
Fluorogenic reaction-based prodrug conjugates as targeted cancer theranostics
Min Hee
Lee†
 *a,
Amit
Sharma†
b,
Min Jung
Chang
a,
Jinju
Lee
a,
Subin
Son
b,
Jonathan L.
Sessler
*c,
Chulhun
Kang
*d and
Jong Seung
Kim
*b
*a,
Amit
Sharma†
b,
Min Jung
Chang
a,
Jinju
Lee
a,
Subin
Son
b,
Jonathan L.
Sessler
*c,
Chulhun
Kang
*d and
Jong Seung
Kim
*b
aDepartment of Chemistry, Sookmyung Women's University, Seoul 04310, Korea. E-mail: minheelee@sookmyung.ac.kr
bDepartment of Chemistry, Korea University, Seoul 02841, Korea. E-mail: jongskim@korea.ac.kr
cDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712-1224, USA. E-mail: sessler@cm.utexas.edu
dThe School of East-West Medical Science, Kyung Hee University, Yongin 17104, Korea. E-mail: kangch@khu.ac.kr
First published on 23rd October 2017
Abstract
Theranostic systems are receiving ever-increasing attention due to their potential therapeutic utility, imaging enhancement capability, and promise for advancing the field of personalized medicine, particularly as it relates to the diagnosis, staging, and treatment of cancer. In this Tutorial Review, we provide an introduction to the concepts of theranostic drug delivery effected via use of conjugates that are able to target cancer cells selectively, provide cytotoxic chemotherapeutics, and produce readily monitored imaging signals in vitro and in vivo. The underlying design concepts, requiring the synthesis of conjugates composed of imaging reporters, masked chemotherapeutic drugs, cleavable linkers, and cancer targeting ligands, are discussed. Particular emphasis is placed on highlighting the potential benefits of fluorogenic reaction-based targeted systems that are activated for both imaging and therapy by cellular entities, e.g., thiols, reactive oxygen species and enzymes, which are present at relatively elevated levels in tumour environments, physiological characteristics of cancer, e.g., hypoxia and acidic pH. Also discussed are systems activated by an external stimulus, such as light. The work summarized in this Tutorial Review will help define the role fluorogenic reaction-based, cancer-targeting theranostics may have in advancing drug discovery efforts, as well as improving our understanding of cellular uptake and drug release mechanisms.
 Min Jung Chang, Min Hee Lee, Jinju Lee, Jonathan L. Sessler (from left to right) | Prof. Min Hee Lee was born in Icheon, Korea, in 1983. She received her PhD from Korea University in 2012 under the supervision of Prof. Jong Seung Kim. After postdoctoral work at The University of Texas at Austin (Prof. Jonathan L. Sessler, supervisor), she began her academic career in 2015 in the Department of Chemistry at Sookmyung Women's University in Seoul. Her research interests are focused on the development of novel fluorescence-based smart molecules for applications in the sensing and imaging of bioactive species and theranostic drug delivery systems. |
Prof. Jonathan L. Sessler was born in Urbana, Illinois, USA, in 1956. He received his PhD from Stanford University in 1982. After postdoctoral work with Prof. Jean-Marie Lehn and Iwao Tabushi, he began his academic career at The University of Texas at Austin in 1984, where he now holds the position of the Doherty-Welch Chair in Chemistry. He is the author of over 650 publications. He was a cofounder of Pharmacyclics, Inc., a company that was acquired by AbbVie for $21B in 2015, and, more recently, Cible, Inc. His research interests include cancer drug development, ion recognition, supramolecular chemistry, sensing, expanded porphyrins, and electron transfer. |
Min Jung Chang received her BS from Sookmyung Women's University in 2017. She is currently a Master's candidate in the Chemistry Department of Sookmyung Women's University. Her scientific interests involve the development of novel fluorescent molecules that can be used as molecular probes and prodrug delivery conjugates. |
Jinju Lee received her BS from Daejin University in 2017. She is currently a Master's candidate in the Chemistry Department of Sookmyung Women's University. Her research interests are focused on the design and synthesis of fluorescent probes that can detect and image biomolecules associated with various human diseases. |
 Chulhun Kang (front left), Jong Seung Kim (front right), Amit Sharma (back left), Subin Son (back right) | Prof. Jong Seung Kim received a PhD from the Department of Chemistry and Biochemistry at Texas Tech University. After a 1 year postdoctoral fellowship at the University of Houston, he joined the faculty at Konyang University in 1994 and transferred to Dankook University in 2003. In 2007, he moved to the Department of Chemistry at Korea University in Seoul as a professor. To date, his research has produced 380 scientific publications and 70 domestic and international patents. He has been a member of Korea Academy of Science and Technology since 2014 and serves as a vice president of the Korean Chemical Society. |
Prof. Chulhun Kang received an MS Degree in Organic Chemistry from the Department of Chemistry at the Seoul National University and a PhD in Biochemistry from the Department of Biochemistry and Biophysics at Iowa State University. Since 1997, he has been a faculty member at Kyung Hee University, where he is currently a Professor in the Department of Medical Science. His research record includes 55 scientific publications and 10 domestic and international patents in the fields of organic chemistry, protein chemistry, and biology. |
Amit Sharma was born in Nagrota Bagwan (H.P.), India, in 1983. He received his PhD from Guru Nanak Dev University, Amritsar, India, under the supervision of Prof. Kamaljit Singh. Thereafter, he joined Sphaera Pharma Pvt. Ltd, India, as a research scientist (2011–2014). His work was mainly focused on designing and developing new kinase inhibitors as potent small molecule-based cancer therapeutics. Later, he joined Prof. Kim's research group in 2014 as a research professor. Currently, his current research interests are focused on the development of smart biomarkers and next generation drug delivery systems for the advancement of cancer therapeutics. |
Subin Son received his BS Chemistry from Korea University in 2017. He is currently a Master's candidate at the Chemistry Department of Korea University. His research interests include development of sonodynamic based cancer therapeutic agents and fluorescent probes to image biomolecules related to tumour microenvironments. |
Key learning points(1) Concept of theranostic drug delivery as achieved using fluorogenic reaction-based prodrug conjugates.(2) Physiological factors that distinguish cancer cells from normal cells that may be exploited for drug delivery. (3) Characteristics of fluorogenic reactions and cleavable linkers that are attractive for drug delivery and imaging. (4) Summary of anticancer drug agents being explored in the context of theranostic prodrug delivery systems. (5) Strategies for targeting cancer cells. |
Introduction
Theranostic agents are dual function systems that offer both therapeutic promise and potential for concurrent diagnosis. They are particularly attractive in the context of personalized cancer therapy, as well as in high precision cancer imaging.1,2 One important approach to theranostic development involves the creation of targeted drug delivery conjugates able to target cancer cells selectively, provide cytotoxic chemotherapeutics, and allow facile monitoring of the location and efficacy of anticancer agents in vitro and in vivo.1,3–5 A desire to attain these objectives is animating research efforts devoted to preparing conjugates composed of imaging reporters, masked chemotherapeutic drugs, cleavable linkers, and cancer targeting ligands. Ideally, the active therapeutic agents should be masked by conjugating with a cleavable linker, allowing reconversion to the active drug form under physiological conditions. Such multifunctional systems must also operate intracellularly and should be activated by cellular components that are associated with cancer states or expressed at higher levels in cancer cells relative to normal cells. Among the drug delivery conjugates currently being explored, fluorogenic reaction-based targeted prodrug conjugates are of particular interest since they are stable in the blood plasma. However, they may be activated efficiently by various cellular constituents, such as thiols, reactive oxygen species (ROS), and enzymes that are overexpressed in tumours.6 Physiological characteristics of cancer cells, e.g., hypoxia and acidic pH, can also be exploited to achieve activation, as can external stimuli, such as light.7 In cases where the reactions are used to generate a fluorogenic response, as well as release an active drug form, the conjugates are effectively self-contained theranostic drug delivery agents. Introducing a tumour-targeting component to the construct allows an even greater degree of control.8In this Tutorial Review, we will summarize a variety of fluorogenic reaction-based prodrug strategies being pursued in an effort to achieve targeted theranostic drug delivery. The systems in question, all of which are at the research stage of development, will be organized on the basis of the trigger used to achieve drug activation, namely thiols, reactive oxygen species (ROS), acidic pH, hypoxia, reduction of platinum(IV) centres, enzymes, and light. Emphasis will be placed on the design, synthesis, spectroscopic characterization, and preliminary in vitro or in vivo biological evaluation of various theranostic conjugates produced in the author's laboratories and elsewhere.
Design of targeted fluorescent prodrug conjugates
Ideally, fluorogenic reaction-based theranostic conjugates provide both targeted therapeutic release and fluorescence imaging. Such systems typically require the following components to be combined in one drug candidate: fluorescent reporters, masked chemotherapeutic agents, cleavable linkers, and cancer targeting ligands. To date, particular effort has been devoted to developing systems that undergo cleavage under physiological conditions, including inter alia via the hydrolysis of esters, amides, and hydrazone linkers, disulfide exchange-based scission, hypoxia-induced activation, enzymatic reactions, photolysis, and thermolysis.6,7 When the cleavable linkers serve to tether a fluorophore to a prodrug in such a way that the fluorescence signal is modulated upon cleavage, it becomes possible to create systems that operate as both therapeutics and diagnostics (Fig. 1). Classic strategies to achieve signal modulation, including cleavage-induced increases in fluorescence intensity (so-called turn-on systems), will be used to illustrate the core concepts. Most of the results obtained to date can be readily explained in terms of classical mechanisms, such as internal charge transfer (ICT), photo-induced electron transfer (PET), aggregation-induced fluorescence enhancement (AIE), etc.9,10 Fluorophores in common use, including naphthalimide, coumarin, BODIPY, rhodol, and Cy7, have been exploited in the generation of conjugates that rely on doxorubicin, camptothecin, paclitaxel, gemcitabine, and cisplatin as the active payload. In addition, tumour targeting of the conjugates has been achieved via the use of specific site-localizing entities (“ligands” in biological parlance), such as folate, biotin, galactose, and RGD (Arg-Gly-Asp) peptide sequences. The key attributes associated with targeted fluorescent prodrug development are illustrated schematically in Fig. 1.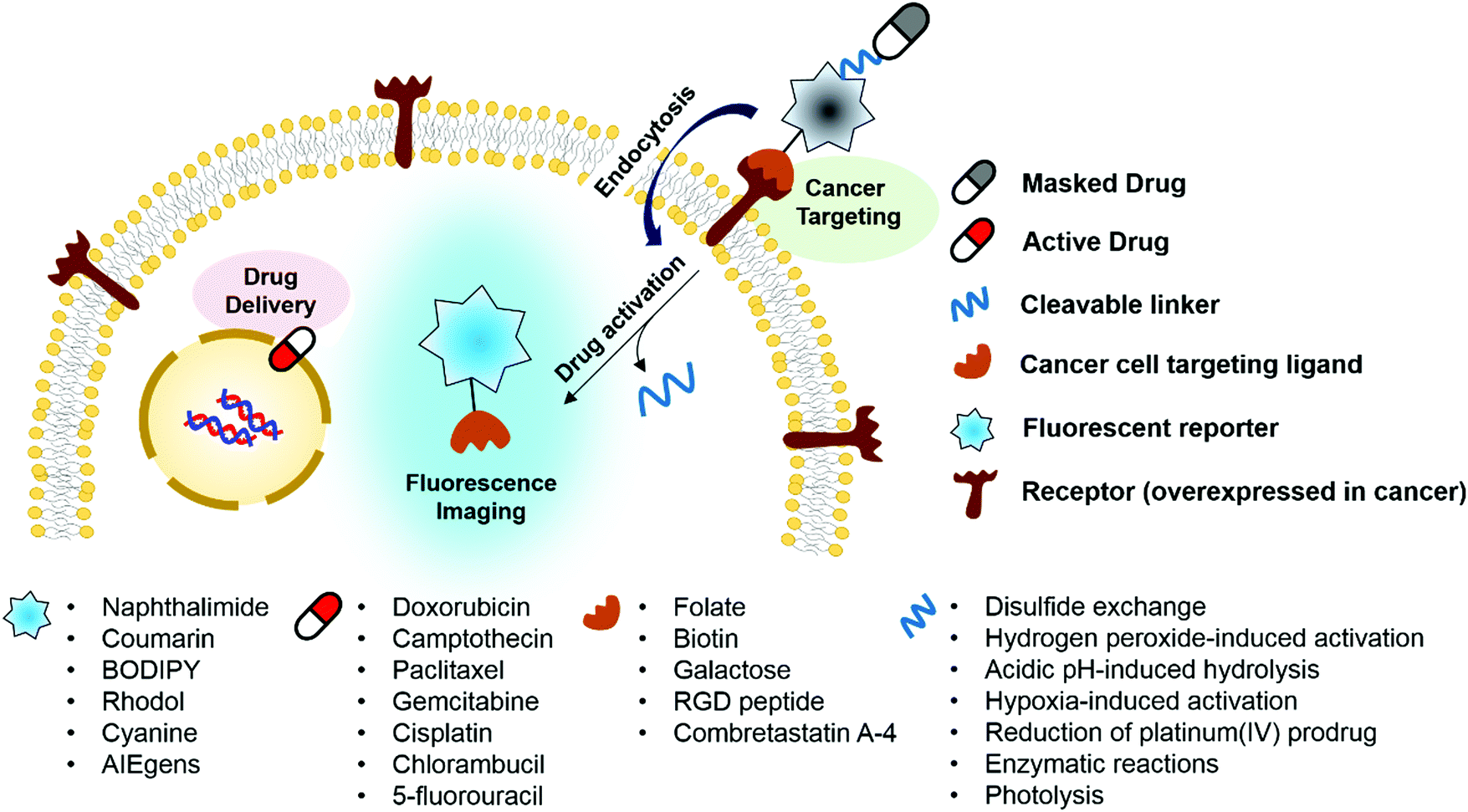 | ||
| Fig. 1 Design principle for achieving fluorogenic reaction-based prodrug conjugates that are able to target cancer cells selectively, provide cytotoxic chemotherapeutics, and produce readily monitored imaging signals in vitro and in vivo. | ||
1. Cellular thiol-activatable theranostic prodrugs
In cancer cells, several endogenous thiols, including glutathione (GSH), thioredoxin (Trx), cysteine (Cys), hydrogen sulfide (H2S), etc., are overexpressed. The associated increase in local concentrations provides a means to distinguish cancer cells from normal cells and is attractive in terms of producing anticancer drug delivery systems (DDS). Distinctions between endogenous thiols can also be exploited for targeting. For instance, it is known that the concentration of intracellular GSH is in the millimolar range, while GSH is typically present only at micromolar levels in common fluids, e.g., blood plasma. This allows thiol-activatable DDS to deliver anticancer agents preferentially into the targeted cancer cells rather than blood vessels. Disulfide bonds are relatively stable in the bloodstream, while in the cancer cells they readily undergo cleavage mediated by intracellular thiols via disulfide–thiol exchange reactions. Not surprisingly, therefore, disulfide bonds have been explored extensively in an effort to generate theranostic DDS that release an anticancer agent and produce a fluorescence change in cancer cells.We reported a RGD peptide-appended naphthalimide pro-camptothecin (CPT) agent 1, which is composed of a cyclic RGD peptide as the tumour targeting ligand, a naphthalimide as a fluorescent reporter, a CPT (an inhibitor of topoisomerase I) as the antitumor payload, and a disulfide bond that was readily cleaved in the presence of certain endogenous thiols (Fig. 2).11 The resulting prodrug conjugate was internalized into the targeted cancer cells by endocytosis mediated by an αvβ3 integrin receptor. The disulfide bond was cleaved by cellular thiols to provide a cytotoxic CPT drug, along with a fluorescence response. The U87 and C6 cell lines were used for the biological evaluation of 1. A strong increase in fluorescence was observed in the U87 cells, which have higher levels of the αvβ3 integrin receptor than the C6 cells. In addition, pre-treatment of the U89 cells with the endocytosis inhibitor, okadaic acid, led to reduced uptake of 1, as inferred from the lower levels of fluorescence intensity observed relative to the inhibitor-free control. Moreover, MTT assays and fluorescence co-localization experiments performed using 1 and various organelle tracking dyes revealed that after cellular uptake, cleavage of the disulfide bond occurs and is actively mediated by cellular thiols mainly in the endoplasmic reticulum (ER) of the cells. The net result is that CPT is released from 1 and then readily diffuses into the nucleus, leading to cancer cell death. These results led to the suggestion that 1 could serve as a theranostic drug delivery agent that provides a cytotoxic chemotherapeutic response while facilitating fluorescence imaging of cancer cells.
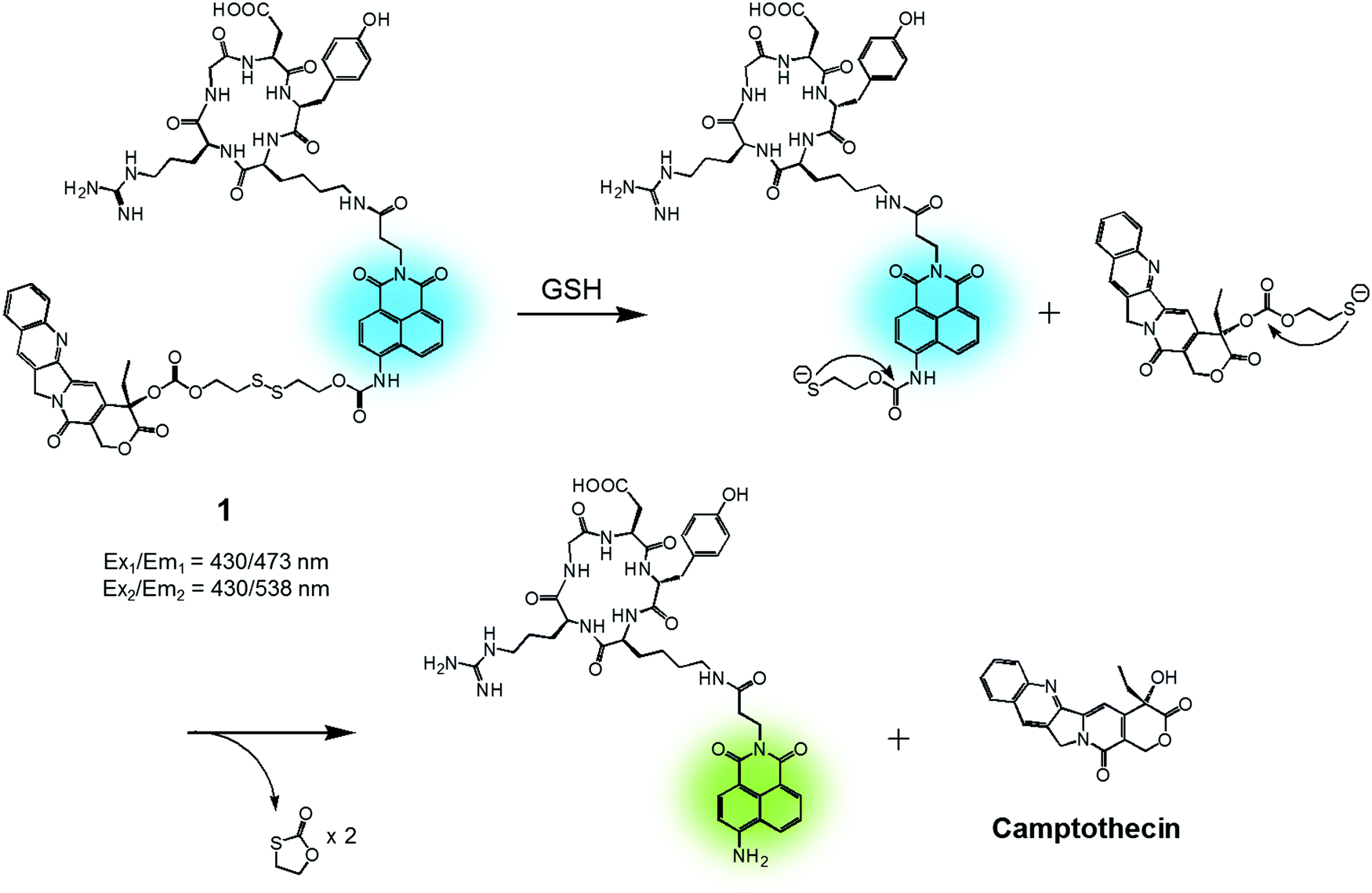 | ||
| Fig. 2 GSH-induced disulfide bond cleavage of theranostic agent 1. Ex1/Em1 and Ex2/Em2 represent, respectively, the excitation and emission wavelengths before and after therapeutic activation. | ||
We recently reported the theranostic agent 2, which is capable of providing both magnetic resonance imaging (MRI) and fluorescence-based imaging for monitoring cellular uptake and prodrug activation processes.12 In this case, a widely used therapeutic agent, doxorubicin (Dox), was conjugated to a gadolinium (Gd3+) texaphyrin via a disulfide bond resulting in theranostic 2. The core gadolinium texaphyrin complex acts as a MR imaging agent that is capable of generating reactive oxygen species (ROS). Conjugation to Dox was expected to enhance the overall potency. To improve the solubility and the tumour targeting ability in vivo, theranostic 2 was loaded into folate-receptor-targeted liposomes producing FL-2 (Fig. 3). As controls, FL-3 (a folate receptor-targeting liposome loaded with analogue 3 but lacking a disulfide bond) and L-2 (a liposome loaded with 2 but without the folate ligand) were also prepared. Theranostic FL-2 was almost non-fluorescent on its own. However, in the presence of thiols, a strong fluorescence signal with a maximum at 592 nm was observed along with the concomitant release of Dox. Evidence for folate receptor-mediated endocytosis in the case of FL-2 came from studies involving the use of both folate receptor positive cell lines (KB and CT26 cells) and folate receptor negative cell lines (HepG2 and NIH3T3 cells). After incubation with FL-2, a strong fluorescence increase was seen in the KB and CT26 cells, whereas a weak fluorescence was observed in the HepG2 and NIH3T3 cells. The therapeutic efficacy of FL-2 was evaluated using both xenograft nude mice and metastatic liver cancer models produced using the KB and CT26 cell lines, respectively. It was found that FL-2 accumulates in the tumour site, as inferred from the build-up of a strong fluorescent signal ascribable to the free Dox released from FL-2. As importantly, this system was found capable of reducing the tumour burden in vivo (Fig. 4A–D). Conjugate FL-2 also produced an enhanced MR signal and could be used to distinguish effectively the tumour area from surrounding normal tissue. In the case of the metastatic liver cancer model, it was found that the mice treated with FL-2 enjoyed a higher survival rate than control animals treated with saline or the non-cleavable conjugate FL-3 (Fig. 4E and F).
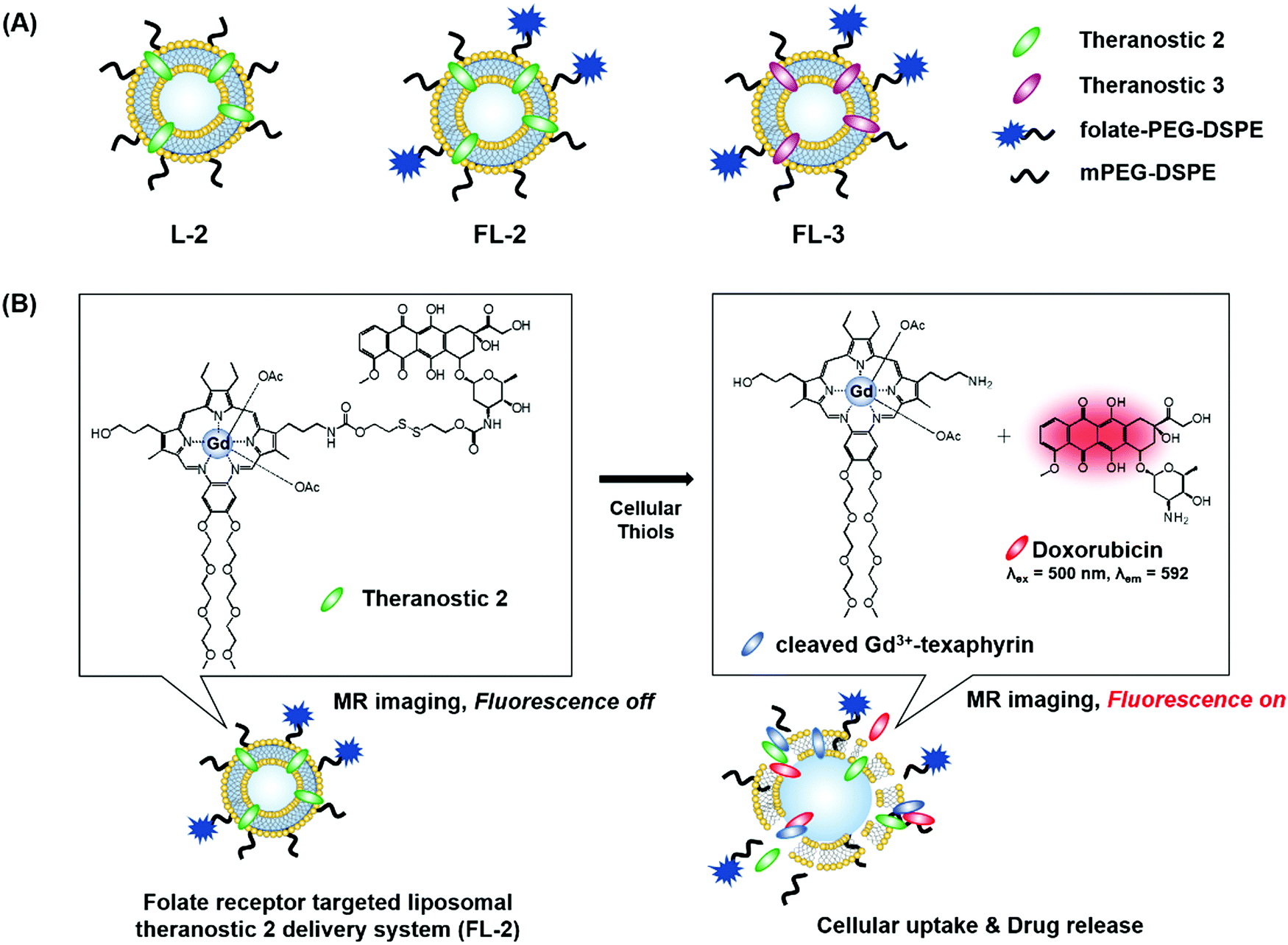 | ||
| Fig. 3 (A) Thiol-activatable theranostic agent FL-2 and controls L-2 and FL-3. (B) Proposed drug release mechanism and fluorescence turn on features expected for FL-2 upon reaction with cellular thiols. | ||
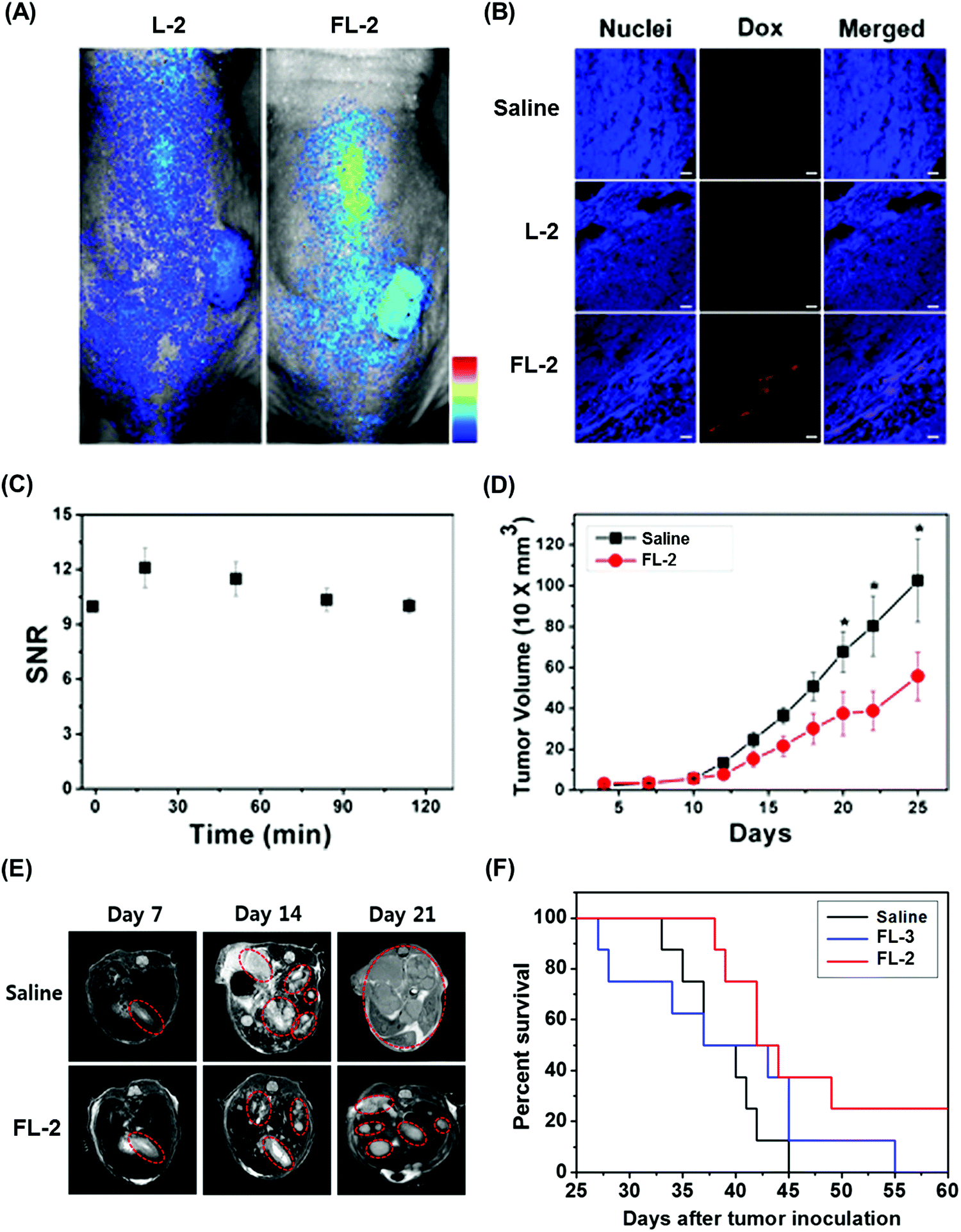 | ||
| Fig. 4 Bioimaging and therapeutic effects of theranostic FL-2 and controls L-2 and FL-3 in (A–D) xenograft tumour nude mice and (E and F) metastatic liver cancer mice. (A–F) are reproduced with permission from ref. 12. Copyright 2016 American Chemical Society. | ||
We also developed theranostic agent 4 which incorporates a rhodol subunit as a fluorescent reporter, biotin as a cancer-targeting ligand, SN-38 as an anticancer drug, and a disulfide linkage to permit cancer cell-based cleavage (Fig. 5).13 The therapeutic efficacy and diagnostic fluorescence changes of 4 were demonstrated by performing both MTT assays in vitro and animal experiments ex vivo. In human derived cancer cells, agent 4 underwent a disulfide cleavage reaction giving rise to an increase in the fluorescence intensity with a concomitant release of SN-38. Ex vivo experimental studies revealed that among various organ tissues, only the tumours produced a strong fluorescence image. These same studies revealed that the tumour volume was significantly diminished compared to the control (an analogue of 4 lacking biotin). On the basis of these results, it was suggested that theranostic 4 could prove useful as a drug delivery system that (1) can selectively enter into tumour tissues via a biotin ligand-related cellular uptake process and (2) provide a source of cytotoxic SN-38 both in vitro and in vivo.
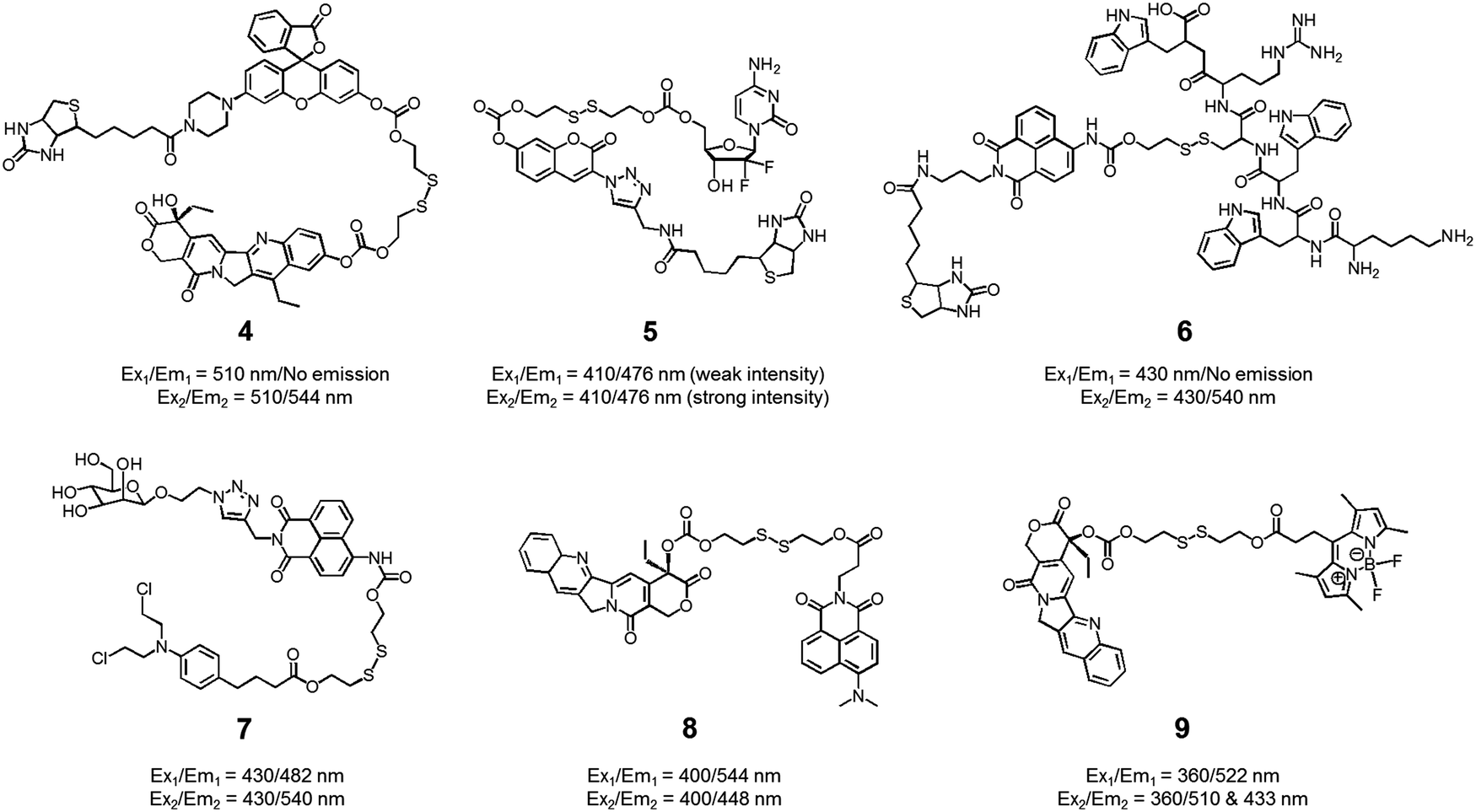 | ||
| Fig. 5 Thiol-activatable theranostic agents 4–9. | ||
In another study, the conjugated biotin–coumarin–gemcitabine system 5 was prepared and tested as a tumour specific theranostic anticancer prodrug (Fig. 5).14 Gemcitabine (GMC) is an excellent anticancer drug but it has several limitations, including a short plasma half-life and an unfavourable toxicity profile. By endowing GMC with a cancer targeting capability it might be possible to improve its chemotherapeutic effect by protecting it from renal clearance, thereby prolonging its circulation half-life. Unfortunately, GMC is essentially non-fluorescent. This makes it difficult to monitor the drug delivery process in vitro and in vivo. The design strategy underlying 5 includes a fluorescent coumarin reporter subunit that is tethered via a disulfide linker. In the presence of GSH, the disulfide linkage is cleaved, giving rise to a readily detectable fluorescence feature at 478 nm and releasing GMC in its cytotoxic free form. The presence of the biotin subunit was designed to allow 5 to target tumours effectively. This expected targeting was demonstrated by confocal microscopic imaging using A549 (biotin receptor positive) and WI38 cell lines (biotin receptor negative). These studies revealed that 5 could be selectively internalized into the lysosome of A549 (human lung cancer) cells through a biotin-associated cellular uptake. When compared to control (a conjugate without biotin), prodrug 5 was found to be a more potent anticancer agent in A549 cells.
The delivery of peptides that can act as antitumor agents is a very active area of research within the DDS field. Peptide-based antitumor agents commonly contain local cationic and anionic charge sites resulting in poor penetration into cancer cells and low therapeutic efficiency. To overcome these perceived bottlenecks, we developed the theranostic agent 6 as a peptide drug delivery system (PDDS). Conjugate 6 contains the Holliday junction inhibitor peptide 2 (KWWCRW) linked to a biotin-naphthalimide moiety via a disulfide linker (Fig. 5).15 Holliday junction (HJ) inhibitor peptide 2 was used as a model peptide drug that shows promise as an antimicrobial and anticancer agent. To investigate the role of the biotin within the conjugate, both biotin-receptor-positive HepG2 cells and biotin-receptor-negative W138 cells were treated with 6. It was found that conjugate 6 gave rise to an enhanced fluorescence emission response in the HepG2 cells. Presumably, this is the result of cell-specific disulfide cleavage. In contrast, no appreciable fluorescence was seen in the case of the W138 cells. In addition, on a per mole basis the anticancer effects of the HJ inhibitor peptide 2 were found to be enhanced when HepG2 cells were incubated with conjugate 6 (cell viability, around 55% at 300 μM) rather than the parent HJ inhibitor peptide 2 (cell viability, around 90% at 300 μM), as confirmed by MTT assays.
Disulfide-based naphthalimide scaffolds have also been used to construct promising theranostic agents. For example, Zhao et al. developed the disulfide-based naphthalimide conjugate 7. This system contains chlorambucil (an anticancer drug) as the active agent and a D-mannose subunit as a tumour targeting group (Fig. 5).16 Because the two parts differ in polarity (i.e., a water-soluble D-mannose group and a water-insoluble chlorambucil unit), prodrug 7 could be used to create self-assembled vesicles. The formation of these vesicles was supported by transmission electron microscopy (TEM) imaging. Their anticancer effects and targeting ability were then assessed in HeLa cells and MCF-7 cells. MCF-7 cells, in contrast to HeLa cells, overexpress a mannose receptor. In accord with design expectations, the vesicles made up of 7 exhibited a higher cytotoxicity in the MCF-7 cells than in the HeLa cells. Support for this enhancement and the underlying localization came from confocal microscopy and flow cytometry studies.
In a separate work, Zeng et al. developed a fluorescence resonance energy transfer (FRET)-based theranostic prodrug 8. This conjugate consists of a disulfide-based naphthalimide as the FRET energy acceptor, a CPT moiety as the anticancer prodrug and FRET energy donor (Fig. 5).17 The design reflects the fact that the emission band of CPT overlaps well with the absorption profile of the naphthalimide unit. As a result, a change in the FRET-based fluorescence features of 8 was expected upon cleavage of the disulfide bond. As with other disulphide-containing theranostics, cleavage should be enhanced in the presence of GSH. Without GSH, prodrug 8 exhibited an emission centred at 544 nm, which is ascribed to the naphthalimide subunit; no fluorescence signal associated with the CPT was observed, presumably due to the FRET-On effect. In contrast, exposure to GSH led to a concentration dependent increase in a new fluorescence feature centred at 448 nm ascribable to free CPT, while a concordant decrease in emission intensity at 544 nm was observed (FRET-Off state). Similar changes were seen in vitro in the HeLa cell line, lending support to the suggestion that system 8 allows for the cellular uptake and GSH-mediated release of CPT. The anticancer effect of 8 was also demonstrated in human cervical cancer cells (HeLa cell line) and normal cells (L292 cell line).
Similar FRET-based theranostic agents have been developed by others. For instance, Xie et al. developed a CPT prodrug 9 linked to a fluorescent BODIPY through a disulfide bond (Fig. 5).18 Upon excitation of this conjugate at 360 nm, a wavelength corresponding to the absorption maximum of CPT, a fluorescence emission feature at 522 nm, ascribable to the BODIPY moiety, was observed. In the presence of thiols, increases in the fluorescence features at 510 and 433 nm, corresponding to the isolated BODIPY and CPT moieties, respectively, were observed. Such a finding is consistent with the individual species being released from the conjugate upon S–S bond scission. Because two emissive species are produced through this bond breaking process, it was suggested that 9 could prove useful as a ratiometric probe system and allow monitoring of both cellular uptake and CPT drug release. The therapeutic efficacy of agent 9 was demonstrated in HeLa cells where an IC50 of 1.27 μM was observed.
To date, the majority of the fluorescent theranostic drug delivery conjugates developed with the goal of achieving both therapy and diagnosis have proved effective in vitro and ex vivo. The development of systems suitable for use in vivo has proved much more challenging. A major bottleneck has been the lack of fluorophores that permit excitation and emission in the far-red visible or near-IR (NIR) spectral regions where tissues are most transparent. This has led to efforts to create theranostic prodrug delivery systems containing near-infrared (NIR) fluorescent dyes. Such systems are attractive because they might (1) allow deep tissue penetration for imaging in vivo, (2) induce minimal tissue damage by virtue of NIR illumination, (3) be subject to lower interference arising from the auto-fluorescence of tissues, and (4) permit real time in vivo monitoring of drug delivery.
With the above putative benefits in mind, we reported a Cy7 NIR dye-based drug delivery agent 10 that contains gemcitabine (GMC) as the active anticancer drug and a folate subunit as a cancer localizing group (Fig. 6).19 The KB and A549 cell lines, which are folate receptor positive and negative, respectively, were used to test the theranostic potential of 10. It was found that conjugate 10 selectively enters KB cells, as evidenced by a strong fluorescence enhancement at 735 nm, as well as cytotoxicity as ascribable to the release of GMC. Evidence for the selective uptake of theranostic 10 into tumour tissues came from NIR fluorescence imaging studies.
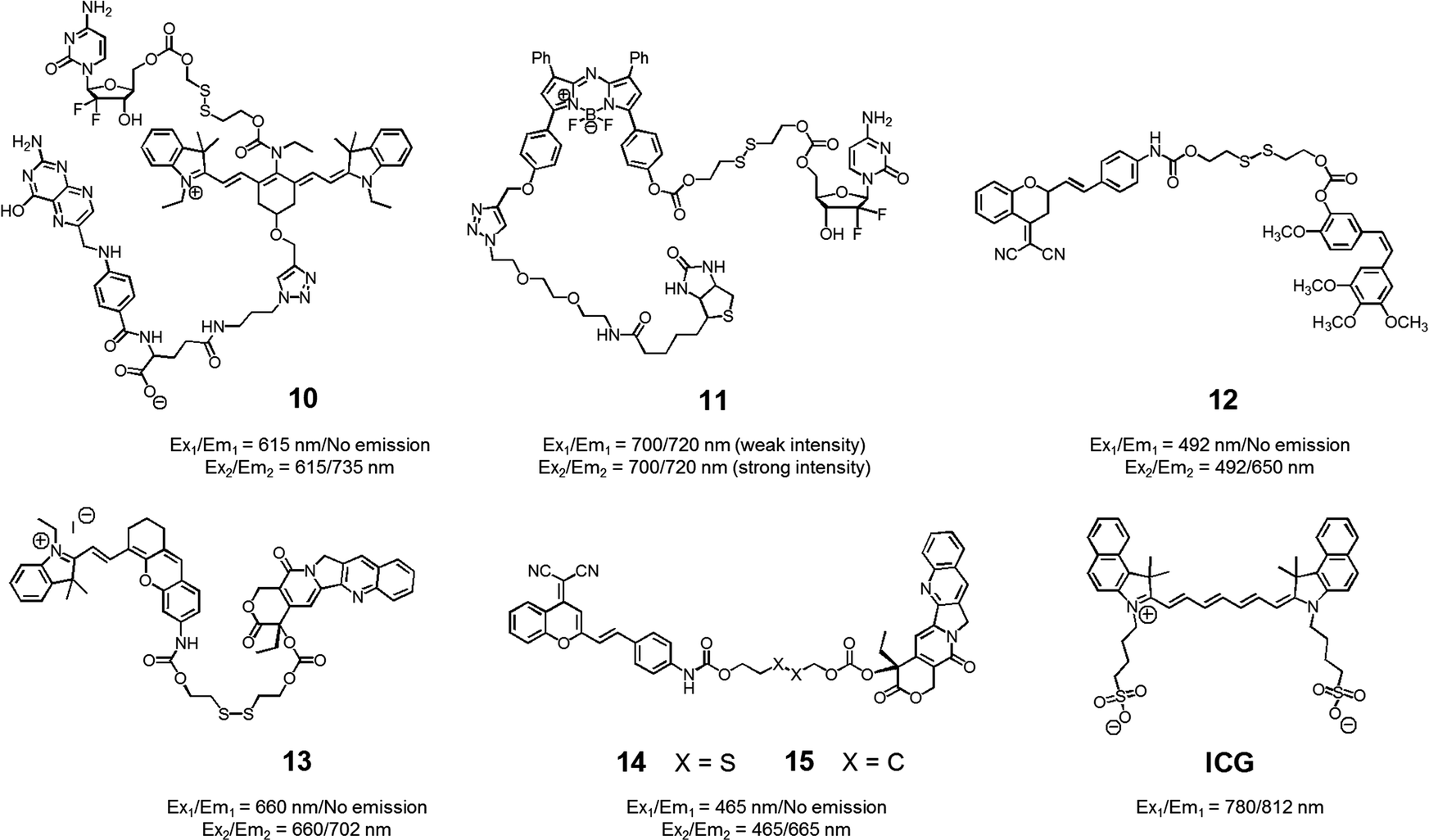 | ||
| Fig. 6 Thiol-activatable NIR dye-based theranostic agents 10–15. Also shown is a NIR dye used for in vivo imaging, ICG. | ||
We also prepared the GMC–BODIPY-biotin conjugate 11 as a theranostic anticancer drug delivery system that contains an appended BODIPY as a NIR fluorescence reporter and GMC in a prodrug form (Fig. 6).20 This conjugate undergoes disulfide cleavage mediated by thiols. This bond scission gives rise to a significant increase in the intensity of the fluorescence feature centred at 720 nm ascribed to isolated BODIPY. It also releases free GMC. Conjugate 11 was found to be taken up well by A549 cells. Disulfide cleavage then occurs predominantly in the ER of these cells. This releases GMC in its free form, which then diffuses into the nucleus where it mediates its cytotoxic effect.
In a separate work, Brown et al. reported the theranostic prodrug 12, which contains a dicyanomethylene-4H-pyran as a NIR-fluorescence reporter, a disulfide bond as a cleavable linker, and combretastatin A-4 (CA-4) as a tumour-targeting therapeutic agent (Fig. 6).21 CA-4 binds to tubulin thus targeting tumour vasculature. It inhibits angiogenesis and promotes cancer cell death. When the disulphide bond present in 12 is cleaved by thiols, free CA-4 is released from the conjugate and a concomitant increase in the NIR fluorescence intensity at 650 nm is observed. To test the potential utility of 12, a triple negative breast cancer (TNBC) cell line was chosen that lacks three major receptors, the estrogen receptor, the progesterone receptor, and the human epidermal growth factor receptor-2. In the case of such TNBC cells, conjugate 12 gave rise to a distinct NIR fluorescence image and considerable therapeutic efficacy was observed. This was not true in the case of normal breast cells (MCF10A). It was thus suggested that 12 could eventually be used as a targeted prodrug delivery agent for cancers, such as TNBC, that are characterized by the absence of currently targetable hormone receptors.
Recently, Tang et al. developed the theranostic prodrug 13. It was obtained via the conjugation of merocyanine as a NIR fluorophore to CPT via a disulfide bond (Fig. 6).22 Prodrug 13 exhibited a significant fluorescence turn-on upon GSH-mediated disulfide bond cleavage in cancer cells. To demonstrate the therapeutic potential of 13, cancer HepG2 and normal HL-7702 cell lines were used. Greater cytotoxicity was seen in the HepG2 cells than in the normal HL-7702 cells. Animal experiments using tumour-bearing nude mice revealed that 13 was predominantly accumulated into tumour tissue where it produced a fluorescence turn-on response. On the basis of these in vivo studies, it was suggested that prodrug 13 shows promise as a tumour-activatable theranostic drug delivery system.
Zhu et al. developed a different CPT-based NIR fluorescent theranostic, namely prodrug 14 that exploits a dicyanomethylene-4H-pyran subunit as a NIR fluorescence reporter, CPT as the anticancer drug, and a GSH-cleavable disulfide bond (Fig. 7).23 Prodrug 14 showed a higher photostability than ICG, a commercially available NIR dye that is often used for in vivo tracking. To investigate whether disulfide cleavage would produce a cytotoxic effect, presumably induced by CPT release, a control system 15, lacking the disulfide linker, was prepared. In an effort to maximize its therapeutic efficacy, prodrug 14 was combined with polyethylene glycol-polylactic acid (PEG-PLA); this resulted in the formation of nanoparticles (NPs) containing 14. Similar loadings were carried out with 15. In cell viability tests performed in cancerous BCap-37 cells, nanoparticle 14 produced a cytotoxic response similar to that of free CPT. In contrast, the nanoparticles made up of 15 were less cytotoxic, presumably because CPT release was not occurring. Animal experiments were carried out using a BCap-37 tumour xenograft mice model. The tumour volume and weight were found to be significantly decreased in the case of treatment with either CPT or 14 relative to what was seen for pure PBS or 15. In addition, a stronger NIR fluorescence signal was seen for 14 compared to 15 following administration to tumour-bearing mice. Such a finding is consistent with the lack of release expected in the case of 15.
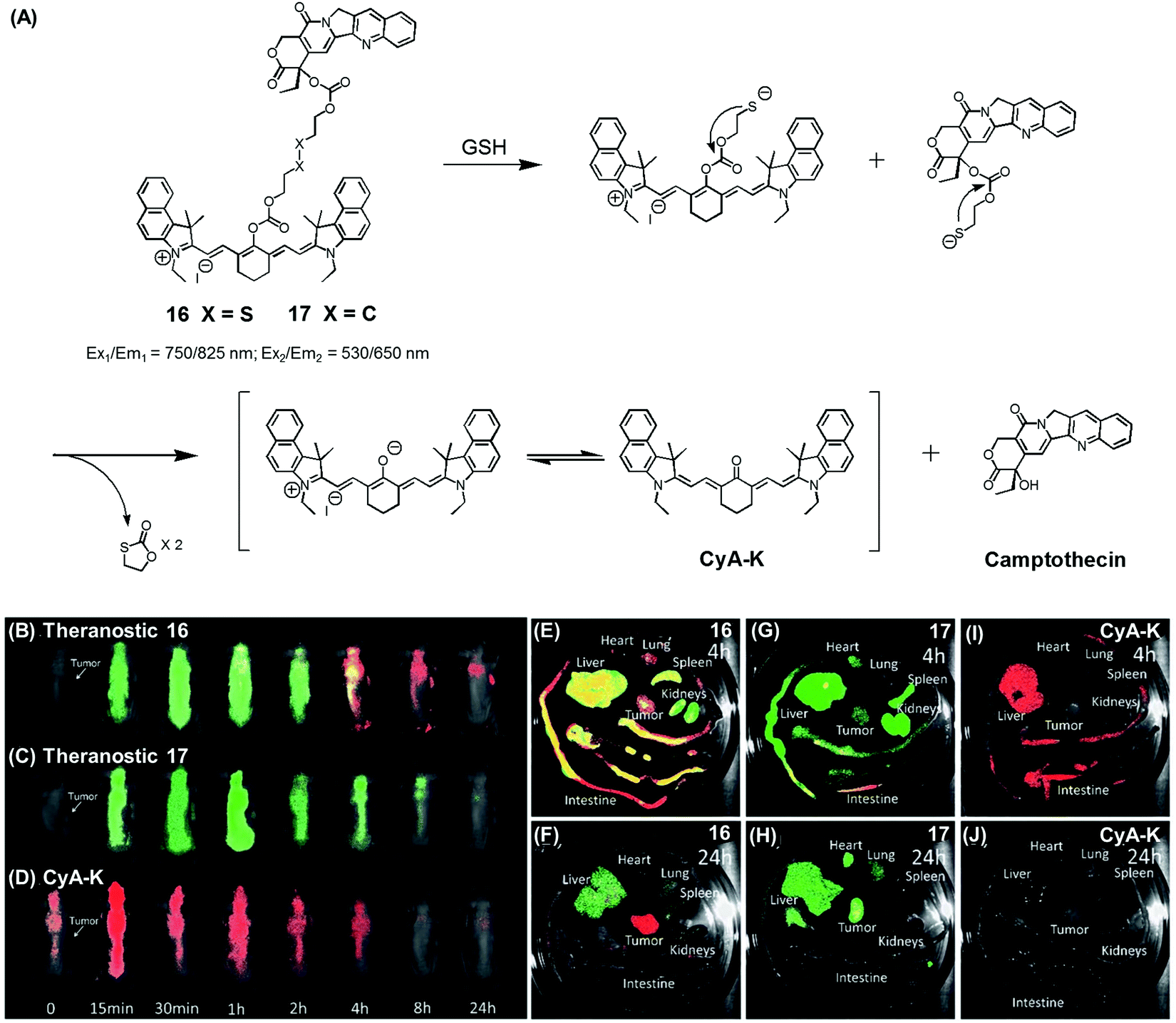 | ||
| Fig. 7 (A) Thiol-activatable NIR dye-based theranostic agents 16 and 17, and their disulfide cleavage reaction. (B–D) In vivo and (E–J) ex vivo biodistribution images of mice treated with agents 16, 17, and CyA-K. (B–J) are reproduced from ref. 24 with permission from the Royal Society of Chemistry. | ||
In another study, Zhu et al. reported a CPT prodrug 16 formally obtained via the conjugation of CPT with a cyanine dye through a disulfide bond (Fig. 7A).24 Prodrug 16 exhibits a NIR fluorescence emission feature at 825 nm. However, upon the GSH-induced disulfide cleavage, a new fluorescence feature is observed at 650 nm with a concomitant colour change from green to purple-red. Thus, the biodistribution and drug activation of 16 could be monitored by comparing the relative intensity of the two fluorescent signals (i.e., at 825 and 650 nm, respectively). As shown in Fig. 7B–D, when the PEG-PLA nanoparticles loaded with 16 were used to treat tumour-bearing mice, the initial disperse green fluorescence seen throughout most of the animal gradually changed to a red fluorescence, from which it was inferred that drug activation occurs via a disulfide cleavage in vivo (Fig. 7B). In contrast, PEG-PLA nanoparticles loaded with 17, a control lacking a disulfide bond, did not produce an appreciable fluorescence change (Fig. 7C). As a control experiment, a CyA-K dye, corresponding to the isolated NIR fluorescent dye in 16, was also used to treat the mice (Fig. 7D). Similar fluorescence changes were also observed in ex vivo studies (Fig. 7E–J). The cytotoxic effect of 16 was monitored in cancer cell BCap-37; the IC50 value of 16 was 1.7 μM that was slightly higher than a parent CPT drug. In vivo, the antitumor activity of PEG-PLA NPs loaded with 16 was evaluated by using BCap-37 tumour xenograft mice. In these studies, the inhibition rate of tumour growth was 94.0%, which proved superior to the 55.8% inhibition rate seen for the clinical anticancer agent (CPT-11). On this basis, it was suggested that 16 could be used as a NIR fluorescence probe to monitor the uptake and distribution of a prodrug that would be released in a cancer specific manner.
Aggregation induced emission (AIE)-based fluorescent reporters have also been exploited for the creation of theranostic prodrug conjugates. Fluorescent reporters, known as AIEgens, display remarkably enhanced fluorescence intensity in their aggregated states as a result of restricted intramolecular rotation.10 This is in contrast to the conventional fluorophores, such as rhodamine, naphthalimide, cyanine, etc., which display self-quenching effects in their aggregated forms. Thus, linking AIEgens to a cancer targeting ligand and an anticancer drug agent via a disulfide bond, an AIE-based fluorescence change would be expected upon thiol-induced disulfide cleavage. To the extent this occurs, it would allow for the real-time monitoring of cellular uptake and drug activation.
To test the potential of this approach, we prepared the mitochondria-targeted AIE-based prodrug 18. In this conjugate, a tetraphenylethene scaffold serves as the AIEgen, a lipophilic cationic triphenylphosphonium is used to target the mitochondria of cancer cells, and a DNA cross-linking agent, chlorambucil, was used as a therapeutic agent (Fig. 8).25 A control system, 19, which lacks the triphenylphosphonium targeting moiety was also synthesized. It was found that prodrug 18 is non-fluorescent in the absence of thiols. However, in the presence of a thiol, disulfide cleavage occurs to give a strong fluorescence signal around 490 nm that is attributed to the AIE and concomitant release of the therapeutic agent. Conjugate 18 was found to be specifically accumulated in the mitochondria of cancer cells, as inferred from fluorescence colocalization experiments carried out using a mitochondria-tracking dye (Mito-tracker deep red). Moreover, from MTT assays carried out using prodrugs 18, 19, and free chlorambucil, it was concluded that 18 is more potent than the recognized therapeutic agent, chlorambucil, both in colon cancer (HCT 116) and cervical cancer (HeLa) cells. It was also found that 18 produced a more-obvious therapeutic effect than 19, which lacks a mitochondria targeting moiety. On the basis of these studies, it was inferred that 18 releases chlorambucil in the mitochondria of cancer cells selectively, resulting in a mitochondria dysfunction and efficient cell death.
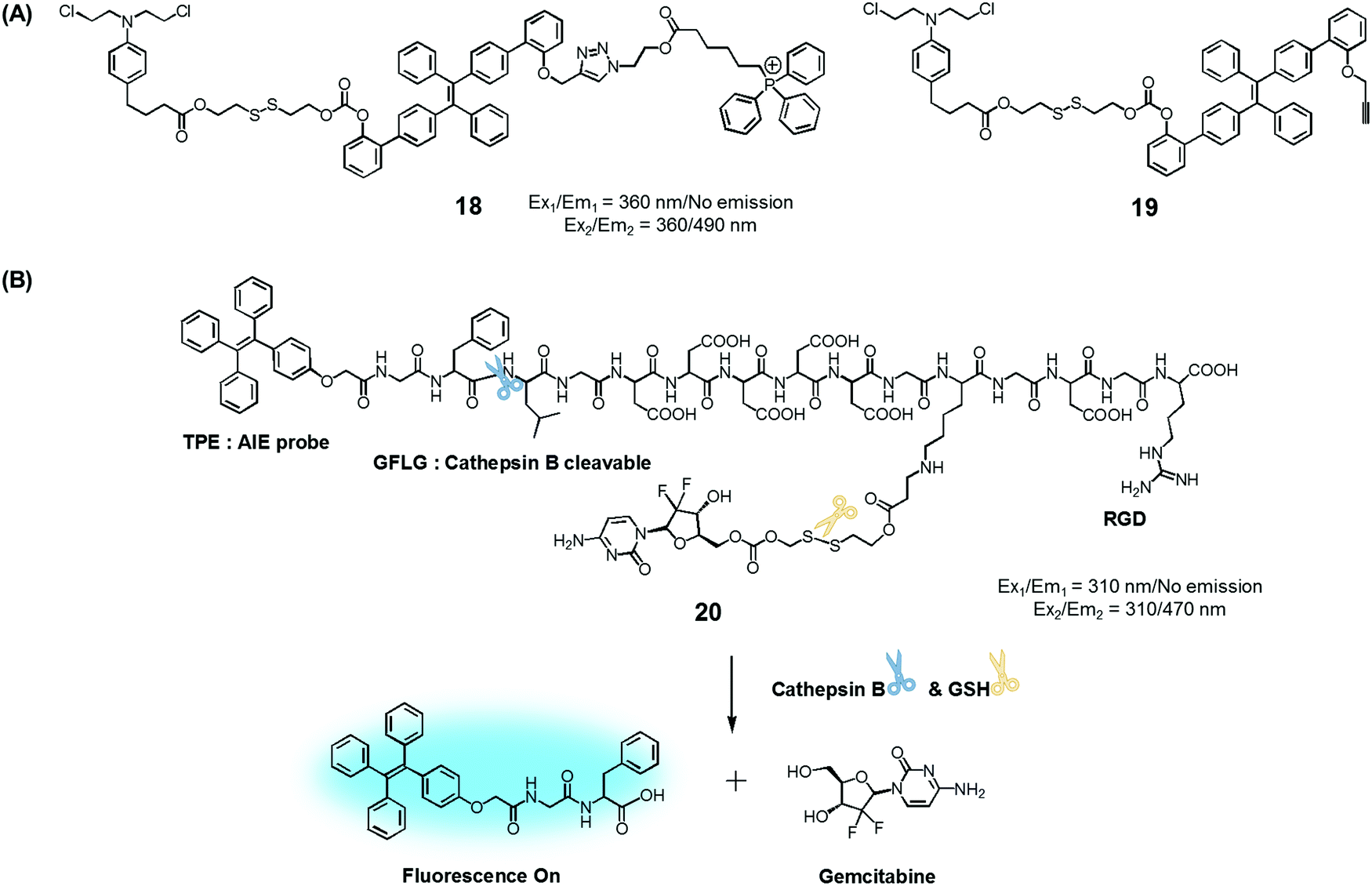 | ||
| Fig. 8 (A) Thiol-activatable AIE-based theranostic agents 18 and 19. (B) Conjugate 20 and its activation by GSH and cathepsin B, respectively. | ||
Ji et al. reported an AIE-based theranostic agent 20 containing tetraphenylethene as an AIEgen, a disulfide-linked gemcitabine (GEM) as a thiol-activatable prodrug, RGD as a tumour targeting peptide, and a hydrophilic peptide with five Asp (D5) units (Fig. 8).26 Here, the hydrophilic peptide D5 was designed to enhance the water solubility of 20 and to restrict the AIE effect. The AIEgen was conjugated to the GEM prodrug via a cathepsin B-cleavable peptide sequence (GFLG). Cathepsin B is known to be a lysosomal protease that is upregulated in several kinds of cancers. Thus, it was expected that, as prepared, the water-soluble prodrug 20 would display little fluorescence intensity. However, in the presence of cathepsin B, the GFLG peptide present in 20 would be cleaved to provide a hydrophobic self-assembled AIEgen with a strong fluorescence feature around 470 nm. Pancreatic cancer cells (BxPC-3) were used for fluorescence imaging because cathepsin B is highly expressed in this cell line. Conjugate 20 was found to be internalized into pancreatic cancer cells through RGD receptor-mediated endocytosis and undergo an intracellular thiol-induced disulfide cleavage to release the cytotoxic agent, GEM, resulting in cancer cell death.
2. Hydrogen peroxide-activated fluorogenic drug release
In normal cell physiology, redox balance is carefully maintained. However, this balance is significantly disturbed during various pathological conditions, including aging, cancer progression, cardiovascular disease, diabetes, and neurodegenerative disorders. Often these perturbations are due to the enhanced production of various reactive oxygen species (ROS), such as hypochlorous acid (HOCl), hydrogen peroxide (H2O2), hydroxyl radicals (OH˙), and singlet oxygen (1O2). The reactive nature of ROS can trigger a number of irreversible functional alterations, including those associated with damaged nucleic acids, oxidized hydrocarbon entities, and modified lipids. These otherwise deleterious oxidizing effects have been explored in the context of drug delivery as a means of releasing covalently linked drugs within cancerous lesions or sites of inflammation. Boronic acids, thioethers, thioketals, polysaccharides, amino acrylates, polyproline, and selenium/tellurium have all been studied in an effort to create cancer-specific diagnostic and therapeutic agents.We developed the boronate ester functionalized coumarin-SN-38 conjugate 21 as a putative hydrogen peroxide responsive therapeutic system that could be used to target metastatic lung cancer (Fig. 9A).27 Upon exposing to cancer cells in the presence of exogenous H2O2, conjugate 21 was found to undergo activation in the lysosome. This resulted in an enhancement in the intensity of the fluorescence emission feature at 450 nm that is ascribed to free coumarin. The release of coumarin from 21 is correlated with the co-release of SN-38, a topoisomerase I inhibitor. Thus, the fluorescence enhancement at 450 nm could be used to monitor the drug release process. Theranostic agent 21 exhibited cytotoxicity in B16F10 (murine metastatic melanoma) and HeLa cell lines that were pre-treated with an ROS inducer, PMA (phorbol 12-myristate 13-acetate). Furthermore, conjugate 21 was successfully used for the treatment of metastatic lung cancer in an in vivo tumour model in mice under conditions of intratracheal administration as determined by non-invasive magnetic resonance imaging (MRI) (Fig. 9Bi–iii) and ex vivo histological assays (Fig. 9Biv–vi). Compared with what was seen in the case of untreated mice, agent 21 produced a statistically significant improvement in the mouse survival rate.
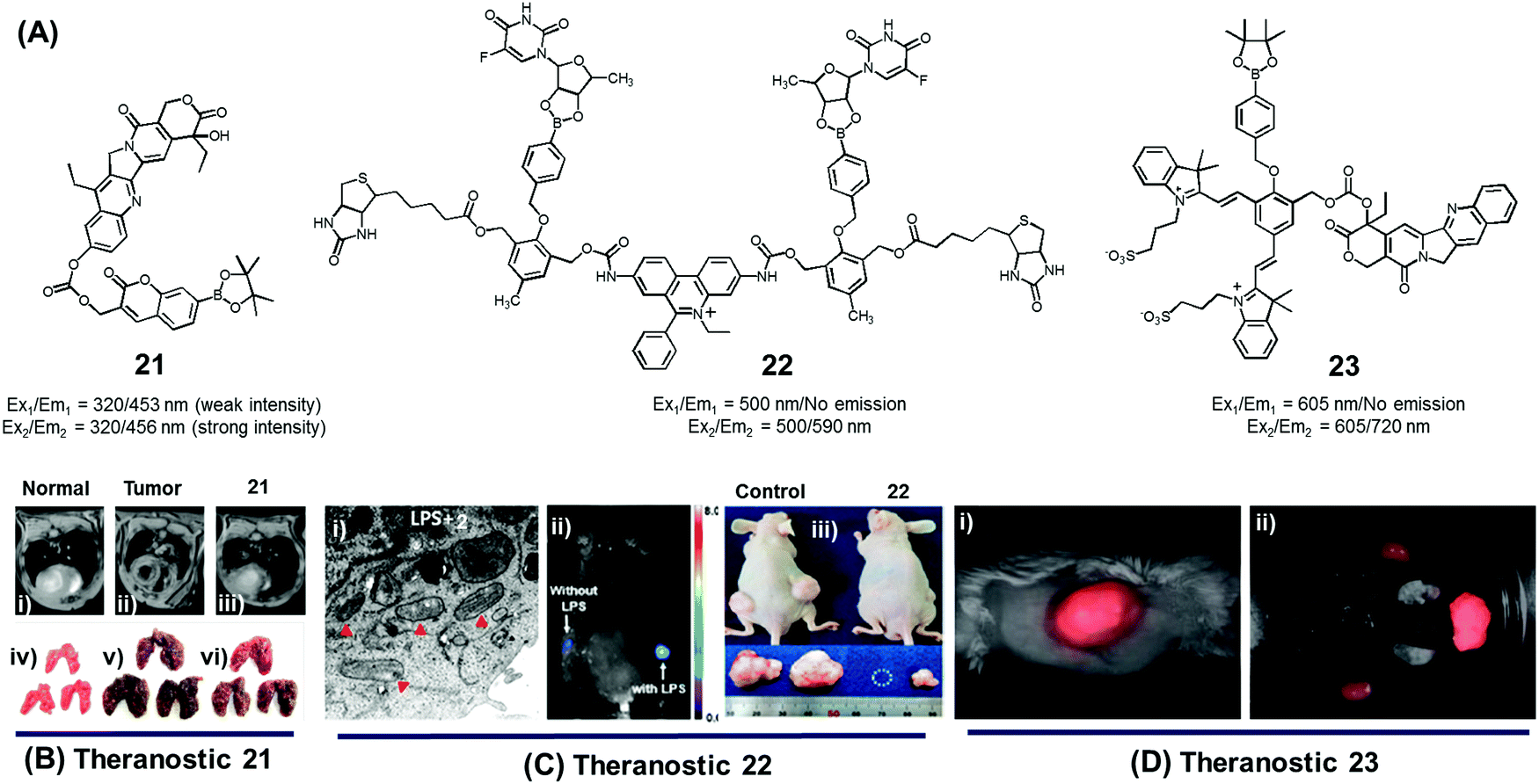 | ||
| Fig. 9 (A) H2O2-triggered theranostic systems (21–23). (B–D) Bio-imaging and therapeutic effects of theranostics 21, 22, and 23, respectively. (Bi–iii) In vivo MRI images of normal mice and saline, 21 treated lung metastasis mice (axial plane views) at day 10 post-inoculation, respectively, and (Biv–vi) images of lungs isolated from the mice. (Ci) Mitochondrial ultrastructure after treatment with 22 in LPS pre-treated cells. (Di) Whole body image of U-87 MG tumour bearing mice after 23 injection (1 min) and (Dii) ex vivo images of dissected organs 5 min post injection. (B and C) are reproduced with permission from ref. 27 and 28. Copyright 2014 American Chemical Society. (D) was adapted from ref. 29. Copyright 2015 Wiley-VCH. | ||
Using the same activation strategy, a theranostic conjugate 22 incorporating two cancer targeting biotin moieties, an ethidium dye (a classic mitochondrial apoptosis marker) and two 5′-deoxy-5-fluorouridine (a prodrug of the active chemotherapeutic, 5-fluorouracil (5-FU)) was reported (Fig. 9A).28 After being internalized into A549 cancer cells as a result of interactions with biotin receptors upregulated in this human lung cancer cell line, it was expected that the positively charged ethidium moiety would further guide the conjugate to the mitochondria preferentially (Fig. 9Ci). Thereafter, upregulated mitochondrial H2O2 would result in prodrug activation, which would release 5′-deoxy-5-fluorouridine to be further converted into 5-FU by thymidine phosphorylase, an enzyme overexpressed in various cancer cell lines. A fluorescence enhancement corresponding to free ethidium could be used as a fluorescence marker to quantify the resulting apoptosis. Western blot analysis further demonstrated that cells treated with conjugate 22 exhibited an enhanced expression of various mitochondrial BAK (Bcl-2 homologous antagonist killer), BAX (Bcl-2 associated X protein), BID (BH3 interacting-domain death agonist), PUMA (p53 upregulated modulator of apoptosis), and NOXA (phorbol-12-myristate-13-acetate-induced protein 1) and cell (caspase-3/-9, cytochrome C) specific apoptosis genes/markers. The in vivo therapeutic efficacy of 22 was monitored in A549-xenograft mice via intravenous tail vein injection. The tumour showed a significant fluorescence enhancement upon treatment with lipopolysaccharide (LPS) (Fig. 9Cii). Compared to analogous animals treated with the control and active drug (5′-FU), conjugate 22 exhibited a preferential tumour accumulation in the mice. It also produced a statistically significant reduction in the tumour burden and led to improved survival rates (Fig. 9Ciii).
In another study, Shabat et al. reported the H2O2-responsive quinone cyanine 7-CPT prodrug conjugate 23 (Fig. 9A).29 In preliminary studies, treatment of conjugate 23 (50 μM) with H2O2 (5 equiv.) resulted in the essentially complete release of the active CPT drug over a 90 minute time period. Incubation of human glioblastoma multiform (GBM) U-87 cells with theranostic 23 produced pronounced cytotoxic effects under both H2O2 pre-treated and untreated conditions. A distinct turn-on fluorescence response at 720 nm, corresponding to free QCy7, was also seen. Evidence for the presumed tumour selective activation of theranostic 23 came from animal model studies. Specifically, it was found that both intratumoral and intravenous tail vein injection of U-87 MG tumour-bearing mice with conjugate 23 produced a strong fluorescence signal in the tumour region that was ascribed to drug activation (Fig. 9Di and ii).
3. Acidic pH-activated fluorogenic drug release
Dysregulated pH is now considered to be an adaptive feature associated with most cancers and, indeed, is widely recognized as being a key cancer hallmark. In normal tissues, the intracellular pH (pHi) ∼7.2, is lower than the extracellular pH (pHe) ∼7.4. Conversely, cancer cells are characterized by a higher pHi (>7.4) and lower pHe (6.6–7.1). The extracellular pH can be substantially lower (pHe ∼ 5) in certain cases. pH dysregulation is thought to play a significant role in various stages of cancer progression, including evasion of apoptosis, faster proliferation, abrupt metabolic adaptation, cell migration, and metastatic spread. Over the past few years, considerable efforts have been devoted to exploiting this distinct feature for both tumour diagnosis and therapeutics.30 Typically, an acid-labile functional group is linked to a fluorescent reporter, anticancer drug, or various targeting functionalities to produce conjugates that are fairly stable under physiological conditions. These conjugates are designed to be readily hydrolysed in acidic tumour environments, as well as endosomes and lysosomes (pH 6.5–5.5), to furnish active optical signals and release the drug in question. This pH-based reactivity can thus provide the basis for tumour specific diagnoses and more precise drug delivery. To date, drug–reporter conjugation through a hydrazone linkage has been widely used to create acid-labile systems. This approach is applicable to a specific class of drugs, possessing aldehyde or ketone moieties, which can further be coupled through a hydrazone linker. In this section, we summarize recently reported cancer-specific, acidic-activated drug delivery systems.Zhang et al. developed an acid-responsive theranostic 24 designed to deliver Dox to cancer cells (Fig. 10A).31 This conjugate contains a Dox subunit tethered to the integrin GRDS-oligopeptide, used as a cancer targeting unit, through an acid labile hydrazone linkage. It also contains a fluorescent coumarin reporter group. The intact system 24 displays reduced fluorescence emission intensity, presumably due to a contact-mediated fluorescence quenching effect involving the coumarin and Dox subunits. The drug release profile of agent 24 was studied in vitro. It was found that about 94% of the Dox was released in free form over a period of 11 h at pH 5, whereas at pH 7.4 only about 41% was released under otherwise identical conditions. Theranostic 24 was also found to display dose-dependent toxicity in integrin-positive U87 cells (human glioblastoma) with an IC50 of 0.19 μg mL−1. Red (Dox) and blue (coumarin) fluorescence enhancement in lysosomes was also observed, providing a means of monitoring drug activation in real time (Fig. 10B).
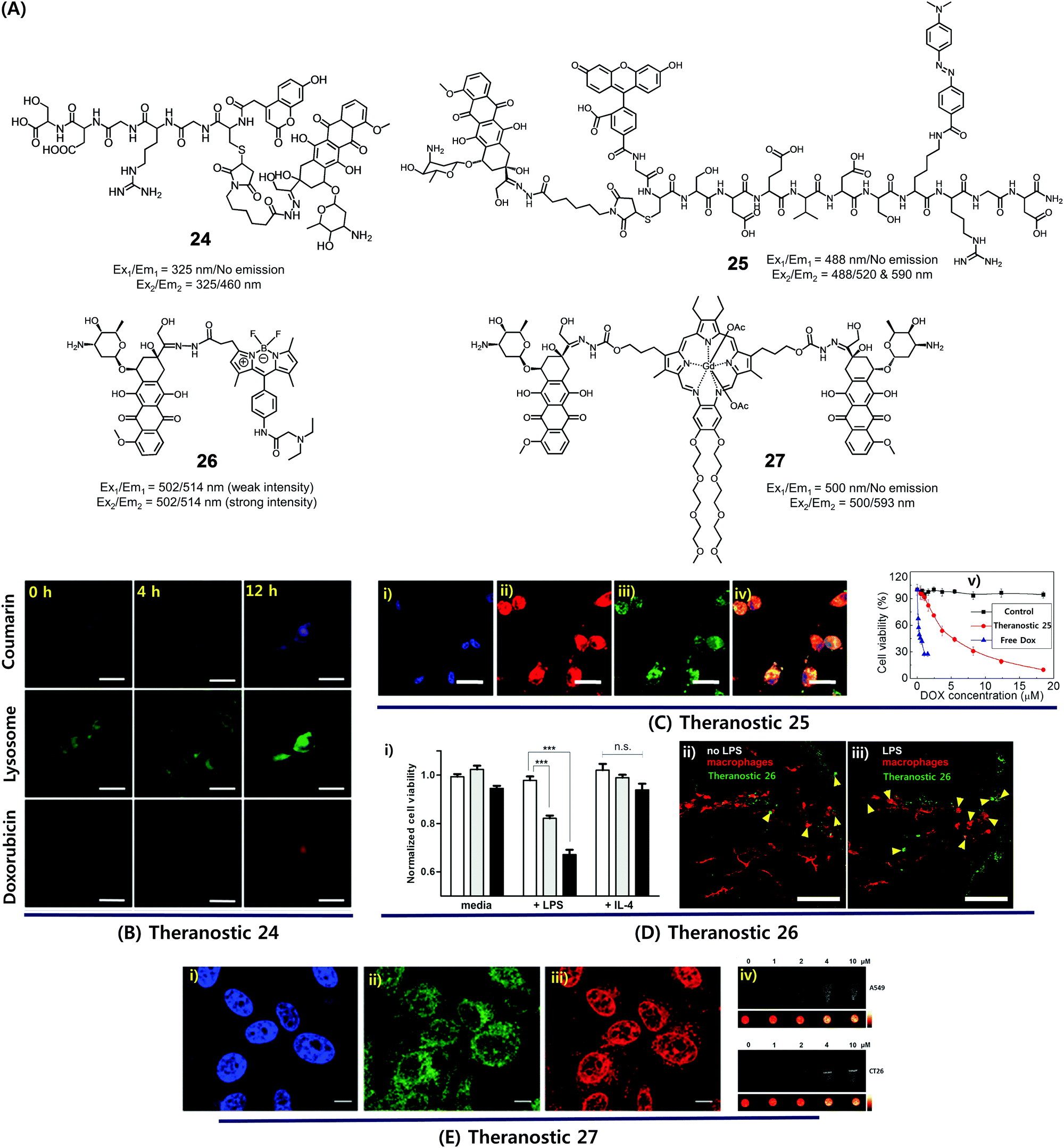 | ||
| Fig. 10 (A) Acid-activated theranostic systems 24–27. (B–E) Bio-imaging and therapeutic effect of theranostic systems 24, 25, 26 and 27, respectively. (Ci–iv) confocal microscopy images of U87 cells upon treatment with 25 (post 42 h) and cell viability at different concentrations of 25, free Dox and control. Cell nuclei stained with Hoechst 33342 (blue), Dox (red fluorescence), FAM (blue fluorescence), and merged image. (Di) Normalized cell viability of 26 to nonactivated (media), LPS-, and IL-4-treated macrophages. (Dii and iii) Live fluorescence confocal microscopic in vivo imaging of macrophages in 26-treated zebrafish without (Dii) and with (Diii) LPS treatment. The arrow heads indicate the Dox activation (green) and surrounding apoptotic macrophages (red). (Ei–iii) Confocal images of CT26 cells pre-treated with Hoechst (blue), Lysotracker (green) and 27 (red). (Eiv) T1-weighted MR images of A549 and CT26 cells co-incubated with 27 at various concentrations at 200 MHz. (B–E) are reproduced with permission from ref. 31–34, respectively. Copyright 2014 Royal Society of Chemistry (for B), Copyright 2015 Wiley-VCH (for C), 2017 American Chemical Society (for D), and Copyright 2016 Royal Society of Chemistry (for E). | ||
Considerable efforts have been made to exploit tumour-specific drug activation to create cancer chemotherapeutics that function in a non-invasive manner. As a result of separate work, a large body of cell apoptosis markers have been developed in an effort to evaluate the response of cancer cells towards particular cytotoxic agents. Combining these two lines of investigation within a single entity could conceivably give systems that allow the concurrent in situ monitoring of prodrug activation and fine-tuning of dosage levels for use in, e.g., personalized medicine. With such considerations in mind, Zhang et al. developed a dual Förster resonance energy transfer (FRET) theranostic 25. This system contains a Dox subunit linked through an acid-labile hydrazone bond to 4-(dimethylamino azo)benzene-4-carboxylic acid (Dabcyl, a potent fluorescent quencher), through a caspase-3 enzyme responsive Asp-Glu-Val-Asp (DEVD) peptide sequence (Fig. 10A).32 A 5(6)-carboxyfluorescein (FAM) moiety was further incorporated between the Dox and Dabcyl subunits to allow for the real-time evaluation of hydrazone-based cleavage and Dox release at the cellular level. For cancer selective targeting, a widely utilized integrin-specific Arg-Gly-Asp (RGD) ligand was employed. Theranostic 25 exhibited nearly 90% active Dox release at pH 5 compared to about 19% at pH 7.4. Further, in vitro studies confirmed that upon incubation in integrin positive U87 cells, conjugate 25 exhibited a time-dependent red fluorescence enhancement (ascribed to Dox release) and gave rise to intracellular caspase-3 activation. This was accompanied by a cleavage of the DEVD peptide sequence to regenerate the FAM-based fluorescence (green) and a significant anticancer effect (IC50 = 4.3 × 10−6 M) (Fig. 10C).
In a separate work, Vendrell et al. developed an acidic pH-activatable theranostic 26. Here, the goal was to monitor stimulus-responsive drug activation kinetics and distribution patterns during intracellular trafficking in immune cells.33 Theranostic agent 26 is made up of fluorophores and a prodrug, i.e. an inherently cytotoxic Dox subunit linked through an acid-labile hydrazone bond to a 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) fluorophore (Fig. 10A). Under physiological conditions, system 26 displays a relatively weak fluorescence intensity. However, in mildly acidic environments, i.e., intracellular acidic phagosomes (pH 4.5–6.5), the hydrazone linkage is hydrolysed resulting in the release of cytotoxic DOX and the turning on of a pH-dependent BODIPY fluorescence. The RAW264.7 macrophages were activated with lipopolysaccharide (LPS) to prepare proinflammatory M1 macrophages that precede phagosomal acidification. In LPS-induced proinflammatory M1 macrophages, theranostic 26 displayed a dose-dependent cytotoxicity, as well as a turn-on fluorescence response allowing for the real-time monitoring of prodrug activation. In contrast, no response was observed in either nonactivated (media) or anti-inflammatory M2 (treatment with IL-4) macrophages (Fig. 10Di). Moreover, 26 was applied to proinflammatory M1 macrophages in zebrafish and could be monitored by live fluorescence confocal microscopy (Fig. 10Dii and iii). A brighter BODIPY fluorescence (green) was seen around or in apoptotic macrophages (red) in LPS-treated zebrafish than in untreated ones. Thus, theranostic 26 was suggested as being a platform for the monitoring of targeted therapies for immune-related diseases.
Recently, we synthesized a multimodal theranostic, conjugate 27, that consists of a paramagnetic, motexafin gadolinium (MGd) texaphyrin unit linked to two Dox subunits through an acid-labile hydrazone linker (Fig. 10A).34 This system was designed to permit monitoring of cellular uptake and prodrug activation through two complementary, but inherently orthogonal, imaging modalities, namely fluorescence emission and magnetic resonance imaging (MRI). As prepared, theranostic 27 displayed little appreciable fluorescence over the 550–700 nm spectral region (corresponding to free Dox). On the other hand, once internalized into A549 (human lung) cancer cells and CT26 (colon carcinoma) cancer cells, a strong fluorescence signal centred at 500 nm was observed upon irradiation. This emission was ascribed to conjugate activation and release of free Dox in the acidic cellular environment. Theranostic 27 displayed significant anticancer efficacy in A549 and CT26 cell lines, while minimal toxicity was observed in NIH3T3 cell line (normal fibroblast). Further, compared with standard Gd3+ based contrast agents, enhanced T1-contrast relaxivities of 20.1 ± 0.4 mM−1 s−1 and 6.1 ± 0.2 mM−1 s−1 at 60 and 200 MHz were observed for agent 27 in PBS buffer (Fig. 10E). Hence, conjugate 27 was suggested as being a promising theranostic agent that could be used to monitor both cellular uptake and drug activation, while being readily detectable in its intact and cleaved forms through MR- and fluorescence-based imaging, respectively.
4. Hypoxia-activated fluorogenic drug release
Over the past couple of years, nitro-appended aromatics have attracted attention as progenitors of non-invasive targeted tumour diagnostic and therapeutic agents. Reduction of aromatic nitro group by overexpressed nitroreductase enzymes in the tumour hypoxia environment can be used to unmask the active form of a prodrug hence providing a tumour-selective chemotherapeutic effect. An example of this approach is embodied in theranostic 28. This system comprises a nitrobenzyl group, a cancer targeting biotin moiety, and a tethered SN38 subunit, which serves both as a chemotherapeutic agent (topoisomerase 1 inhibitor) and a fluorescence marker (Fig. 11A).35 Per design expectations, elevated nitroreductase activity in the tumour served to reduce the nitrobenzyl group to the corresponding aniline derivative. This latter species is inherently unstable and releases the active form of SN38 as a result of an electronic rearrangement as illustrated in Fig. 11. This release was accompanied by a readily observed enhancement in the fluorescence emission. Theranostic 28 offers the possibility of monitoring selective drug delivery to tumours since the active SN38 drug form cannot be released unless the prodrug reaches the hypoxic tumour environment. Analyses of the in vitro efficacy of 28 were carried out using MTT-based viability assays using biotin receptor-positive cancer cells (A549, HeLa) and biotin receptor-negative cells (WI-38, BJ) under normoxic and hypoxic conditions. Prodrug 28 was found to exhibit high cytotoxicity in the biotin receptor-rich A549 and HeLa cells under hypoxic conditions, but not under normoxic conditions. Little cytotoxicity was observed in the biotin receptor-negative cells. Animal experiments using HeLa cell-inoculated xenograft mice revealed that 28 was specifically accumulated in the solid tumour in vivo and reduced the tumour burden, presumably as a result of hypoxia-induced activation of the nitrobenzene moiety and concomitant drug release. The tumour regression induced by 28 was greater than that produced by various control systems (e.g., 28a and 28b) or free SN38 (Fig. 11B–E).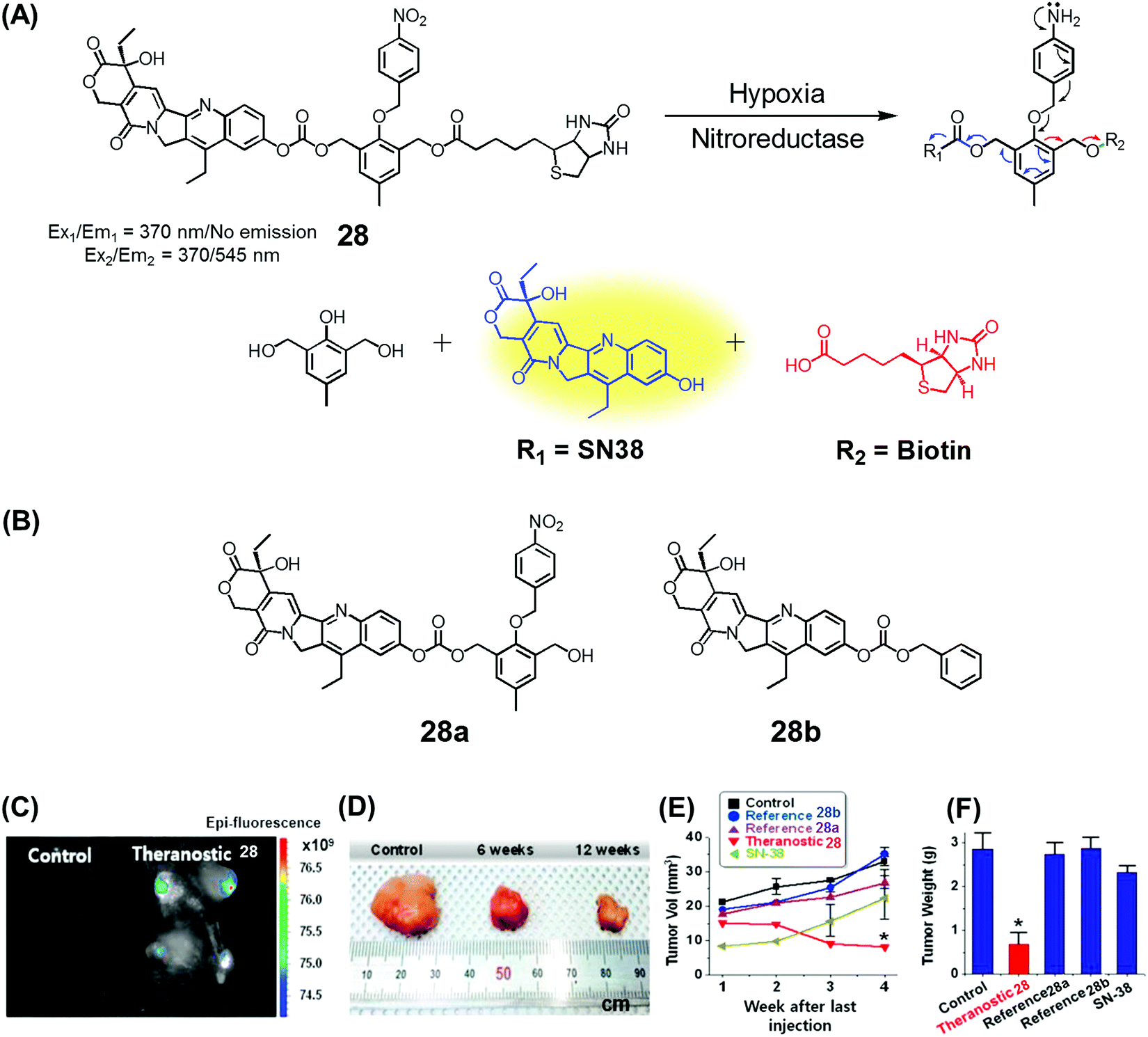 | ||
| Fig. 11 (A) Hypoxia-activated theranostic agent 28 and its cleavage reaction. (B) Chemical structure of the reference compounds 28a and 28b. (C–F) Bio-imaging and therapeutic effects of theranostic 28. (C–F) are reproduced with permission from ref. 35. Copyright 2016 Elsevier. | ||
In another study, a nitrobenzyl hypoxic release trigger was used to create a construct that would be activated by UV-irradiation.36 The species in question, conjugate 29 (Fig. 12), was composed of a gemcitabine (GMC), a nitrobenzyl group, and a trans-cinnamic acid. These subunits were expected to act as the anticancer prodrug, a hypoxia-sensitive reactive element, and a photoinducible fluorogenic drug release trigger, respectively. As is true for 28, the nitrobenzyl group in 29 was expected to be reduced by nitroreductase under hypoxic conditions to expose a hydroxyl group at the ortho position of the trans-cinnamic ester. Under UV-irradiation, the trans configuration of the cinnamic ester was isomerized to the cis form. This rearrangement places the ortho-hydroxyl group in a position ideal for nucleophilic attack on the ester bond. This nucleophilic attack leads to cyclization and production of a fluorescence coumarin moiety. Cyclization serves to release the erstwhile masked GMC in its active drug form. Thus, theranostic 29 offers the possibility of controlling drug release via locus-specific UV-irradiation under hypoxic conditions.
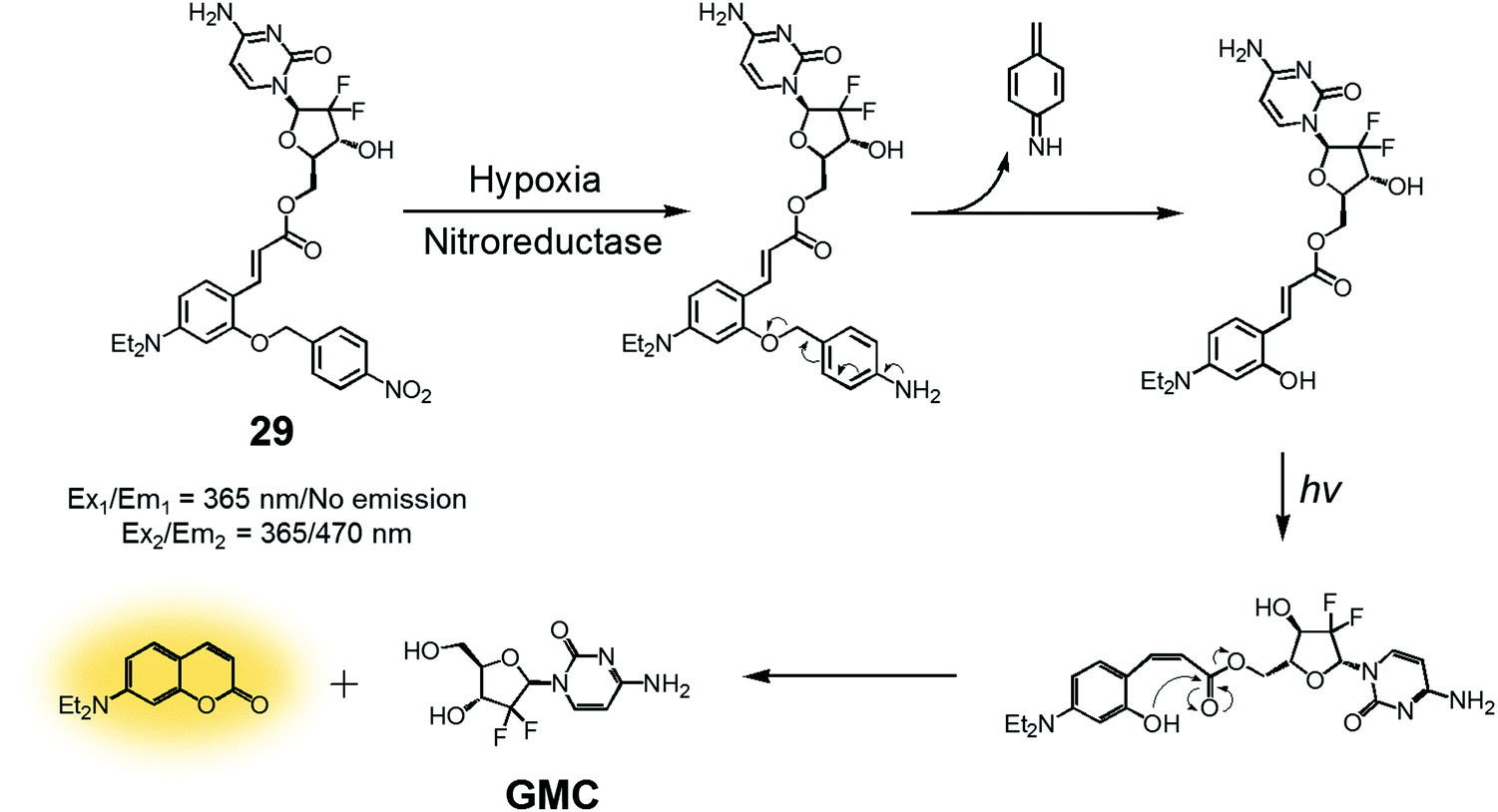 | ||
| Fig. 12 Design strategy underlying theranostic 29, an agent designed to release the cytotoxic species GMC via controlled UV-irradiation under hypoxic conditions. An active coumarin fluorophore is also produced. | ||
Recently, we developed a drug delivery system (DDS), conjugate 30, that contains an azobenzene scaffold. Overall, theranostic 30 embodies a diazo motif as a hypoxia-responsive cleavable group, a fluorescent asymmetric rhodamine 123/B with a lipophilic cationic triphenylphosphonium group as a mitochondria-targeting moiety, and a masked form of nitrogen mustard (a classic alkylating agent) as the anticancer drug (Fig. 13).37 This nitrogen mustard delivery system was designed to target mitochondrial DNA instead of nuclear DNA, the canonical site of action for alkylating agents in current clinical usage. It was found that theranostic 30 was preferentially localized in the mitochondria upon cellular uptake and its diazo subunit was reduced by reductase activity in the hypoxic tumour environment. Upon reduction, the fluorescence signal of rhodamine is enhanced and the nitrogen mustard is released. Agent 30 exhibited an efficient fluorogenic response and concurrent drug release in several cancer cell lines (Huh7, A549, MDA-MB-231, DU145) under hypoxic conditions, but not under normoxic ones. Xenograft mice were treated with 30, with analogues lacking the triphenylphosphonium group (30a), and control systems lacking the nitrogen mustard (30b). These studies revealed that treatment with 30 leads to a statistically significant reduction in tumour growth compared to the controls. On this basis, it was proposed that theranostic 30 embodies a new drug delivery strategy that could be used to overcome the cellular resistance often seen for nitrogen mustard.
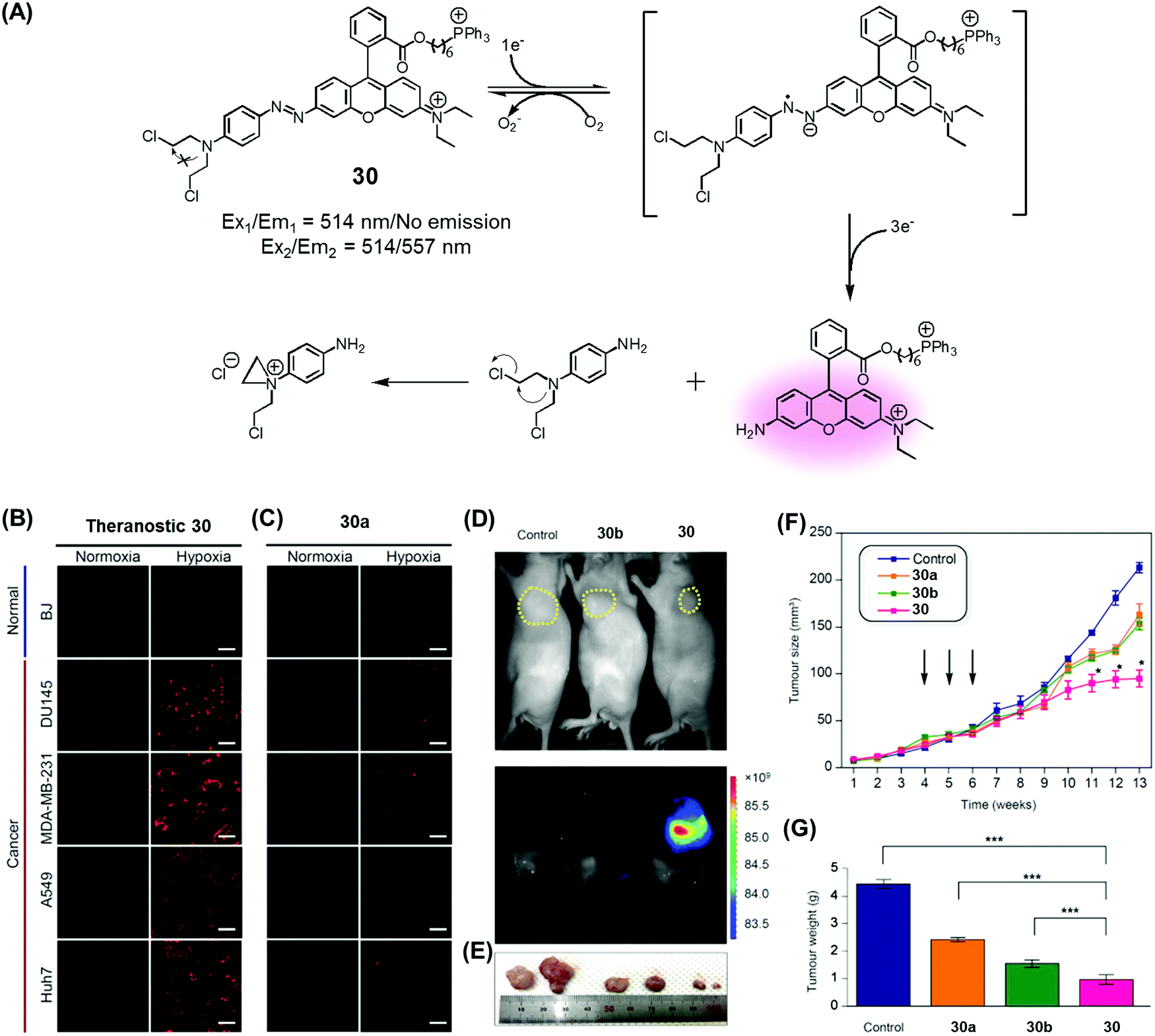 | ||
| Fig. 13 (A) Hypoxia-activated theranostic agent 30 and its mode of cleavage. (B–G) Bio-imaging and therapeutic effects of theranostic 30. (B–G) are reproduced with permission from ref. 37. Copyright 2017 Elsevier. | ||
5. Platinum reduction-based fluorogenic drug release
Since the serendipitous discovery of cis-platinum and its clinical benefits as an anticancer agent were recognized, continuous efforts have been devoted to the search for improved platinum-based drugs. Currently, there are three FDA-approved platinum-based drugs, cisplatin, oxaliplatin, and carboplatin. Together, they are used in approximately 50% of all cancer chemotherapeutic regimens. Relative to organic-based drugs, platinum agents are endowed with certain inherent advantages, such as the possibility of adjusting the geometry, coordination number, and redox state (+2 vs. +4). In principle, these adjustments allow the mode of action to be fine-tuned and the inherent toxicity to be modulated. To date, several promising platinum-based theranostics have been reported. Most have been constructed by tagging an imaging component, such as a fluorophore or an active MRI agent to a platinum complex so as to allow prodrug activation and tumour specific accumulation to be monitored in real time.Liu and Tang et al. developed the platinum(IV)-based drug delivery system 31 in an effort to follow conjugate activation and drug release in cancer cells (Fig. 14A).38 The Pt(IV) core in 31 serves two functions. First, it was expected to be less toxic than the corresponding Pt(II) species due to reduced ligand exchange. Second, it would allow release of an active cytotoxin via in situ reduction to the Pt(II) form. In theranostic 31, the Pt(IV) centre is combined with a cancer targeting cyclic RGD peptide and an AIEgen reporter, which are linked through aspartate residues through the axial positions. In aqueous media, theranostic agent 31 remained non-emissive. Once internalized in integrin positive MDA-MB-231 cancer cells, the Pt(IV) centre undergoes reduction to the corresponding active Pt(II) form. This reduction leads to loss of the axial ligands and production of a high emissive AIE fluorescence signal (Fig. 14Bi–iv). This signal was used to quantitate the active drug concentration inside the cells. Further, cytotoxicity studies revealed that theranostic agent 31 exhibited greater potency in MCF-MDA-231 breast cancer cells (IC50 = 30.2 × 10−6 M) than in MCF-7 integrin-negative cancer cells (Fig. 14Bv).
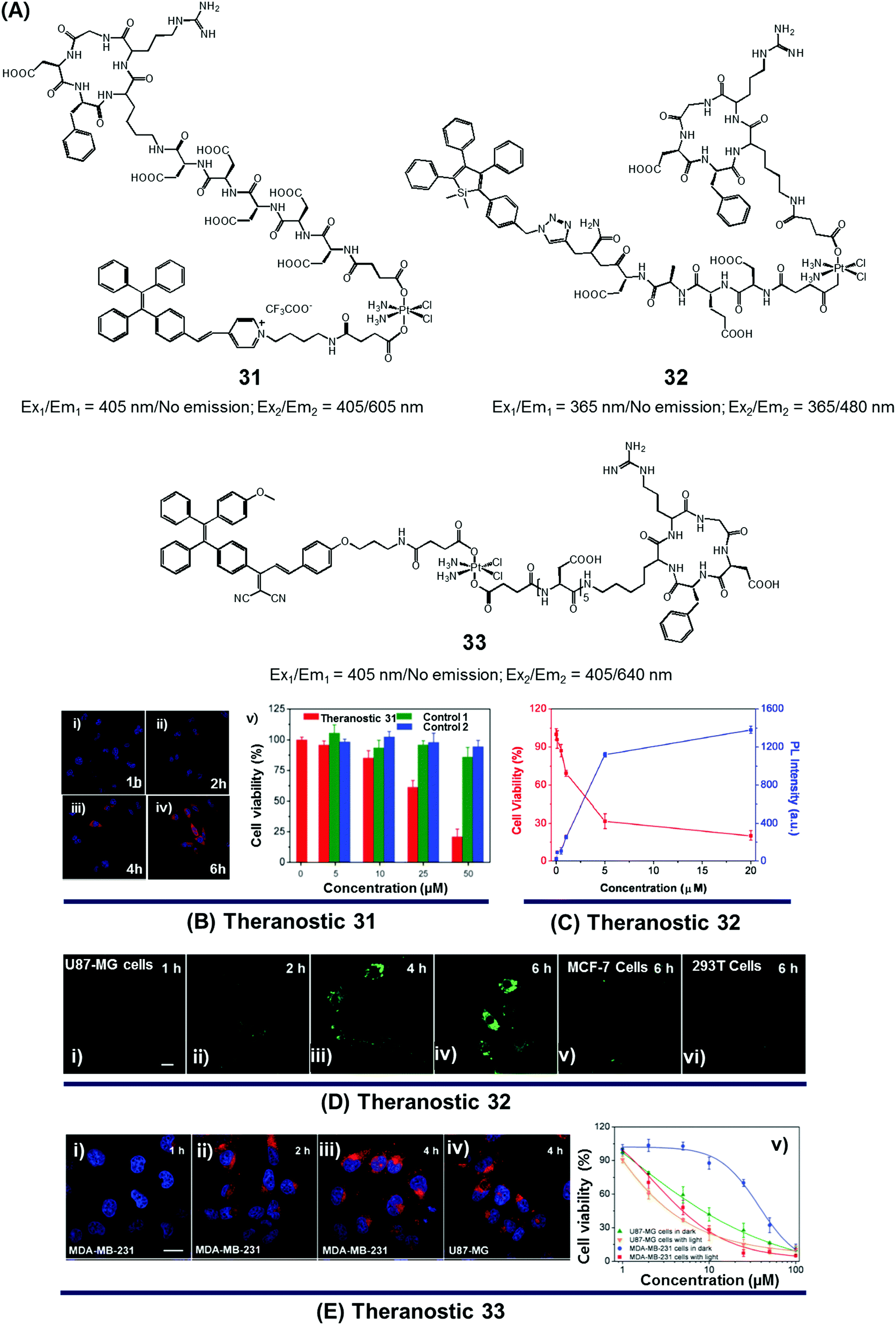 | ||
| Fig. 14 (A) Platinum-based theranostic agents (31–33). (B–E) Bio-imaging and therapeutic response of theranostic agents 31, 32, and 33, respectively. (Bi–iv) Confocal images of MDA-MB-231 cells pre-treated with 31 at 1 h, 2 h, 4 h and 6 h, respectively, and (Bv) cell viability upon treatment with 31 and other controls. (Di–iv) Real time confocal images for apoptosis progress in U87-MG cells stained with 32 and MCF-7 cells (Dv) and 293T cells (Dvi). (Ei–iv) Confocal images of MDA-MB-231 cells and U87-MG cells recorded after incubation with 33 at different time intervals. (Ev) Viability of U87-MG and MDA-MB-231 cells upon incubation with 33 in the dark and with light illumination (0.25 W cm−2 for 1 min). (B–E) are reproduced with permission from ref. 38, 39, and 40, respectively. Copyright 2014 Royal Society of Chemistry (for B), Copyright 2014 American Chemical Society (for C and D), and Copyright 2015 Royal Society of Chemistry (for E). | ||
In designing an efficient therapeutic system, it is important to take into consideration various desired features, such as tumour-specific drug accumulation, controlled activation, and an ability to monitor tumour response at early treatment stages so as to guide clinical treatment decisions. Fluorescence imaging is attractive as a non-invasive reporting modality for monitoring cancer response at early stages of therapy. Recognizing these desiderata, Liu et al. developed a cancer-selective Pt(IV) based theranostic, 32 (Fig. 14A).39 Theranostic 32 is based on a relatively nontoxic Pt(IV) centre linked to a cancer-selective integrin receptor (αvβ3)-targeting cyclic arginine-glycine-aspartic acid (RGD) motif, as well as an AIE-based tetraphenylsilole (TPS) reporter group linked through a caspase-3 specific Asp-Glu-Val-Asp (DEVD) peptide sequence at the axial positions. Upon internalization in integrin-positive U87-MG human glioblastoma cancer cells, theranostic agent 32 is reduced to furnish an active Pt(II) drug form, which induces cytotoxicity (Fig. 14Di–iv). Reduction is accompanied by caspase-3 activation and the simultaneous release of the apoptosis marker TPS-DEVD with a concurrent enhancement of the fluorescence intensity at 480 nm. As shown in Fig. 14C and D, compared with the control cell lines (MCF-7 breast and normal 293T cells), a direct correlation between the fluorescence changes and its early stage toxicity profile is seen for 32 in U87-MG cancer cells. It was thus considered to be a promising drug delivery system (DDS) that might permit the simultaneous monitoring of drug activation and initial therapeutic efficacy assessments.
While good progress has been made in developing theranostic systems that allow prodrug activation and its visualization, the problem of overcoming drug resistance remains a largely unmet challenge. Combining chemo- and phototherapeutic modalities represents an attractive approach to meeting this clinical need. An initial test of this strategy was reported by Zhang and Liu et al., who developed the targeted platinum(IV) theranostic 33 (Fig. 14A).40 Theranostic 33 contains an AIEgen-based photosensitizer (PS) for real-time monitoring of prodrug activation, as well as a tumour-targeting cyclic RGD peptide linked to the Pt(IV) axial positions. As shown in Fig. 14E, theranostic 33 is non-emissive and displays minimal dark toxicity. However, once internalized in MDA-MB-231 and U87-MG cancer cells, presumably through receptor-mediated endocytosis, prodrug activation occurs as a result of Pt(IV) reduction mediated by intracellular GSH. An enhanced AIE-based red fluorescence signal is seen upon reduction, allowing for the real-time monitoring of drug activation. The free chromophore also allows for phototherapy under conditions of visible light irradiation. The anti-proliferative properties of 33 were tested via standard MTT assays. Compared to cisplatin (IC50 = 37.1 μM), theranostic 33 exhibited improved cytotoxicity upon illumination in the cisplatin-resistant MDA-MB-231 cancer cell line (IC50 = 4.2 μM; Fig. 14E).
6. Enzymatic cleavage-based fluorogenic drug release
Amide and ester linkers have been widely used to construct drug conjugates due to their functional compatibility and synthetic convenience. The ester linker is susceptible to hydrolysis catalysed by acids, bases, metals, and hydrolytic proteins, such as esterases, under physiological conditions. However, the amide linker is much stable and less sensitive to chemical hydrolysis. Nevertheless, amide linker-based prodrug systems can be activated by specific enzymes allowing release of a tethered cytotoxic drug. Several amide-cleaving enzymes, including glycosidases, peptidases, and bioreductive DT diaphorases, are overexpressed in cancer cells. This overexpression and the resulting amide scission chemistry have been exploited recently to produce cancer-selective theranostic drug delivery agents.7,10An early example of an enzyme-cleavage release strategy is embodied in the theranostic 34 reported by Shabat et al. (Fig. 15A and B).3 This system is based on 7-hydroxycoumarin linked with “end unit chemotherapeutic drugs” and enzymatic active site “triggers”. When the “trigger” is cleaved, either chemically or enzymatically, the “end unit” is released from 34 with a concomitant formation of the fluorescent coumarin derivative through a spontaneous 1,8-elimination reaction (Fig. 15A). In the case of 34a, the 7-hydroxycoumarin is linked to a phenylacetamide moiety that serves as the “trigger”, as well as to the chemotherapeutic drug melphalan (Fig. 15B). The phenylacetamide group is a known substrate for penicillin-G-amidase (PGA). It was demonstrated that theranostic 34a exhibited a high cytotoxicity (IC50 = 2.5 × 10−6 M) and strong fluorescence ascribed to the coumarin derivative in the presence of PGA in MOLT-3 cells (Human T-lineage acute lymphoblastic leukemia). This cytotoxicity was found to be equivalent to that of free melphalan. In contrast, in the absence of PGA, 34a showed a very low cytotoxicity (IC50 > 100 × 10−6 M) in MOLT-3 cells (IC50 = 2.5 × 10−6 M). In the related system, 34b, the 7-hydroxycoumarin is linked to a melphalan subunit and a dipeptide Phe-Lys, which is a known substrate for cathepsin B that is overexpressed in cancer cells and tumor endothelial cells (Fig. 15B). Cell starvation was exploited to elevate the expression of cathepsin B in MOLT-3 cells. It was found that theranostic 34b was more potent in starved MOLT-3 cells (IC50 = 4 × 10−6 M) than in normal MOLT-3 cells (IC50 = 30 × 10−6 M). Finally, analogue 34c, having a non-toxic tryptophan instead of the cytotoxic drug, was found to be non-toxic to MOLT-3 cells.
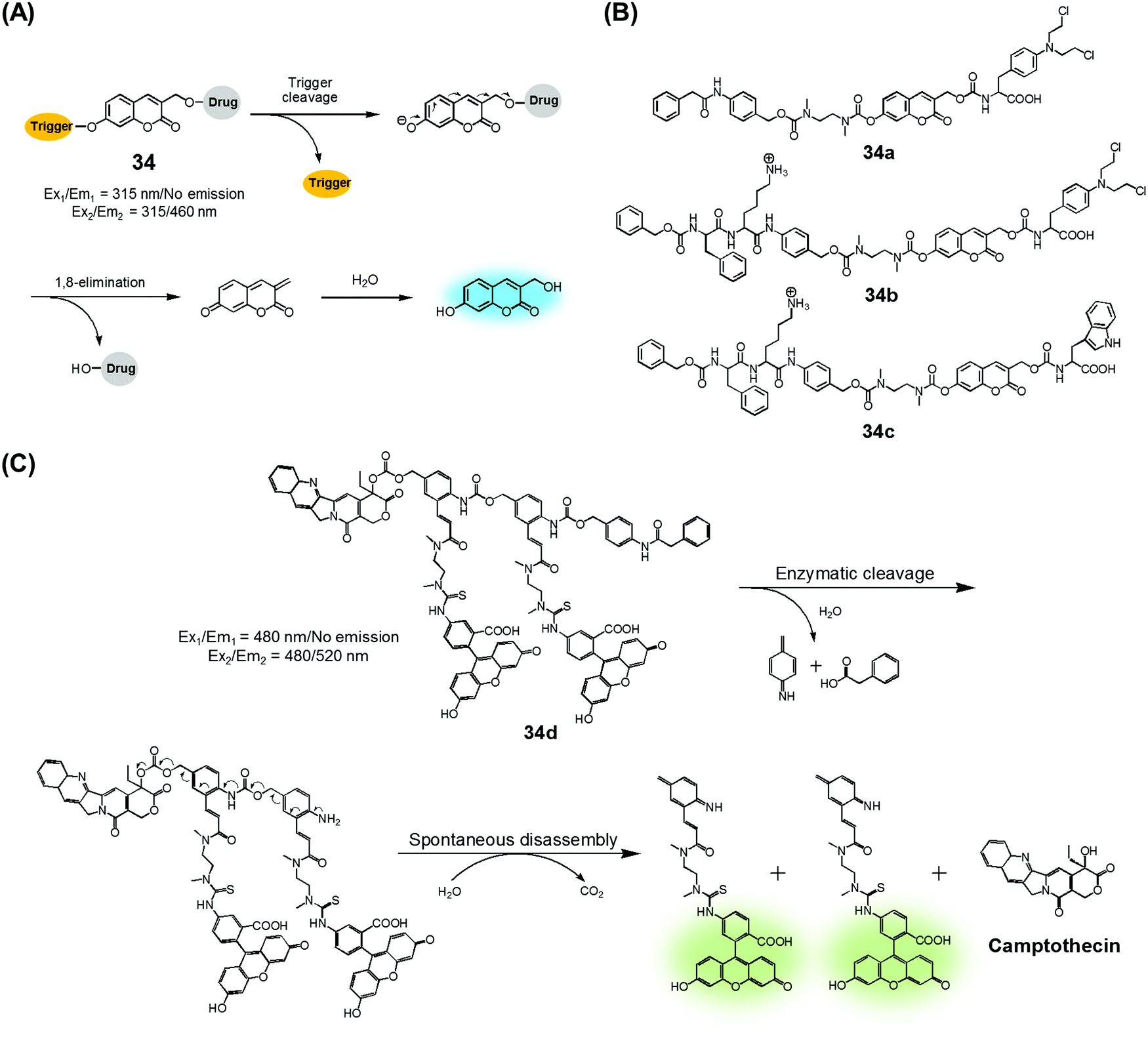 | ||
| Fig. 15 (A) Design of theranostic agent 34 and its activation. (B) Enzyme-activatable theranostic agents (34a–34c). (C) Enzymatic activation of theranostic agent 34d. | ||
Similarly, Shabat et al. reported PGA-activatable prodrug 34d.41 Here, a PGA-cleavable phenylacetamide group was conjugated to the anticancer agent, CPT, via a self-immolative linker decorated with two fluorescein moieties (a pair of FRET dyes). As detailed in Fig. 15C, the phenylacetamide unit of 34d can be cleaved by PGA. It then undergoes a sequence of rapid 1,6-azaquinone-methide eliminations to liberate two fluorescein moieties and CPT in its free cytotoxic form. This process gives rise to an enhanced fluorescence emission feature centred at 520 nm, presumably because the FRET-mediated self-quenching between the two fluorescent dyes in 34d is shut off upon enzyme-induced cleavage. Analysis using HPLC and fluorescence spectroscopy revealed an excellent correlation between the extent of CPT release and the fluorescence enhancement. This was taken as evidence that in 34d PGA-induced drug release occurs at a rate similar to that of the overall conjugate disassembly. On this basis, it was suggested that the design strategy embodied in 34d could be potentially adopted to probe other hydrolytic enzymes that have relevance to cancer biology.
Quinone groups have also been incorporated into prodrug conjugates. Typically, quinones are sensitive to reduction to the corresponding hydroquinones by DT-diaphorase (also called NAD(P)H:quinone oxidoreductase or NQO1). DT-diaphorase is a cytoplasmic flavoenzyme that is upregulated in a number of cancer cells (up to 50-fold) relative to normal cells. Wu et al. developed a DT-diaphorase-activatable theranostic agent, 35, consisting of a CPT subunit and a quinone moiety (Fig. 16).42 Prodrug 35 was found to undergo enzymatic reduction to the corresponding hydroquinone, which, in turn, triggered intramolecular amide hydrolysis to release the cytotoxic CPT drug in its free form. Prior to cleavage, photo-induced electron transfer (PET) from the CPT to the quinone moiety serves to quench much of the fluorescence of the CPT unit in 35 (centred at 436 nm upon 365 nm excitation). On the other hand, the relatively weak fluorescence of conjugate 35 dramatically increased (by ca. 6-fold) under physiological conditions. This was taken as support for the proposed DT-diaphorase-catalysed reductive release. Tests of 35 for cell imaging and its possible therapeutic effect were then evaluated in A549 (DT-diaphorase over-expressing lung carcinoma) cells and L929 (normal) cell lines. After incubation with 35, a strong fluorescence was seen in the A549 cells. However, an essentially undetectable fluorescence signal was observed in the case of the L929 cells. Additionally, MTT assays revealed that prodrug 35 was very potent in lung carcinoma A549 cells that exhibit a high level of DT-diaphorase expression (IC50 = 1.18 μM). In contrast, little cytotoxicity was seen in the L929 cells (IC50 > 80 μM). It was thus proposed that this strategy could provide theranostic drug delivery systems that are activated by the DT-diaphorase enzymes overexpressed in certain cancers.
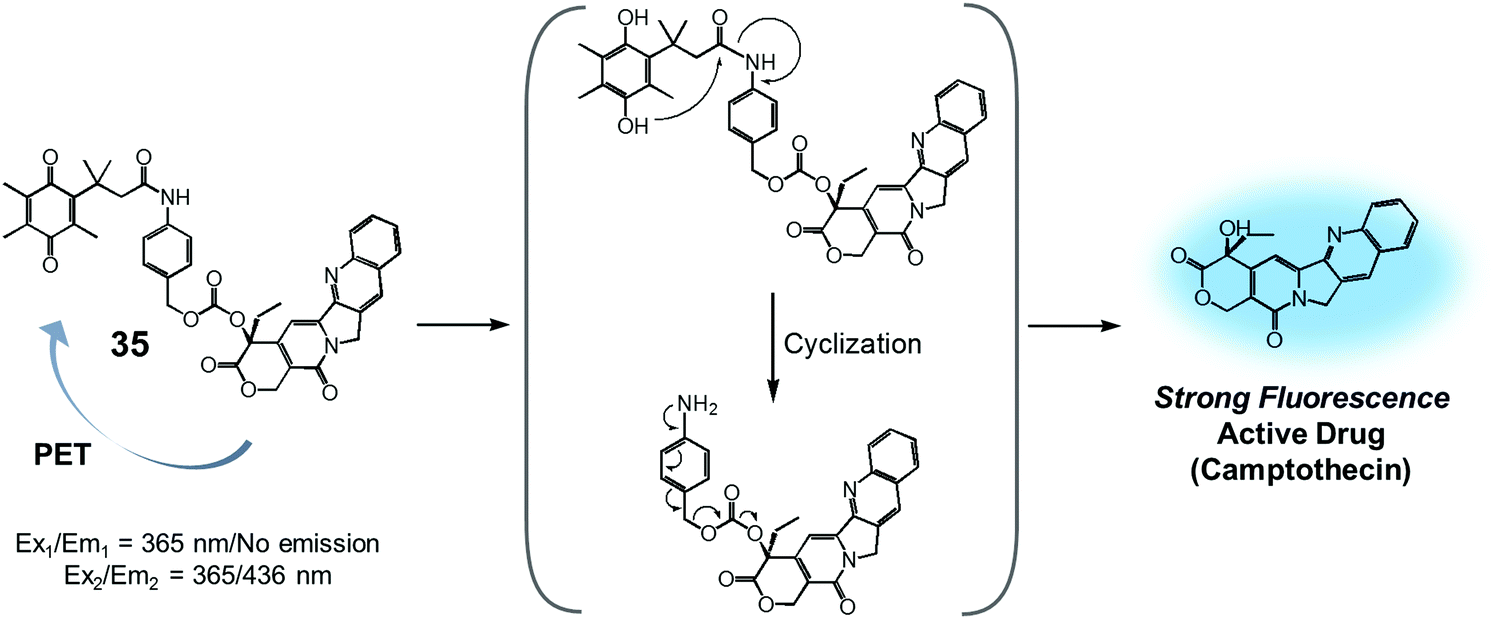 | ||
| Fig. 16 Proposed enzymatic activation of theranostic agent 35. | ||
We recently reported a DT-diaphorase-activatable theranostic that contains a cancer-guiding unit. The system in question, 36, contains tethered biotin, quinone, and SN-38 subunits. These subunits act as the cancer-guiding group, a DT-diaphorase activated release trigger, and an anticancer topoisomerase I inhibitor, respectively (Fig. 17A).43 Enzyme-mediated activation of 36 serves to release SN-38 in its free cytotoxic form and provide an enhanced fluorescence signal centred at 550 nm (Fig. 17B). By monitoring the fluorescence change, the cellular uptake of 36 and its ability to deliver an active drug form could be monitored in real time. The chemotherapeutic effect of prodrug 36 was assessed in vitro using four cell lines. Two were cancer cell lines (A549, HeLa) that express a high level of biotin transporter. The other two were normal fibroblast cell lines (WI-38, BJ). Prodrug 36 exhibited excellent cytotoxicity in the cancer cell lines, but not in the normal cell lines (Fig. 17C). On this basis, it was concluded that 36 is internalized into cancer cells more effectively than into normal cells, presumably as a result of biotin-mediated transport, and that the quinone moiety is reduced by DT-diaphorase to release SN-38 in its free cytotoxic form, thus promoting efficient cancer cell death. Prodrug 36 was further tested using A549 cell-inoculated xenograft mice. Under conditions of tail vein injection, administration of conjugate 36 resulted in a significant reduction in the tumour burden compared to what was seen for the vehicle control and free SN-38 (Fig. 17D and E). Evidence for specific tumour localization was also seen (Fig. 17F and G).
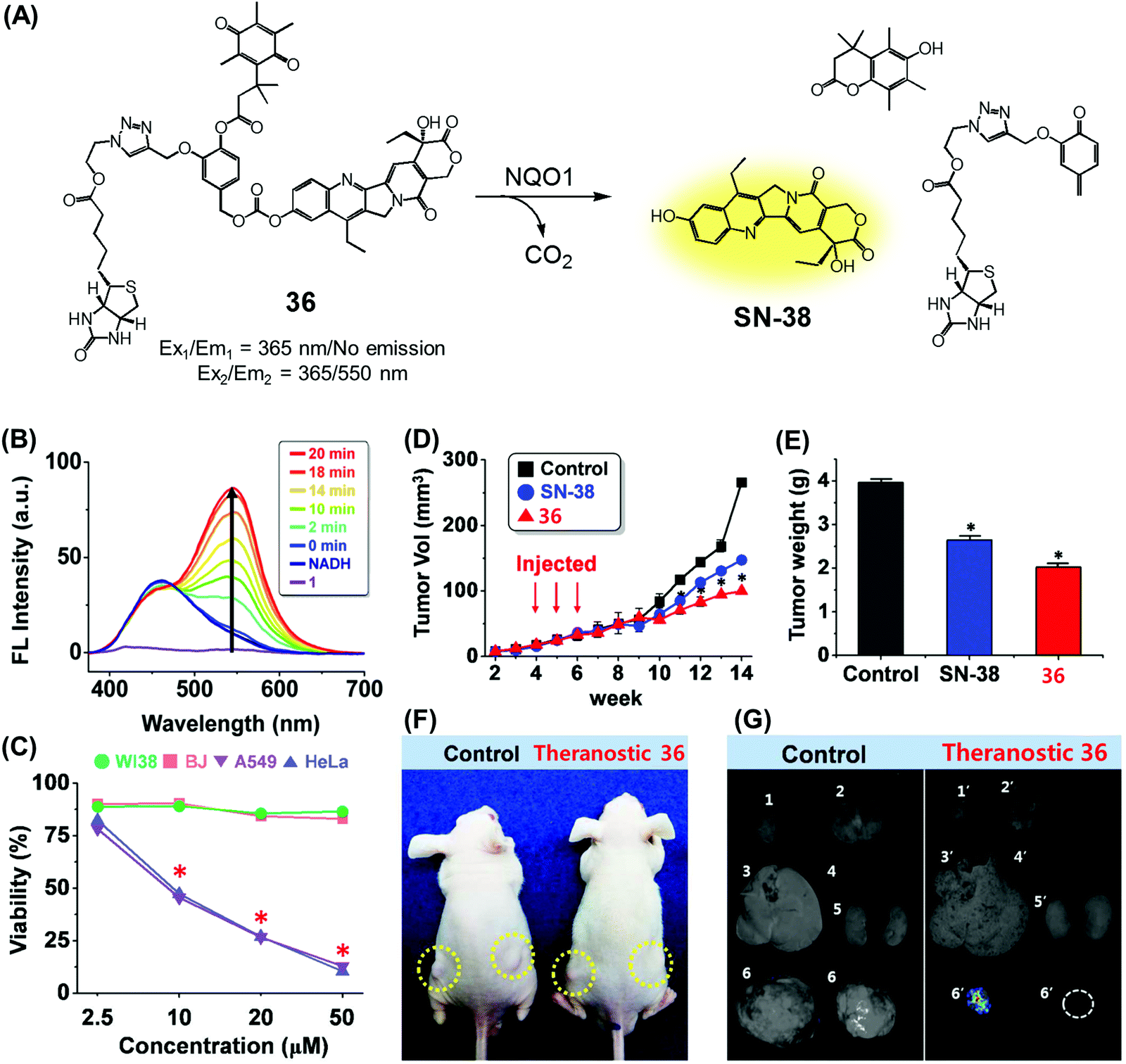 | ||
| Fig. 17 (A) Enzymatic activation of theranostic agent 36. (B) Fluorescence response seen when 36 was allowed to react with DT-diaphorase (NQO1). (C–G) Bioimaging and therapeutic effects of 36. (B–G) are reproduced with permission from ref. 43. Copyright 2016 American Chemical Society. | ||
We were also curious to explore whether a sequence involving activation by two different enzymes upregulated in cancer cells could be exploited to produce more precise theranostic drug delivery systems. To test this hypothesis, we designed and developed theranostic 37 (Fig. 18). This system contains an indomethacin tag as a cancer guiding group, an acetylated lysine moiety that is a potential substrate for two different enzymes, namely histone deacetylase (HDAC) and cysteine cathepsin L (CTSL). It also contains a DOX core, which was expected to function as a topoisomerase II inhibitor and comprise the cytotoxic payload (Fig. 18).44 Indomethacin is an agent that is well recognized for its ability to bind to cyclooxygenase (COX), an enzyme that is overexpressed in tumours and known to play a vital role in promoting tumour growth and metastasis.45 HDAC and CTSL are enzymes that are also involved in cancer pathogenesis and which are perceived as being markers for cancer metastasis.46,47 In the presence of HDAC and CTSL, it was found that theranostic 37 first undergoes HDAC-mediated deacetylation (to expose a lysine moiety) and then CTSL-catalysed amide bond cleavage to release the active Dox. This release, which occurs under physiological conditions, leads to an increase in the fluorescence emission feature centred around 590 nm. However, neither Dox release nor an increase in fluorescence intensity was seen in the presence of the individual enzymes.
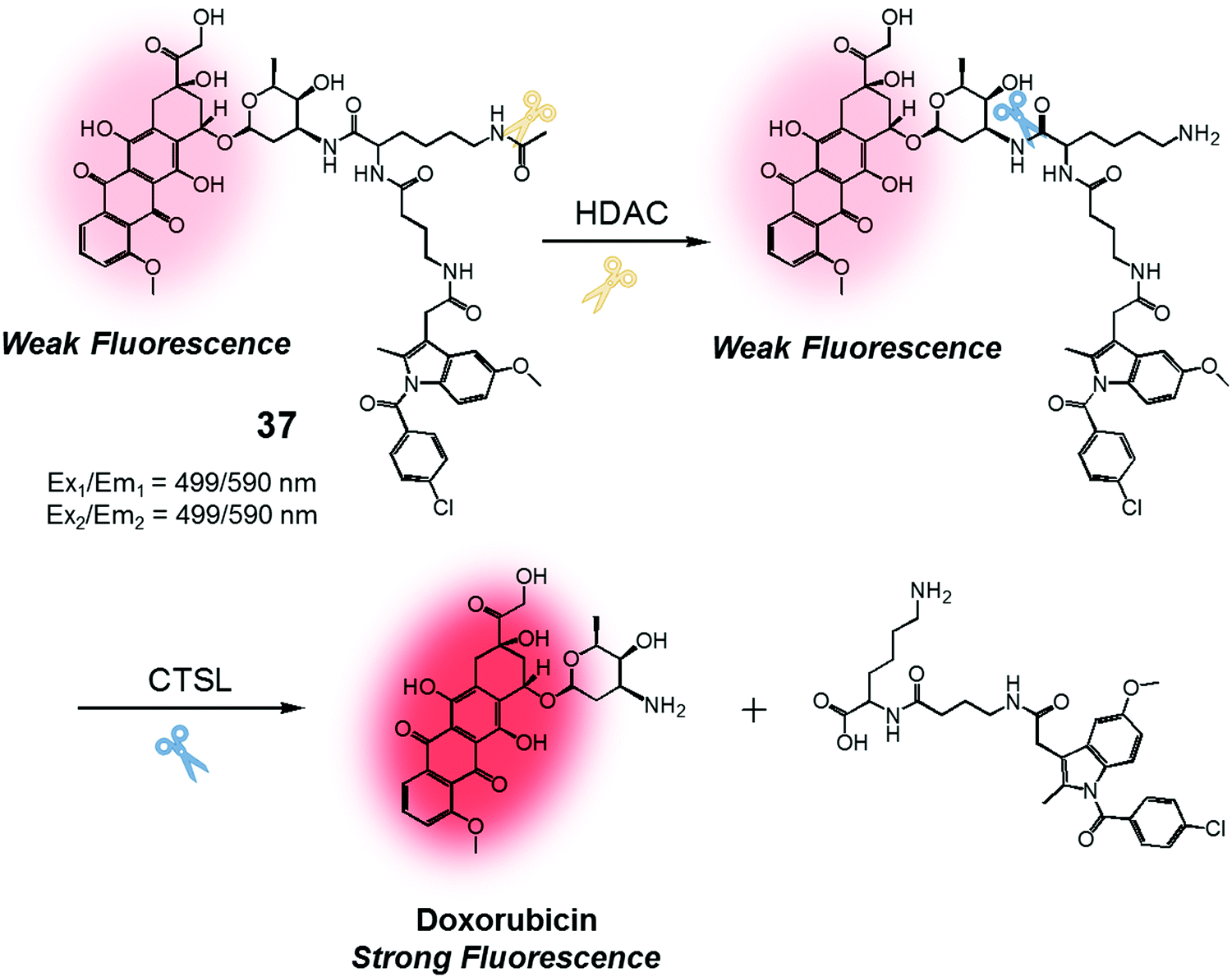 | ||
| Fig. 18 Enzymatic activation of theranostic agent 37. | ||
The biological imaging and therapeutic potential of 37 were tested in COX-2 positive cell lines (HeLa, HepG2) and a COX-2 negative cell line (HCT 116). Prodrug 37 displayed higher fluorescence intensity in the COX-2 positive cells (relative fluorescence intensity per cell >4 after 2 min of incubation) than in the COX-2 negative cell line (relative fluorescence intensity per cell <3 after 2 min of incubation). Theranostic 37 was also found to mediate a greater cytotoxic response in COX-2 positive cells (cell viability; around 50% at 100 μM of 37) than in COX-2 negative cells (cell viability; over 90% at 200 μM of 37). The in vivo therapeutic potential of theranostic 37 was evaluated in xenograft mice inoculated with COX-2 positive cells (HeLa or HepG2) or COX-2 negative cells (HCT 116) via tail vein injection. It was found that theranostic 37 localizes preferentially in the tumour mass, presumably as a result of selective binding to COX-2, while providing a significant reduction in tumour volume as compared to appropriate controls.
7. Light-activated fluorogenic drug release
In the context of DDS, the ability to control precisely the drug release kinetics once a prodrug is successfully taken up by cancerous tissues holds tremendous appeal. In previous sections, we discussed various drug release strategies that are based either on chemical or enzymatic triggers. As a general rule, the underlying mechanisms require external factors or stimuli. This can introduce unwanted complexities. Light, on the other hand, can provide potentially more facile spatiotemporal control. To date, most light-based drug activation processes have been based upon the concept of photocaging. Here, the antitumor drug is temporarily protected or “caged” by linking it to a photocage (light-responsive functional group), which releases the active drug in a controlled manner once subjected to photoillumination.The theranostic analogues 38a–e, developed by You et al., were designed to highlight the therapeutic advantages of combining photodynamic damage with photo-released chemotherapy (Fig. 19A).48 Conjugates 38a–e contain a phthalocyanine photocage unit linked to the anticancer drug combretastatin A-4 through an amino acrylate moiety designed to function as a singlet oxygen responsive cleavable linker. Conjugates 38b–d contain folic acid groups and polyethylene glycol chains to modulate the overall hydrophilicity, while 38a,e were designed to serve as control compounds. Relative to the other members of the series, conjugates 38c,d exhibited preferential cellular uptake in colon 26 cancer cells and displayed improved potency (IC50 values = 2.71 × 10−8 and 1.65 × 10−8 M, respectively) upon illumination with 690 nm laser light (5.6 mW cm−2) for 30 min as compared to analogues 38a,b (IC50 = 4.47 × 10−8 and 4.03 × 10−8 M, respectively) and 38e (IC50 = 4.85 × 10−8 M), an analogue lacking the folic acid group. Taken in concert, these data serve to highlight the benefits of both folic acid-mediated tumour active targeting and overall hydrophilicity for efficient tumour uptake. The therapeutic efficacy of agents 38a–e was also studied in Balb/c mice bearing SC colon 26 tumours. Theranostic 38d exhibited preferential tumour uptake (tumour/skin ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in mice 7 h post injection with significant tumour inhibition (i.e., tumour-free mice at day 75 following start of treatment) being observed upon illumination (690 nm laser light) with minimal surrounding damage, even in the illuminated areas (Fig. 19B). However, theranostic 38e, a system with a lower tumour/skin accumulation ratio (1:3), produced more photodamage to the skin (Fig. 19Biii).
:1) in mice 7 h post injection with significant tumour inhibition (i.e., tumour-free mice at day 75 following start of treatment) being observed upon illumination (690 nm laser light) with minimal surrounding damage, even in the illuminated areas (Fig. 19B). However, theranostic 38e, a system with a lower tumour/skin accumulation ratio (1:3), produced more photodamage to the skin (Fig. 19Biii).
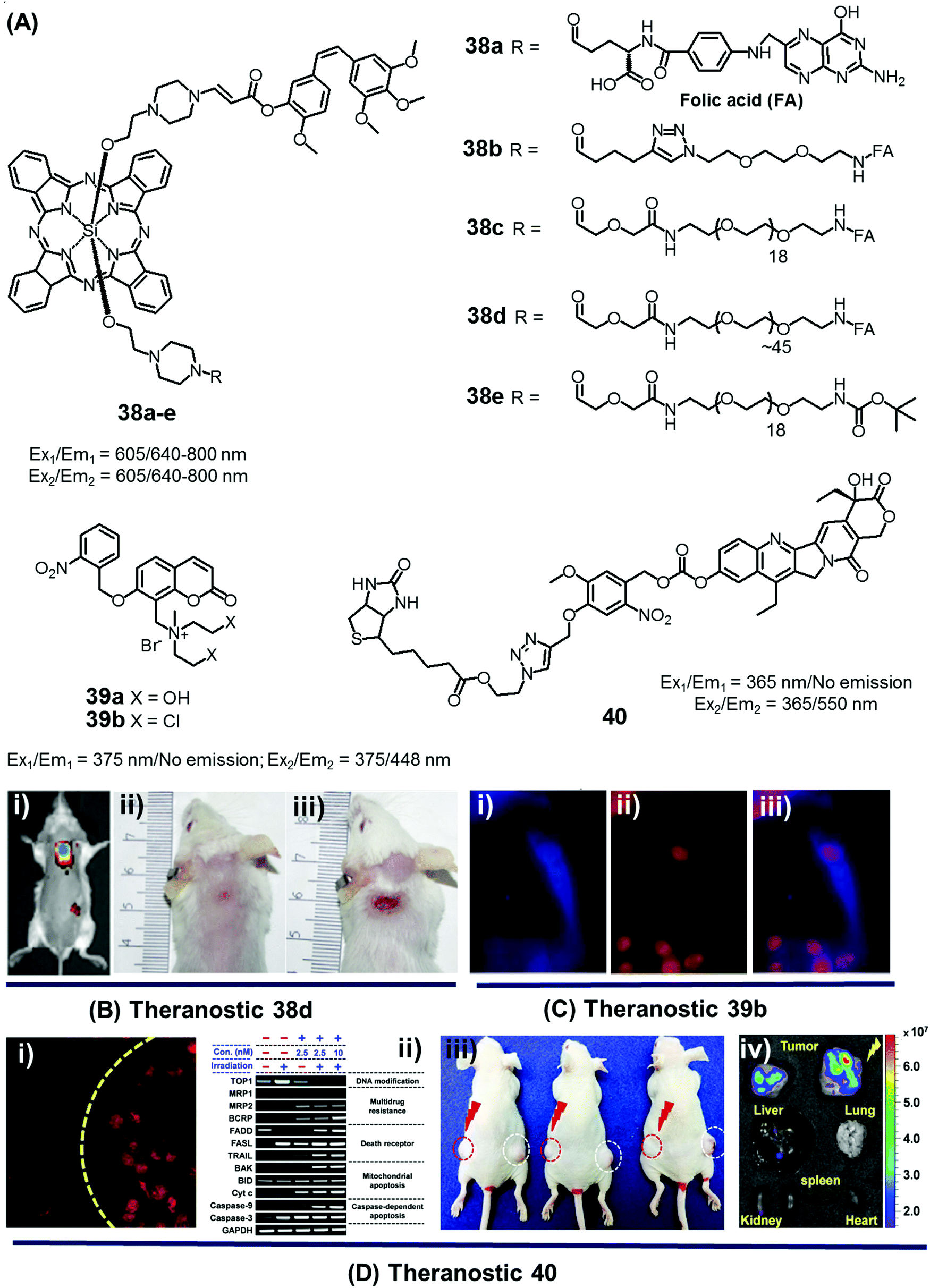 | ||
| Fig. 19 (A) Light-activated prodrugs 38–40. (B and C) Bio-imaging and therapeutic effects of 38d, 39b, and 40. (Bi) Fluorescence image of SC colon cancer bearing Balb/c mice after 38d treatment (7 h post injection) and (Bii and iii) photographic images of mice treated with 38d and 38e and illumination (690 nm, 100 mW cm−2, 30 min) at day 15 post-illumination. (Ci–iii) Fluorescence images of HeLa cells recorded after UV irradiation (30 min) and pre-treated with 39b showing blue fluorescence (activated 39b), red (propidium iodide stained nuclei), and merged images respectively. (Di) Fluorescence image of A549 cells pre-treated with 40 before and after illumination at 405 nm (1 h). (Dii) Enhanced expression of various apoptosis gene/markers upon treatment with 40 and illumination under different conditions in A549 cells. (Diii and iv) In vivo images of A549 bearing mice injected with 40 and control with/without illumination and ex vivo images of tumour and organs after treatment with 40 and photoillumination. (B–D) are reproduced with permission from ref. 48–50, respectively. Copyright 2014 American Chemical Society (for B), Copyright 2015 Royal Society of Chemistry (for C), and Copyright 2016 Nature Publishing Group (for D). | ||
Wang et al. reported a light triggered theranostic, 39b, that contains an o-nitrophenyl ethyl derivative as a photolabile masking group, an inherent fluorescent coumarin moiety as a reporter and a mechlorethamine group as a DNA alkylating agent (Fig. 19A).49 Conjugate 39b displayed a low dark toxicity in normal (Hekn) skin cells. Once illuminated with UV radiation at 365 nm for 30 min, however, it exhibited a turn-on fluorescence response at 448 nm and produced significant toxicity in HeLa cells. Compared to the control compound 39a, theranostic 39b was found to give rise to increased DNA cross-linking upon exposure to UV irradiation as measured by agarose gel electrophoresis. These researchers also monitored the cellular uptake and drug activation ability of 39b following light illumination by recording the change in fluorescence intensity with time in vitro. After 2 h incubation in HeLa cells, followed by UV exposure for 30 min, a shift in the fluorescence signal from the cytoplasm to the nucleus was observed. These results serve to highlight the dual role that theranostic 39b can play, namely allowing for the photoactivated release of an active drug form and monitoring its delivery to a desired site of action (Fig. 19C).
We used a related strategy to develop the theranostic agent 40, a system designed to produce light-triggered cytotoxicity within cancer cells while sparing normal cells and other tissues.50 It relies on a nitrovanillin subunit as a phototrigger moiety. This latter group is linked to a 7-ethyl-10-hydroxycamptothecoin (topoisomerase 1 inhibitor) through a covalent carbonate bond. A biotin moiety was further incorporated into conjugate 40 to provide for cancer targeting. Preliminary studies revealed that 40 was effectively stable in the absence of light. However, upon irradiation with 405 nm laser light, a strong fluorescence emission feature at 550 nm, corresponding to the free drug, was observed (Fig. 19Di). Agent 40 was then tested in various biotin (+ve) and (−ve) cell lines. Upon irradiation, agent 40 was found to produce a cytotoxic response in the biotin (+ve) A549 and HeLa cell lines with diminished expression of topoisomerase 1 as inferred from a WST (water-soluble tetrazolium salt)-based cell proliferation assay. Additionally, enhanced expression of various cancer death receptors like FADD (Fas-associated protein with death domain), FasL (Fas ligand), TRAIL (TNF-related apoptosis-inducing ligand), and apoptotic genes like BAK (Bcl-2 homologous antagonist killer), BID (BH3 interacting-domain death agonist), CytC (Cytochrome C) was observed (Fig. 19Dii). The in vivo therapeutic potency was tested using an A549 inoculated xenograft murine model (via tail vein injection). Theranostic agent 40 was found to possess sufficient plasma stability to allow for a near-maximal serum concentration for 6 h. After administration, preferential tumour accumulation along with light-induced tumour suppression was observed in these mouse xenografts compared to what was seen in the absence of irradiation and with various controls (Fig. 19Diii and iv).
Conclusion and outlook
Theranostics, a fusion of specialized diagnosis and therapy, is an emerging area of drug discovery. It is attractive because it might allow for the development of targeted diagnostic modalities and individual therapeutic regimens through rational design. Although a number of theranostic systems are currently being studied, in this Tutorial Review we have focused on experimental fluorogenic theranostic conjugates that can be activated by specific stimuli (endogenous biomolecule-promoted reactions, enzymes, and light) for use in cancer therapy. These theranostics are able to target cancer cells preferentially, provide cytotoxic chemotherapeutics after activation, and offer the possibility of monitoring both the location and therapeutic response produced as a result of activation. As inferred from spectroscopic analyses, confocal microscopic imaging studies, MTT assays, and in some instances preliminary in vivo studies, there is a mounting body of evidence supporting the notion that fluorescent prodrug conjugates can be produced that are not only selectively recognized and internalized by specific tumour cells, but also undergo stimulus-promoted cleavage to produce both a fluorogenic response and release an active cytotoxic drug. This theranostic strategy is likely to be generalizable to produce new drug leads that are effective in a variety of disease areas beyond the cancer focus of this Tutorial Review. It is also expected to improve the knowledge and understanding of various factors, such as cellular uptake and drug activation that can help guide advanced drug design.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Korean National Research Foundation (NRF) (2015R1C1A2A01054496, M. H. L.), (2017R1A2A2A05069805, C. K.), the Ministry of Science, ICT & Future Planning in Korea (CRI project no. 2009-0081566, J. S. K.), the US National Institutes of Health (CA 68682, J. L. S.), and the Robert A. Welch Foundation (F-1018, J. L. S.).Notes and references
- R. Kumar, W. S. Shin, K. Sunwoo, W. Y. Kim, S. Koo, S. Bhuniya and J. S. Kim, Chem. Soc. Rev., 2015, 44, 6670 RSC.
- E. K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2015, 115, 327 CrossRef CAS PubMed.
- R. Weinstain, E. Segal, R. Satchi-Fainaro and D. Shabat, Chem. Commun., 2010, 46, 553 RSC.
- M. H. Lee, J. L. Sessler and J. S. Kim, Acc. Chem. Res., 2015, 48, 2935 CrossRef CAS PubMed.
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2014, 47, 2970 CrossRef CAS PubMed.
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071 CrossRef CAS PubMed.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388 CrossRef CAS PubMed.
- T. M. Allen, Nat. Rev. Cancer, 2002, 2, 750 CrossRef CAS PubMed.
- M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185 RSC.
- Y. Yuan and B. Liu, Chem. Sci., 2017, 8, 2537 RSC.
- M. H. Lee, J. Y. Kim, J. H. Han, S. Bhuniya, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2012, 134, 12668 CrossRef CAS PubMed.
- M. H. Lee, E.-J. Kim, H. Lee, H. M. Kim, M. J. Chang, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 16380 CrossRef CAS PubMed.
- S. Bhuniya, S. Maiti, E.-J. Kim, H. Lee, J. L. Sessler, K. S. Hong and J. S. Kim, Angew. Chem., Int. Ed., 2014, 53, 4469 CrossRef CAS PubMed.
- S. Maiti, N. Park, J. H. Han, H. M. Jeon, J. H. Lee, S. Bhuniya, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 4567 CrossRef CAS PubMed.
- T. Kim, H. M. Jeon, H. T. Le, T. W. Kim, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 7690 RSC.
- H. Chen, H. P. Tham, C. Y. Ang, Q. Qu, L. Zhao, P. Xing, L. Bai, S. Y. Tan and Y. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 24319 CAS.
- Y. Hu and F. Zeng, Mater. Sci. Eng., C, 2017, 72, 77 CrossRef CAS PubMed.
- Y. Liu, Q. Pei, L. Chen, Z. Li and Z. Xie, J. Mater. Chem. B, 2016, 4, 2332 RSC.
- Z. Yang, J. H. Lee, H. M. Jeon, J. H. Han, N. Park, Y. He, H. Lee, K. S. Hong, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 11657 CrossRef CAS PubMed.
- S. Bhuniya, M. H. Lee, H. M. Jeon, J. H. Han, J. H. Lee, N. Park, S. Maiti, C. Kang and J. S. Kim, Chem. Commun., 2013, 49, 7141 RSC.
- Y. Kong, J. Smith, K. Li, J. Cui, J. Han, S. Hou and M. L. Brown, Bioorg. Med. Chem., 2017, 25, 2226 CrossRef CAS PubMed.
- F. Kong, Z. Liang, D. Luan, X. Liu, K. Xu and B. Tang, Anal. Chem., 2016, 88, 6450 CrossRef CAS PubMed.
- X. Wu, X. Sun, Z. Guo, J. Tang, Y. Shen, T. D. James, H. Tian and W. Zhu, J. Am. Chem. Soc., 2014, 136, 3579 CrossRef CAS PubMed.
- M. Ye, X. Wang, J. Tang, Z. Guo, Y. Shen, H. Tian and W.-H. Zhu, Chem. Sci., 2016, 7, 4958 RSC.
- W. S. Shin, S. K. Park, P. Verwilst, S. Koo, J. H. Lee, S.-G. Chi and J. S. Kim, Chem. Commun., 2017, 53, 1281 RSC.
- H. Han, Q. Jin, Y. Wang, Y. Chen and J. Ji, Chem. Commun., 2015, 51, 17435 RSC.
- E.-J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888 CrossRef CAS PubMed.
- R. Kumar, J. Han, H.-J. Lim, W. X. Ren, J.-Y. Lim, J.-H. Kim and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 17836 CrossRef CAS PubMed.
- O. Redy-Keisar, S. Ferber, R. Satchi-Fainaro and D. Shabat, ChemMedChem, 2015, 10, 999 CrossRef CAS PubMed.
- J. T. Hou, W. X. Ren, K. Li, J. Seo, A. Sharma, X. Q. Yu and J. S. Kim, Chem. Soc. Rev., 2017, 46, 2076 RSC.
- S.-Y. Li, L.-H. Liu, H.-Z. Jia, W.-X. Qiu, L. Rong, H. Cheng and X.-Z. Zhang, Chem. Commun., 2014, 50, 11852 RSC.
- S.-Y. Li, L.-H. Liu, L. Rong, W.-X. Qiu, H.-Z. Jia, B. Li, F. Li and X.-Z. Zhang, Adv. Funct. Mater., 2015, 25, 7317 CrossRef CAS.
- A. Fernandez, M. Vermeren, D. Humphries, R. Subiros-Funosas, N. Barth, L. Campana, A. MacKinnon, Y. Feng and M. Vendrell, ACS Cent. Sci., 2017, 3, 995 CrossRef CAS PubMed.
- M. H. Lee, E.-J. Kim, H. Lee, S. Y. Park, K. S. Hong, J. S. Kim and J. L. Sessler, Chem. Commun., 2016, 52, 10551 RSC.
- R. Kumar, E. J. Kim, J. Han, H. Lee, W. S. Shin, H. M. Kim, S. Bhuniya, J. S. Kim and K. S. Hong, Biomaterials, 2016, 104, 119 CrossRef CAS PubMed.
- W. Feng, C. Gao, W. Liu, H. Ren, C. Wang, K. Ge, S. Li, G. Zhou, H. Li, S. Wang, G. Jia, Z. Li and J. Zhang, Chem. Commun., 2016, 52, 9434 RSC.
- P. Verwilst, J. Han, J. Lee, S. Mun, H. G. Kang and J. S. Kim, Biomaterials, 2017, 115, 104 CrossRef CAS PubMed.
- Y. Yuan, Y. Chen, B. Z. Tang and B. Liu, Chem. Commun., 2014, 50, 3868 RSC.
- Y. Yuan, R. T. K. Kwok, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2014, 136, 2546 CrossRef CAS PubMed.
- Y. Yuan, C.-J. Zhang and B. Liu, Chem. Commun., 2015, 51, 8626 RSC.
- O. Redy and D. Shabat, J. Controlled Release, 2012, 164, 276 CrossRef CAS PubMed.
- P. Liu, J. Xu, D. Yan, P. Zhang, F. Zeng, B. Li and S. Wu, Chem. Commun., 2015, 51, 9567 RSC.
- W. S. Shin, J. Han, P. Verwilst, R. Kumar, J. H. Kim and J. S. Kim, Bioconjugate Chem., 2016, 27, 1419 CrossRef CAS PubMed.
- J. H. Jang, H. Lee, A. Sharma, S. M. Lee, T. H. Lee, C. Kang and J. S. Kim, Chem. Commun., 2016, 52, 9965 RSC.
- Z. Khan, N. Khan, R. P. Tiwari, N. K. Sah, G. B. Prasad and P. S. Bisen, Curr. Drug Targets, 2011, 12, 1082 CrossRef CAS PubMed.
- J. E. Bolden, M. J. Peart and R. W. Johnstone, Nat. Rev. Drug Discovery, 2006, 5, 769 CrossRef CAS PubMed.
- B. Goulet, L. Sansregret, L. Leduy, M. Bogyo, E. Weber, S. S. Chauhan and A. Nepveu, Mol. Cancer Res., 2007, 5, 899 CrossRef CAS PubMed.
- G. Nkepang, M. Bio, P. Rajaputra, S. G. Awuah and Y. You, Bioconjugate Chem., 2014, 25, 2175 CrossRef CAS PubMed.
- Y. Cao, R. Pan, W. Xuan, Y. Wei, K. Liu, J. Zhou and W. Wang, Org. Biomol. Chem., 2015, 13, 6742 CAS.
- W. S. Shin, J. Han, R. Kumar, G. G. Lee, J. L. Sessler, J.-H. Kim and J. S. Kim, Sci. Rep., 2016, 6, 29018 CrossRef CAS PubMed.
Footnote |
| † Contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |