, K. He, Y. Han, Z. Zhang, M. Yu, H. Wang, Y. Huang, Z. Nie and S. Yao, Anal. Chem., 2015, 87, 9998–10005 CrossRef CAS PubMed.
- C. Wang, H. Lin, Z. Xu, Y. Huang, M. G. Humphrey and C. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 6621–6628 CAS.
- J. Liu, Y. Wu, Y. Yu, K. Li, Y. Ji and D. Wu, Biosens. Bioelectron., 2017, 98, 119–125 CrossRef CAS PubMed.
- E. Ju, Z. Liu, Y. Du, Y. Tao, J. Ren and X. Qu, ACS Nano, 2014, 8, 6014–6023 CrossRef CAS PubMed.
- F. W. Pratiwi, C.-H. Hsia, C. W. Kuo, S.-M. Yang, Y.-K. Hwu and P. Chen, Biosens. Bioelectron., 2016, 84, 133–140 CrossRef CAS PubMed.
- L. Lu, G. Yang and Y. Xia, Anal. Chem., 2014, 86, 6188–6191 CrossRef CAS PubMed.
- W. Gu, S. Gong, Y. Zhou and Y. Xia, Biosens. Bioelectron., 2017, 90, 487–493 CrossRef CAS PubMed.
- Y. Wang, L. Lu, H. Peng, J. Xu, F. Wang, R. Qi, Z. Xu and W. Zhang, Chem. Commun., 2016, 52, 9247–9250 RSC.
- J. Shangguan, D. He, X. He, K. Wang, F. Xu, J. Liu, J. Tang, X. Yang and J. Huang, Anal. Chem., 2016, 88, 7837–7843 CrossRef CAS PubMed.
- F. Liu, T. Bing, D. Shangguan, M. Zhao and N. Shao, Anal. Chem., 2016, 88, 10631–10638 CrossRef CAS PubMed.
- C.-H. Hsia, A. Wuttig and H. Yang, ACS Nano, 2011, 5, 9511–9522 CrossRef CAS PubMed.
- E. J. McLaurin, V. A. Vlaskin and D. R. Gamelin, J. Am. Chem. Soc., 2011, 133, 14978–14980 CrossRef CAS PubMed.
- S. Cao, J. Zheng, J. Zhao, Z. Yang, M. Shang, C. Li, W. Yang and X. Fang, Adv. Funct. Mater., 2016, 26, 7224–7233 CrossRef CAS.
- A. M. Derfus, W. C. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS PubMed.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165–172 CrossRef PubMed.
- V. Linko, A. Ora and M. A. Kostiainen, Trends Biotechnol., 2015, 33, 586–594 CrossRef CAS PubMed.
- Y.-J. Chen, B. Groves, R. A. Muscat and G. Seelig, Nat. Nanotechnol., 2015, 10, 748–760 CrossRef CAS PubMed.
- P. Yin, H. M. Choi, C. R. Calvert and N. A. Pierce, Nature, 2008, 451, 318–322 CrossRef CAS PubMed.
- S. Beyer and F. C. Simmel, Nucleic Acids Res., 2006, 34, 1581–1587 CrossRef CAS PubMed.
- C. Mao, W. Sun, Z. Shen and N. C. Seeman, Nature, 1999, 397, 144–146 CrossRef CAS PubMed.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef CAS PubMed.
- H. Pei, X. Zuo, D. Zhu, Q. Huang and C. Fan, Acc. Chem. Res., 2013, 47, 550–559 CrossRef PubMed.
- O. I. Wilner and I. Willner, Chem. Rev., 2012, 112, 2528–2556 CrossRef CAS PubMed.
- H. Lee, A. K. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko and M. Jayaraman, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed.
- C. Y. Tay, L. Yuan and D. T. Leong, ACS Nano, 2015, 9, 5609–5617 CrossRef CAS PubMed.
- J. H. Nuli Xie, X. Yang, Y. Yang, K. Quan, H. Wang, M. O. Le Ying and K. Wang, Chem. Commun., 2016, 52, 2346–2349 RSC.
- W. Zhou, D. Li, C. Xiong, R. Yuan and Y. Xiang, ACS Appl. Mater. Interfaces, 2016, 8, 13303–13308 CAS.
- S. Saha, V. Prakash, S. Halder, K. Chakraborty and Y. Krishnan, Nat. Nanotechnol., 2015, 10, 645–651 CrossRef CAS PubMed.
- P. J. Santangelo, B. Nix, A. Tsourkas and G. Bao, Nucleic Acids Res., 2004, 32, e57 CrossRef PubMed.
- S. Modi, M. Swetha, D. Goswami, G. D. Gupta, S. Mayor and Y. Krishnan, Nat. Nanotechnol., 2009, 4, 325–330 CrossRef CAS PubMed.
- M. Yang, X. Zhang, H. Liu, H. Kang, Z. Zhu, W. Yang and W. Tan, Anal. Chem., 2015, 87, 5854–5859 CrossRef CAS PubMed.
- Y. Wu, J. Liu, Y. Wang, K. Li, L. Li, J. Xu and D. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 11073–11081 CAS.
- H. Pei, L. Liang, G. Yao, J. Li, Q. Huang and C. Fan, Angew. Chem., Int. Ed., 2012, 124, 9154–9158 CrossRef.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
- M. Giovanni, M. I. Setyawati, C. Y. Tay, H. Qian, W. S. Kuan and D. T. Leong, Adv. Funct. Mater., 2015, 25, 3840–3846 CrossRef CAS.
- R. Hu, X. Zhang, Z. Zhao, G. Zhu, T. Chen, T. Fu and W. Tan, Angew. Chem., Int. Ed., 2014, 126, 5931–5936 CrossRef.
- M. You, G. Zhu, T. Chen, M. J. Donovan and W. Tan, J. Am. Chem. Soc., 2015, 137, 667–674 CrossRef CAS PubMed.
- J. Li, C. Zheng, S. Cansiz, C. Wu, J. Xu, C. Cui, Y. Liu, W. Hou, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2015, 137, 1412–1415 CrossRef CAS PubMed.
- H. Shigeto, K. Nakatsuka, T. Ikeda, R. Hirota, A. Kuroda and H. Funabashi, Anal. Chem., 2016, 88, 7894–7898 CrossRef CAS PubMed.
- Y. Liu, Q. Chen, J. Liu, X. Yang, Q. Guo, L. Li, W. Liu and K. Wang, Anal. Chem., 2017, 89, 3590–3596 CrossRef CAS PubMed.
- N. Xie, J. Huang, X. Yang, X. He, J. Liu, J. Huang, H. Fang and K. Wang, Anal. Chem., 2017, 89, 12115–12122 CrossRef CAS PubMed.
- N. Xie, J. Huang, X. Yang, Y. Yang, K. Quan, M. Ou, H. Fang and K. Wang, ACS Sens., 2016, 1, 1445–1452 CrossRef CAS.
- X. Zheng, R. Peng, X. Jiang, Y. Wang, S. Xu, G. Ke, T. Fu, Q. Liu, S.-Y. Huan and X. Zhang, Anal. Chem., 2017, 89, 10941–10947 CrossRef CAS PubMed.
- K. E. Bujold, J. C. Hsu and H. F. Sleiman, J. Am. Chem. Soc., 2016, 138, 14030–14038 CrossRef CAS PubMed.
- L. He, D.-Q. Lu, H. Liang, S. Xie, C. Luo, M. Hu, L. Xu, X. Zhang and W. Tan, ACS Nano, 2017, 11, 4060–4066 CrossRef CAS PubMed.
- Z.-M. Ying, Z. Wu, B. Tu, W. Tan and J.-H. Jiang, J. Am. Chem. Soc., 2017, 139, 9779–9782 CrossRef CAS PubMed.
- S. Harmsen, R. Huang, M. A. Wall, H. Karabeber, J. M. Samii, M. Spaliviero, J. R. White, S. Monette, R. O’Connor and K. L. Pitter, Sci. Transl. Med., 2015, 7, 271ra277 Search PubMed.
- W. C. Chan, D. J. Maxwell, X. Gao, R. E. Bailey, M. Han and S. Nie, Curr. Opin. Biotechnol., 2002, 13, 40–46 CrossRef CAS PubMed.
- Y. Liang, X. Huang, R. Yu, Y. Zhou and Y. Xiong, Anal. Chim. Acta, 2016, 936, 195–201 CrossRef CAS PubMed.
- X. Huang, S. Zhan, H. Xu, X. Meng, Y. Xiong and X. Chen, Nanoscale, 2016, 8, 9390–9397 RSC.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- R. T. Borlinghaus, Microsc. Res. Tech., 2006, 69, 689–692 CrossRef CAS PubMed.
- R. Hoebe, C. Van Oven, T. Gadella Jr, P. Dhonukshe, C. Van Noorden and E. Manders, Nat. Biotechnol., 2007, 25, 249 CrossRef CAS PubMed.
- Y. Han, C. Ding, J. Zhou and Y. Tian, Anal. Chem., 2015, 87, 5333–5339 CrossRef CAS PubMed.
- J. Liu, C. Zhang, J. Dong, J. Zhu, C. Shen, G. Yang and X. Zhang, New J. Chem., 2017, 41, 4733–4737 RSC.
- W. Xu, S. Lu, M. Xu, Y. Jiang, Y. Wang and X. Chen, J. Mater. Chem. B, 2016, 4, 292–298 RSC.
- X.-d. Wang, J. A. Stolwijk, T. Lang, M. Sperber, R. J. Meier, J. Wegener and O. S. Wolfbeis, J. Am. Chem. Soc., 2012, 134, 17011–17014 CrossRef CAS PubMed.
- H. Huang, F. Dong and Y. Tian, Anal. Chem., 2016, 88, 12294–12302 CrossRef CAS PubMed.
- J. Chen, C. Zhang, K. Lv, H. Wang, P. Zhang, P. Yi and J. Jiang, Sens. Actuators, B, 2017, 251, 533–541 CrossRef CAS.
- H. Sun, A. M. Scharff-Poulsen, H. Gu and K. Almdal, Chem. Mater., 2006, 18, 3381–3384 CrossRef CAS.
- S. Laing, L. E. Jamieson, K. Faulds and D. Graham, Nat. Rev. Chem., 2017, 1, 0060 CrossRef.
- I. P. Santos, E. M. Barroso, T. C. B. Schut, P. J. Caspers, C. G. van Lanschot, D.-H. Choi, M. F. van der Kamp, R. W. Smits, R. van Doorn and R. M. Verdijk, Analyst, 2017, 142, 3025–3047 RSC.
- C. Krafft, I. Schie, T. Meyer, M. Schmitt and J. Popp, Chem. Soc. Rev., 2016, 45, 1819–1849 RSC.
- G. Bodelón, V. Montes-García, C. Fernández-López, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Small, 2015, 11, 4149–4157 CrossRef PubMed.
- Y. Wang, B. Yan and L. Chen, Chem. Rev., 2013, 113, 1391–1428 CrossRef CAS PubMed.
- D. Cialla-May, X.-S. Zheng, K. Weber and J. Popp, Chem. Soc. Rev., 2017, 46, 3945–3961 RSC.
- J. Kneipp, ACS Nano, 2017, 11, 1136–1141 CrossRef CAS PubMed.
- S. Harmsen, M. A. Wall, R. Huang and M. F. Kircher, Nat. Protoc., 2017, 12, 1400–1414 CrossRef CAS PubMed.
- O. Stevens, I. E. I. Petterson, J. C. Day and N. Stone, Chem. Soc. Rev., 2016, 45, 1919–1934 RSC.
- M. Jermyn, K. Mok, J. Mercier, J. Desroches, J. Pichette, K. Saint-Arnaud, L. Bernstein, M.-C. Guiot, K. Petrecca and F. Leblond, Sci. Transl. Med., 2015, 7, 274ra219 Search PubMed.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- A. Pallaoro, M.
R. Hoonejani, G. B. Braun, C. D. Meinhart and M. Moskovits, ACS Nano, 2015, 9, 4328–4336 CrossRef CAS PubMed.
- A. Pallaoro, G. B. Braun and M. Moskovits, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16559–16564 CrossRef CAS PubMed.
- A. Oseledchyk, C. Andreou, M. A. Wall and M. F. Kircher, ACS Nano, 2017, 11, 1488–1497 CrossRef CAS PubMed.
- L. Sinha, Y. Wang, C. Yang, A. Khan, J. G. Brankov, J. T. Liu and K. M. Tichauer, Sci. Rep., 2015, 5, 8582 CrossRef PubMed.
- S. Keren, C. Zavaleta, Z. Cheng, A. de La Zerda, O. Gheysens and S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5844–5849 CrossRef CAS PubMed.
- E. Garai, S. Sensarn, C. L. Zavaleta, D. Van de Sompel, N. O. Loewke, M. J. Mandella, S. S. Gambhir and C. H. Contag, J. Biomed. Opt., 2013, 18, 096008 Search PubMed.
- Y. W. Wang, A. Khan, M. Som, D. Wang, Y. Chen, S. Y. Leigh, D. Meza, P. Z. McVeigh, B. C. Wilson and J. T. Liu, Technology, 2014, 2, 118–132 CrossRef PubMed.
- R. J. Mallia, P. Z. McVeigh, C. J. Fisher, I. Veilleux and B. C. Wilson, Nanomedicine, 2015, 10, 89–101 CrossRef CAS PubMed.
- Y. Wang, S. Kang, A. Khan, G. Ruttner, S. Y. Leigh, M. Murray, S. Abeytunge, G. Peterson, M. Rajadhyaksha and S. Dintzis, Sci. Rep., 2016, 6, 21242 CrossRef CAS PubMed.
- E. Garai, S. Sensarn, C. L. Zavaleta, N. O. Loewke, S. Rogalla, M. J. Mandella, S. A. Felt, S. Friedland, J. T. Liu and S. S. Gambhir, PLoS One, 2015, 10, e0123185 Search PubMed.
- S. Kang, Y. Wang, N. P. Reder and J. T. Liu, PLoS One, 2016, 11, e0163473 Search PubMed.
- S. Ye, X. Li, M. Wang and B. Tang, Anal. Chem., 2017, 89, 5124–5130 CrossRef CAS PubMed.
- A. Trojanowska, N. Pazos-Perez, C. Panisello, T. Gumi, L. Guerrini and R. A. Alvarez-Puebla, J. Colloid Interface Sci., 2015, 460, 128–134 CrossRef CAS PubMed.
- Y. Wu, F. Xiao, Z. Wu and R. Yu, Anal. Chem., 2017, 89, 2852–2858 CrossRef CAS PubMed.
- H. Shi, N. Chen, Y. Su, H. Wang and Y. He, Anal. Chem., 2017, 89, 10279–10285 CrossRef CAS PubMed.
- P. Chen, Z. Wang, S. Zong, D. Zhu, H. Chen, Y. Zhang, L. Wu and Y. Cui, Biosens. Bioelectron., 2016, 75, 446–451 CrossRef CAS PubMed.
- L. Tian, M. Fei, S. Tadepalli, J. J. Morrissey, E. D. Kharasch and S. Singamaneni, Adv. Healthcare Mater., 2015, 4, 1502–1509 CrossRef CAS PubMed.
- D. Ma, J. Zheng, P. Tang, W. Xu, Z. Qing, S. Yang, J. Li and R. Yang, Anal. Chem., 2016, 88, 11852–11859 CrossRef CAS PubMed.
- A. Pallaoro, G. B. Braun, N. Reich and M. Moskovits, Small, 2010, 6, 618–622 CrossRef CAS PubMed.
- S. Hanif, H. Liu, M. Chen, P. Muhammad, Y. Zhou, J. Cao, S. A. Ahmed, J. Xu, X. Xia and H. Chen, Anal. Chem., 2017, 89, 2522–2530 CrossRef CAS PubMed.
- L.-L. Qu, Y.-Y. Liu, S.-H. He, J.-Q. Chen, Y. Liang and H.-T. Li, Biosens. Bioelectron., 2016, 77, 292–298 CrossRef CAS PubMed.
- R. Peng, Y. Si, T. Deng, J. Zheng, J. Li, R. Yang and W. Tan, Chem. Commun., 2016, 52, 8553–8556 RSC.
- X. Gu, H. Wang, Z. D. Schultz and J. P. Camden, Anal. Chem., 2016, 88, 7191–7197 CrossRef CAS PubMed.
- Y. Cao, D.-W. Li, L.-J. Zhao, X.-Y. Liu, X.-M. Cao and Y.-T. Long, Anal. Chem., 2015, 87, 9696–9701 CrossRef CAS PubMed.
- P. Rivera-Gil, C. Vazquez-Vazquez, V. Giannini, M. P. Callao, W. J. Parak, M. A. Correa-Duarte and R. A. Alvarez-Puebla, Angew. Chem., Int. Ed., 2013, 52, 13694–13698 CrossRef CAS PubMed.
- Q. Xu, W. Liu, L. Li, F. Zhou, J. Zhou and Y. Tian, Chem. Commun., 2017, 53, 1880–1883 RSC.
- J. Cui, K. Hu, J.-J. Sun, L.-L. Qu and D.-W. Li, Biosens. Bioelectron., 2016, 85, 324–330 CrossRef CAS PubMed.
- D. W. Li, L. L. Qu, K. Hu, Y. T. Long and H. Tian, Angew. Chem., Int. Ed., 2015, 54, 12758–12761 CrossRef CAS PubMed.
- W. Wang, L. Zhang, L. Li and Y. Tian, Anal. Chem., 2016, 88, 9518–9523 CrossRef CAS PubMed.
- C. Kim, C. Favazza and L. V. Wang, Chem. Rev., 2010, 110, 2756 CrossRef CAS PubMed.
- L. V. Wang and S. Hu, Science, 2012, 335, 1458–1462 CrossRef CAS PubMed.
- Y. Lyu, Y. Fang, Q. Miao, X. Zhen, D. Ding and K. Pu, ACS Nano, 2016, 10, 4472–4481 CrossRef CAS PubMed.
- K. Pu, J. Mei, J. V. Jokerst, G. Hong, A. L. Antaris, N. Chattopadhyay, A. J. Shuhendler, T. Kurosawa, Y. Zhou and S. S. Gambhir, Adv. Mater., 2015, 27, 5184–5190 CrossRef CAS PubMed.
- X. Deán-Ben, S. Gottschalk, B. Mc Larney, S. Shoham and D. Razansky, Chem. Soc. Rev., 2017, 46, 2158–2198 RSC.
- B. Wang, Q. Zhao, N. M. Barkey, D. L. Morse and H. Jiang, Med. Phys., 2012, 39, 2512–2517 CrossRef CAS PubMed.
- X. Zhen, X. Feng, C. Xie, Y. Zheng and K. Pu, Biomaterials, 2017, 127, 97–106 CrossRef CAS PubMed.
- S. Wang, J. Lin, T. Wang, X. Chen and P. Huang, Theranostics, 2016, 6, 2394–2413 CrossRef CAS PubMed.
- J. E. Lemaster and J. V. Jokerst, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1404 CrossRef PubMed.
- V. Ntziachristos and D. Razansky, Chem. Rev., 2010, 110, 2783–2794 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, S.-R. Kothapalli, G. A. Tikhomirov, J. Rao and S. S. Gambhir, J. Am. Chem. Soc., 2013, 135, 11015–11022 CrossRef CAS PubMed.
- J. Levi, S. R. Kothapalli, T.-J. Ma, K. Hartman, B. T. Khuri-Yakub and S. S. Gambhir, J. Am. Chem. Soc., 2010, 132, 11264–11269 CrossRef CAS PubMed.
- J. Levi, S.-R. Kothapalli, S. Bohndiek, J.-K. Yoon, A. Dragulescu-Andrasi, C. Nielsen, A. Tisma, S. Bodapati, G. Gowrishankar and X. Yan, Clin. Cancer Res., 2013, 19, 1494–1502 CrossRef CAS PubMed.
- H. Li, P. Zhang, L. P. Smaga, R. A. Hoffman and J. Chan, J. Am. Chem. Soc., 2015, 137, 15628–15631 CrossRef CAS PubMed.
- K. J. Cash, C. Li, J. Xia, L. V. Wang and H. A. Clark, ACS Nano, 2015, 9, 1692–1698 CrossRef CAS PubMed.
- Q. Miao, Y. Lyu, D. Ding and K. Pu, Adv. Mater., 2016, 28, 3662–3668 CrossRef CAS PubMed.
- Q. Chen, X. Liu, J. Chen, J. Zeng, Z. Cheng and Z. Liu, Adv. Mater., 2015, 27, 6820–6827 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233–239 CrossRef CAS PubMed.
- C. Yin, X. Zhen, Q. Fan, W. Huang and K. Pu, ACS Nano, 2017, 11, 4174–4182 CrossRef CAS PubMed.
- J. Zhang, X. Zhen, P. K. Upputuri, M. Pramanik, P. Chen and K. Pu, Adv. Mater., 2017, 29, 1604764 CrossRef PubMed.
- K. Yang, L. Zhu, L. Nie, X. Sun, L. Cheng, C. Wu, G. Niu, X. Chen and Z. Liu, Theranostics, 2014, 4, 134–141 CrossRef CAS PubMed.
-
C. Liu, Y. Yang, Z. Qiu, Y. Huang and L. Sun, In Vivo Assessment of Protease Activity in Colorectal Cancer by Using Activatable Molecular Photoacoustic Imaging, 2015 IEEE International Ultrasonics Symposium (IUS), 2015, p. 1.
- T. D. Horvath, G. Kim, R. Kopelman and S. Ashkenazi, Analyst, 2008, 133, 747–749 RSC.
- A. Ray, H. K. Yoon, Y. E. K. Lee, R. Kopelman and X. Wang, Analyst, 2013, 138, 3126–3130 RSC.
- J. Jo, C. H. Lee, R. Kopelman and X. Wang, Nat. Commun., 2017, 8, 471 CrossRef PubMed.
- Y. Liu, S. Wang, Y. Ma, J. Lin, H. Y. Wang, Y. Gu, X. Chen and P. Huang, Adv. Mater., 2017, 29, 1606129 CrossRef PubMed.
- S. Guha, G. K. Shaw, T. M. Mitcham, R. R. Bouchard and B. D. Smith, Chem. Commun., 2016, 52, 120–123 RSC.
- Q. Chen, X. Liu, J. Zeng, Z. Cheng and Z. Liu, Biomaterials, 2016, 98, 23–30 CrossRef CAS PubMed.
- S. Roberts, M. Seeger, Y. Jiang, A. Mishra, F. Sigmund, A. Stelzl, A. Lauri, P. Symvoulidis, H. Rolbieski, M. Preller, X. Deán-Ben, D. Razansky, T. Orschmann, S. Desbordes, P. Vetschera, T. Bach, V. Ntziachristos and G. Westmeyer, J. Am. Chem. Soc., 2018, 140, 2718–2721 CrossRef CAS PubMed.
- K. K. Ng, M. Shakiba, E. Huynh, R. A. Weersink, A. Roxin, B. C. Wilson and G. Zheng, ACS Nano, 2014, 8, 8363–8373 CrossRef CAS PubMed.
- D.-B. Cheng, G.-B. Qi, J.-Q. Wang, Y. Cong, F.-H. Liu, H. Yu, Z.-Y. Qiao and H. Wang, Biomacromolecules, 2017, 18, 1249–1258 CrossRef CAS PubMed.
- L.-L. Li, Q. Zeng, W.-J. Liu, X.-F. Hu, Y. Li, J. Pan, D. Wan and H. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 17936–17943 CAS.
- Y. Zhou, D. Wang, Y. Zhang, U. Chitgupi, J. Geng, Y. Wang, Y. Zhang, T. R. Cook, J. Xia and J. F. Lovell, Theranostics, 2016, 6, 688–697 CrossRef CAS PubMed.
- J. Wu, L. You, L. Lan, H. J. Lee, S. T. Chaudhry, R. Li, J. X. Cheng and J. Mei, Adv. Mater., 2017, 29, 1703403 CrossRef PubMed.
- K. Suzuki and T. Nagai, Curr. Opin. Biotechnol., 2017, 48, 135–141 CrossRef CAS PubMed.
- J. Cao, R. Lopez, J. Thacker, J. Moon, C. Jiang, S. Morris, J. Bauer, P. Tao, R. Mason and A. Lippert, Chem. Sci., 2015, 6, 1979–1985 RSC.
- X. Zhen, C. Zhang, C. Xie, Q. Miao, K. L. Lim and K. Pu, ACS Nano, 2016, 10, 6400–6409 CrossRef CAS PubMed.
- G. C. Van de Bittner, C. R. Bertozzi and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 1783–1795 CrossRef CAS PubMed.
- K. Saito, N. Hatsugai, K. Horikawa, K. Kobayashi, T. Matsu-ura, K. Mikoshiba and T. Nagai, PLoS One, 2010, 5, e9935 Search PubMed.
- J. O. Lee, B. Lim and Y.-P. Kim, IEEE J. Sel. Top. Quantum Electron., 2014, 20, 57–66 CrossRef.
- J. Yang, D. Cumberbatch, S. Centanni, S.-Q. Shi, D. Winder, D. Webb and C. H. Johnson, Nat. Commun., 2016, 7, 13268 CrossRef PubMed.
- L. Mezzanotte, M. van‘t Root, H. Karatas, E. A. Goua and C. W. Löwik, Trends Biotechnol., 2017, 35, 640–652 CrossRef CAS PubMed.
- T. Yoshida, A. Kakizuka and H. Imamura, Sci. Rep., 2016, 6, 39618 CrossRef CAS PubMed.
- Y. Zhang, Q. Xie, J. B. Robertson and C. H. Johnson, PLoS One, 2012, 7, e43072 CAS.
- T. Thestrup, J. Litzlbauer, I. Bartholomäus, M. Mues, L. Russo, H. Dana, Y. Kovalchuk, Y. Liang, G. Kalamakis and Y. Laukat, Nat. Methods, 2014, 11, 175–182 CrossRef CAS PubMed.
- M. Takeuchi, Y. Nagaoka, T. Yamada, H. Takakura and T. Ozawa, Anal. Chem., 2010, 82, 9306–9313 CrossRef CAS PubMed.
- A. den Hamer, P. Dierickx, R. Arts, J. S. M. de Vries, L. Brunsveld and M. Merkx, ACS Sens., 2017, 2, 729–734 CrossRef CAS PubMed.
- A. De, A. M. Loening and S. S. Gambhir, Cancer Res., 2007, 67, 7175–7183 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, C. T. Chan, A. De, T. F. Massoud and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12060–12065 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *bc,
Bryant C.
Yung
b,
Xiaohua
Huang
*bc,
Bryant C.
Yung
b,
Xiaohua
Huang