
Quinone-based fluorophores for imaging biological processes
Gleiston G.
Dias
a,
Aaron
King
b,
Fabio
de Moliner
b,
Marc
Vendrell
 *b and
Eufrânio N.
da Silva Júnior
*a
*b and
Eufrânio N.
da Silva Júnior
*a
aInstitute of Exact Sciences, Department of Chemistry, Federal University of Minas Gerais, Belo Horizonte, 31270-901, MG, Brazil. E-mail: eufranio@ufmg.br; Web: http://www.eufraniolab.com Tel: +55 31 34095720
bMRC/UoE Centre for Inflammation Research, The University of Edinburgh, EH16 4TJ Edinburgh, UK. E-mail: marc.vendrell@ed.ac.uk; Web: http://www.dynafluors.co.uk Tel: +44 (0)131 242 6685
First published on 3rd November 2017
Abstract
Quinones are privileged chemical structures playing crucial roles as redox and alkylating agents in a wide range of processes in cells. The broad functional array of quinones has prompted the development of new chemical approaches, including C–H bond activation and asymmetric reactions, to generate probes for examining their activity by means of fluorescence imaging. This tutorial review covers recent advances in the design, synthesis and applications of quinone-based fluorescent agents for visualizing specific processes in multiple biological systems, from cells to tissues and complex organisms in vivo.
 Gleiston G. Dias | Gleiston G. Dias received his degree in Chemistry in 2014. In 2017 he concluded his MSc at Federal University of Minas Gerais under the supervision of Prof. Eufrânio N. da Silva Júnior and in the same year he started his PhD studies in da Silva Júnior group working with new methods involving C–H bond activation and synthesis of fluorescent compounds. His research interests are focused on organic synthesis of new fluorophores for biological applications and synthetic methods by using click chemistry, C–H bond activation and cross coupling reactions. He is also interested on Medicinal Chemistry aiming at synthesis of bioactive compounds. |
 Aaron King | Aaron King studied Chemistry at the University of Edinburgh, attaining his Master's degree in Medicinal and Biological Chemistry in 2017. During his undergraduate studies, he cultivated a keen interest in the development of chemical agents for biological imaging, and worked in bioorganometallic chemistry in the lab of Prof. Leong Weng Kee at the Nanyang Technological University (Singapore) in 2016 and in fluorescent chemical probes in the group of Dr Marc Vendrell at the University of Edinburgh in 2017. |
 Fabio de Moliner | Fabio de Moliner obtained his PhD in Organic Chemistry from the University of Genova in 2010 under Prof. Andrea Basso on the development of combinatorial syntheses for chemical libraries. Afterwards, he joined the group of Prof. Christopher Hulme at the University of Arizona (Tucson) as a postdoctoral assistant to design new chemical methodologies based on multicomponent and domino processes. After some industrial experience in England, he joined the Vendrell lab at the University of Edinburgh in 2015. His main research interests are the use of fluorophores to investigate the transport of xenobiotics and the development of in vivo imaging probes. |
 Marc Vendrell | Marc Vendrell obtained his PhD in Chemistry at the University of Barcelona in 2007. He then joined the Singapore Bioimaging Consortium to work with Young-Tae Chang in synthetic fluorophores for optical imaging. In 2012 he started his independent career as an MRC Academic Fellow at the University of Edinburgh. He is a Senior Lecturer in Biomedical Imaging at the University of Edinburgh and his main research interest is the development of activatable fluorophores for imaging cancer and inflammation. He has over 70 publications in chemical biology and imaging, including 8 international patents, and been recognised with several international awards. |
 Eufrânio N. da Silva Júnior | Eufrânio N. da Silva Júnior obtained his PhD in Chemistry at the University of Brasilia in 2009. In 2010, he started his independent career as Professor at the Federal University of Minas Gerais (UFMG). He is a member of the SBQ and RSC. He has received the RSC/BMOS Young Investigator Award (2015) and Jones Travelling Fellowship (2016). His research interests are focused on C–H activation reactions, organocatalysis, mechanistic investigations, and synthesis of heterocyclic and naphthoquinoidal bioactive compounds. Currently, he is also interested in obtaining fluorescent substances for bioimaging studies. He has 70 publications related to Organic Chemistry and Medicinal Chemistry. |
Key learning points1. Recent advances in chemical strategies to synthesize small reactive fluorophores containing quinoidal moieties.2. Reactivity of quinone-based fluorophores as activatable probes for the detection of multiple biomarkers. 3. Trimethyl lock quinones as privileged scaffolds to monitor enzymatic processes in biological systems. 4. The fluorescence derivatisation of quinones with therapeutic action to study their mechanism of action and to monitor drug release. 5. Perspectives and challenges of quinone-based chemical probes in biological imaging. |
1. Introduction
Quinones and their derivatives have been studied for over a century due to their electronic and chemical properties. In particular, quinones present several unique features associated with their redox potential, which allow them to undergo both one- and two-electron reductions and form semiquinones and hydroquinones, respectively. Their reactivity and pharmacological properties can be also tuned through different structural modifications, which have been described in many publications. However, although the earliest reports dealing with the chemistry of quinones date back to the late 19th century, it was not until the 1950s that their abundance amongst biomolecules started to be understood. A milestone in this research area was the discovery in 1957 of coenzyme Q, which was found to be ubiquitous in animals and many bacteria and to play an essential role in mitochondrial electron transport.1 Ever since, naturally occurring quinones have been subsequently found to be involved in multiple biological contexts, ranging from bone formation in mammals to self-defense mechanisms in insects.A wide array of molecules containing quinone moieties show remarkable pharmacological activity. For instance, the plant-derived naphthoquinone plumbagin has been found to induce cell death in human cervical cancer cells.2 Perhaps more notably, atovaquone (Mepron) is a commercially available medication for the treatment of pneumocystis pneumonia, toxoplasmosis and malaria. Besides, the redox properties of quinones can be advantageously exploited for the preparation of prodrugs. In fact, their conjugation to known therapeutic agents can render drugs in an inactive yet bioavailable form so that they are activated in vivo in cells undergoing reductive metabolism. Using this concept, novel benzoquinone derivatives of the anti-angiogenic compounds biochanin A and semaxanib have been synthesized and evaluated as potential prodrug systems.3 In these molecules, it has been shown that the reduction of the benzoquinone unit leads to the release of active drugs in targeted tissues.
Another field in which quinones are being investigated is the development of probes for sensing processes in biological systems, ranging from cells to whole intact organisms. Quinones have shown excellent performance in combination with gold nanoparticles for the sensing of redox potentials in biological environments by means of surface-enhanced Raman spectroscopy (SERS). For instance, the functionalisation of gold nanoshells with naphthoquinone derivatives has been described as a useful approach to monitor redox potentials in real time.4 Another family of widely used analytical tools regards the design and assembly of fluorescent probes. Fluorescence-based assays have consolidated as a highly sensitive methodology for interrogating non-invasively cellular events, including the localisation and dynamics of gene expression, protein expression, and molecular interactions in living cells and tissues. Combinations of fluorescent probes also allow us to sensitively detect multiple biological molecules when suitable optical filters are used. In addition to being low-cost, safe, and displaying long shelf-lives, small molecule fluorophores can produce images at molecular resolution with excellent sensitivity. One of the main challenges currently being pursued in this research area is the synthesis of activatable fluorescent probes, which emit fluorescence upon activation via interaction with a specific analyte, or by a change in the surrounding microenvironment. These activatable fluorophores have been reported for numerous different applications.5 Driven by the continued interest in the preparation of such sophisticated probes, considerable advances in overcoming the inherent difficulties in their synthesis have been attained. Such achievements have been possible with the design of novel and effective synthetic strategies that had been traditionally limited to medicinal chemistry research programmes,6,7 such as multicomponent reactions, C–H activation processes and cycloadditions, among others.8
While the chemistry of quinones and most aspects related to their synthesis, properties and usage have been surveyed in several review articles over the last decade, to date there were no reports covering the integration of quinones within fluorescent probes and their application for bioimaging studies. This tutorial review aims to summarize the growing number of innovative techniques relying upon the unique electronic and chemical properties of quinone derivatives for biological imaging and theranostic purposes.
2. Activatable fluorescent probes based on quinoidal systems
The unique chemistry of the quinone scaffold makes it an excellent building block for the synthesis of activatable fluorescent probes. Smart quinoidal probes only fluorescing upon recognition of target analytes have been described using different activation mechanisms. Such structures typically consist of a fluorophore, which acts as the reporter, linked to a quenching quinone, which undergoes a chemical transformation upon activation. This transformation typically abates its quenching activity and thus results in an increased fluorescence emission. In particular, photoinduced electron transfer (PeT) plays a pivotal role in the activation of these fluorophores. PeT is an excited state electron transfer process in which an electron is transferred from a donor orbital to an acceptor orbital, typically leading to a quenching effect.9 The PeT mechanism has been widely exploited to produce molecules that, upon binding to the analyte, present a donor orbital with lower energy to reduce the electron transfer and increase the fluorescence emission. In this section, examples in which quinones have been an essential part of such activatable fluorescent probes for a range of biomarkers will be surveyed.Biothiols such as cysteine, homocysteine, and glutathione (GSH) are present in nearly all cells and play a critical role as mediators in different cellular processes. In particular, they are key components of the endogenous antioxidant system that maintains the intracellular redox potential. For example, GSH prevents cellular damage from free radicals as well as heavy metals. Abnormal levels of GSH have been found in patients with Alzheimer's disease and different cardiovascular conditions. As a result, highly sensitive tools for the in situ detection of thiols in cells have important applications in biological and biomedical research.
Quinones are well suited for the imaging of biothiols as they are excellent Michael acceptors, and therefore prone to nucleophilic attack by thiol groups. Since this nucleophilic reaction can occur in biological conditions, it represents an ideal activation strategy for the design of activatable probes for the detection of biothiols in vivo. The first example of quinone-fluorescent reporters for biothiols was reported by Huang et al., who successfully synthesized a red fluorescent probe (λem.: 595 nm) consisting of a trimethyl benzoquinone moiety directly linked to a red-shifted coumarin scaffold through an ether moiety.10 Advantageously, these fluorophores possessed remarkably long Stokes shifts (i.e., around 90 nm), with minimal overlapping between absorbance and emission spectra and high signal-to-background ratios. In this case, the coumarin core acted as the reporter upon activation of the probe, displaying strong fluorescence emission in the low energy region of the spectrum, thus minimizing background interference from biological samples. Notably, the fluorescence ‘turn-on’ effect was triggered by a tandem reaction, where the initial reduction of the quinone moiety by thiol addition was followed by a quinone-methide rearrangement that released the fluorogenic coumarin core. This mechanism was further confirmed spectroscopically by the linear relationship between fluorescence intensity and thiol concentration, and competitive inhibition with N-ethylmaleimide. Importantly, the probe was shown to be insensitive to various non-thiol amino acids and other biological reductants.
Subsequently, the same concept was demonstrated in vitro by Chi et al., by means of a tetraphenylethylene-based fluorophore linked to a benzoquinone.11 Using a slightly different chemical design, the reporter was directly connected to the reactive site and the fluorophore was not released upon reaction with the analytes. Instead, the activation of the probe proceeded through a thiol–ene click reaction, which led to the reduction of the quinone structure towards the hydroquinoidal form. Remarkably, good selectivity for thiol-containing amino acids and rapid response times (i.e., shorter than 1 min) were achieved. These properties enabled the use of the probe for in vitro imaging of murine nerve cells by confocal fluorescence microscopy. In those experiments, the probe was found to be cell-permeable and displayed strong blue fluorescence emission upon reaction with intracellular biothiols, without being activated by other amino acids lacking free thiol groups.
Although selective and effective in different biological environments, the above mentioned probes displayed limits of detection in the low μM range, which might not be optimal for their in vivo application. The first quinone-based fluorescent probe capable of detecting submicromolar levels of biothiols was prepared by McCarley et al.12 The compound contained a reactive trimethyl benzoquinone and a napthalimide reporter (λem.: 540 nm), and was shown to detect low concentrations (i.e., in the nM range) of GSH, Cys and Hcy under physiological conditions and with a rapid response (i.e., around 5 min). Similarly to the above mentioned coumarin-based probe, the quinoidal moiety was eliminated upon reduction, leading to the release of the free fluorophore. The main advantages of this probe (e.g. rapid and selective reaction with biothiols, low limit of detection) were demonstrated by performing imaging experiments in human cells. High-magnification images of human H596 lung cancer cells were acquired and compared to those incubated with the thiol scavenger N-ethylmaleimide, corroborating the selective activation mechanism.
Another challenge in the field of biothiol sensing is the selective detection of single thiol-containing analytes in the complex cellular environment. In this regard, Zhao et al. synthesized coumarin-quinone probes (1) with high selectivity for GSH over other biothiols.13 The diethylamino-containing coumarin fluorophore (λem.: 485 nm) was directly linked to a benzoquinone unit, which underwent reduction upon GSH addition to block its quenching ability (Fig. 1A). Selectivity was first attributed to the intramolecular cyclisation of the cysteine amino group of GSH onto a carbonyl group on the GSH-probe adduct to afford a six-membered thiazine heterocycle, the formation of which was confirmed by mass spectral analysis. However, at physiologically relevant temperatures (i.e., 37 °C) the intermediate tautomerised to the phenol form as the thermodynamically stable product, being equally responsive to both GSH and Cys. Therefore, the ability to discriminate between the two analytes was the result of a discrepancy in the molecular energies of the two different adducts. The probe demonstrated excellent response times (i.e., shorter than 1 min) with a limit of detection in the submicromolar range. The authors also demonstrated the applicability of the probe for live cell imaging by confirming marginal cytotoxicity and acquiring fluorescence confocal microscope images in human HeLa cells. These examples highlight the broad applicability of the benzoquinone structure as a reactive moiety for the development of fluorescent probes for imaging biothiols. Overall, benzoquinone adducts display quick response times, high sensitivity and selectivity, as well as good permeability for cell imaging applications.
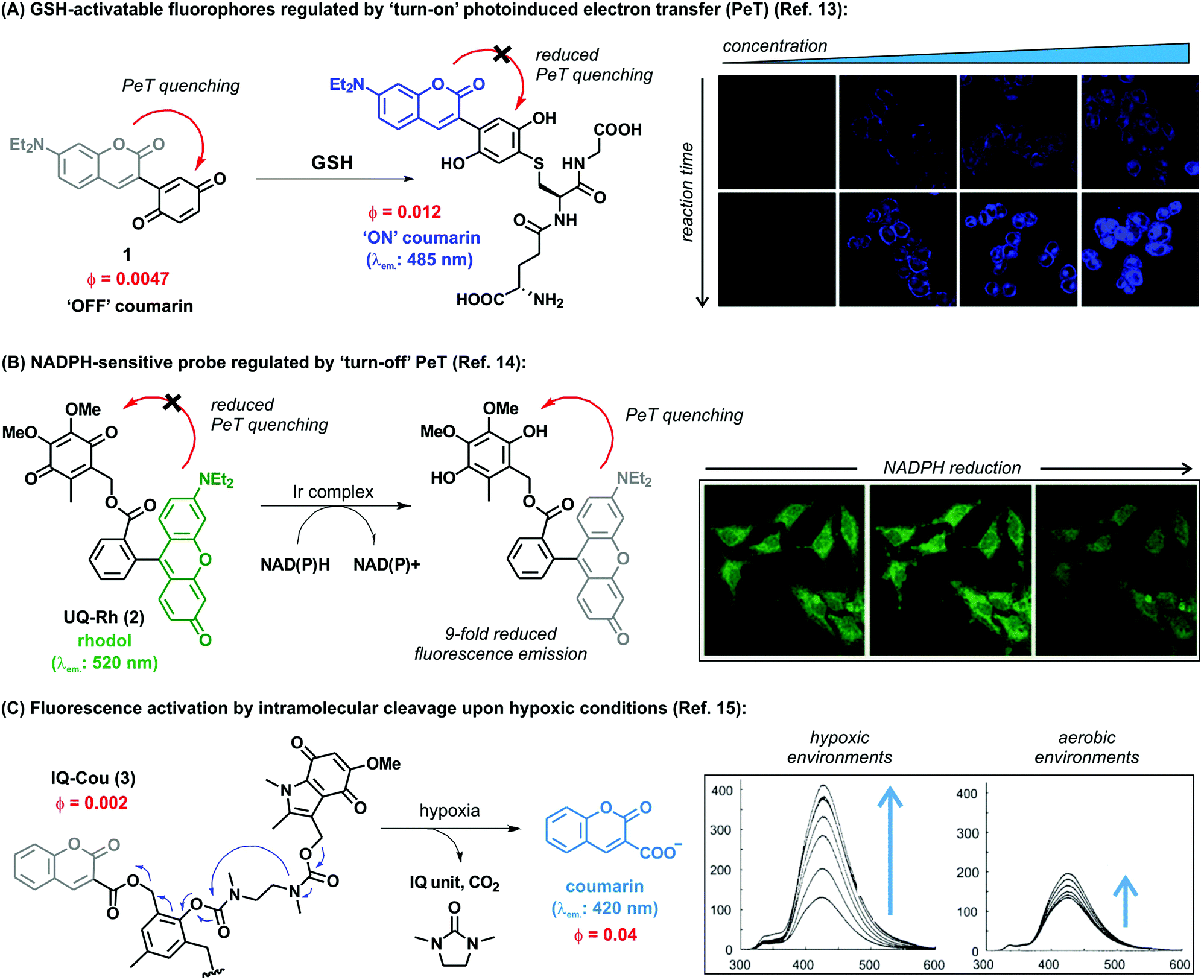 | ||
| Fig. 1 (A) Proposed fluorescence activation mechanism for the coumarin-benzoquinone probe (1) upon reaction with GSH. Confocal microscope fluorescence images of HeLa cells upon incubation with increasing concentrations of the probe and for different incubation times. (B) The ubiquinone-rhodol UQ-Rh (2) and its reactivity for NAD(P)H, leading to a ‘turn-off’ decrease in fluorescence emission. Confocal microscope fluorescence images of HeLa collected over the time-course of 4 minutes, where a 9-fold decrease in fluorescence intensity is observed upon deactivation of the rhodol fluorophore. (C) Simplified activation mechanism of the indolequinone-coumarin probe IQ-Cou in hypoxic conditions, resulting in the release of a fluorescent coumarin scaffold. Fluorescence spectra of IQ-Cou (3) upon X-ray irradiation in hypoxic (left) and aerobic (right) conditions. Images reproduced with permission from Elsevier (ref. 13), and Wiley-VCH (ref. 14 and 15). | ||
Alongside the recognition of biothiols, quinone-based fluorophores have been also constructed for the detection of the nicotinamide adenine dinucleotide derivatives, such as NADH and NADPH, which play key roles in cellular metabolism as electron carriers. NADH is generated by the tricarboxylic acid cycle in a ratio of three molecules per cycle, while NADPH is the phosphorylated form involved in photosynthesis and glycolysis. In addition to their important physiological function, these dinucleotides have been also investigated as potential therapeutic agents for the treatment of neurodegenerative diseases. Upon activation, NAD(P)H act as reducing agents. Consequently, quinone activatable fluorescent probes have been designed by exploiting the reduction of quinone moieties for their detection in biological environments. Unlike the above described thiol-sensitive probes, these probes displayed a decrease in their emission upon NAD(P)H recognition and generation of the hydroquinones, since these attenuate the fluorescence via photoinduced electron transfer (PeT). One remarkable example was reported by Komatsu et al. with a rhodol fluorophore (λem.: 520 nm) conjugated to a reactive ubiquinone.14 Since NAD(P)H requires the presence of a promoter to work as a reducing agent, the [(η5-C5Me5)Ir(phen)(H2O)]2+ Ir complex was used to allow the activation of NAD(P)H. The ubiquinone-rhodol fluorescent probe UQ-Rh (2) responded to intracellular NAD(P)H with around 9-fold decrease in fluorescence intensity in the presence of the Ir promoter (Fig. 1B). NAD(P)H activation was imaged in situ in HeLa cells following addition of the Ir complex, after which the fluorescence intensity rapidly decreased. This work represents an interesting example of quinone-based probes for analysing the concentration of NAD(P)H in cells and its implications in tissue homeostasis.
Most cellular functions rely on the continuous and adequate supply of oxygen from blood vessels. A steady oxygen level is preserved in normal tissues by the so-called oxygen homeostasis, whose alteration can lead to hypoxia. Hypoxic cells are characteristic by having a deficiency in oxygen supply and relevant in multiple diseases, including malignant solid tumours, inflammatory diseases and ischemia. In particular, hypoxic tumours have been found to show enhanced resistance to conventional therapeutic approaches and their targeting constitutes a highly unmet clinical need. As a result, there has been an increasing demand for hypoxia-specific fluorescent probes that can activate specifically in those environments. Notably, indolequinones can undergo one-electron reductive elimination under hypoxic conditions. Given the ability of quinones to quench fluorescence emission, the combination of indolequinones and fluorophores has been exploited to generate fluorescent probes to detect and visualize hypoxic microenvironments. This application was first described by Nishimoto et al., who conjugated an indolequinone to two coumarin chromophores via 2,6-bis(hydroxymethyl)-p-cresol linkers.15 The resulting IQ-Cou (3) probe showed weak fluorescence due to efficient quenching by the indolequinone via intramolecular PeT. However, upon reduction of the indolequinone in hypoxic conditions, the coumarin unit was released from the quencher and an intense fluorescence ‘turn-on’ effect at 420 nm was observed (Fig. 1C). The one-electron reduction of IQ-Cou (3) was shown to be suppressed by molecular oxygen in aerobic conditions, resulting in weak fluorescence emission. On the other hand, 4-fold fluorescence emission enhancement was observed in hypoxic conditions in vitro using HT-1080 human fibrosarcoma cells. In these experiments, the authors demonstrated that IQ-Cou (3) could be activated by intracellular reductase enzymes, representing a promising candidate for imaging hypoxia-specific biological environments. However, limitations of IQ-Cou (3) such as poor solubility in water and short absorption and emission wavelengths (380 nm and 420 nm, respectively) hinder the use for in vivo imaging applications. Improvements over these drawbacks were achieved by the groups of Tanabe and Nishimoto, who developed an alternative indolequinone probe utilising rhodol as the fluorophore.16 In fact, rhodol displays good water solubility as well as longer excitation wavelengths (i.e., around 550 nm) and relatively high fluorescence quantum yields. The rhodol-based probe IQ-R was used in A549 human lung adenocarcinoma cells, which are known to express high levels of NAD(P)H cytochrome P450 reductase. Cells were incubated with IQ-R under both hypoxic and aerobic conditions. While hypoxic cells displayed a bright fluorescence readout upon treatment with the probe, cells incubated in aerobic conditions displayed only minimal changes in fluorescence over the background. This different behaviour allowed them to distinguish between these two closely-related cell populations, being the first example of hypoxic cell imaging by means of a quinone-derived activatable fluorophore.
The variety of analytes that can be detected by using quinone-based fluorophores demonstrates their versatility, which extends beyond the biomarkers and fluorophores described in this section. In fact, in addition to numerous small fluorescent molecules, quinones have been also successfully incorporated to quantum dots (QDs) to generate bright and photostable fluorescent agents. QDs are small semiconductor particles with excellent potential for optical imaging due to their unique photophysical properties. In particular, QDs exhibit narrow and tunable emission profiles as well as excellent photostability and brightness. One important example is represented by ubiquinone–QD bioconjugates for in vitro detection of the intracellular complex I, an enzyme whose impaired activity has been associated with the progression of Parkinson's disease.17 In this example, ubiquinones were endowed with disulphide groups and conjugated to CdSe/ZnS QDs to afford molecular probes for imaging complex I in human neuroblastoma SH-SY5Y cells. Upon reduction of the terminal ubiquinone moiety by the NAD(P)H:complex I, the quenching ubiquinone was ‘switched off’ and an enhanced fluorescence signal was detected (more details in Section 5). The application of quinone-functionalised QDs for the detection of specific enzymes has been also demonstrated by Wu et al.,18 who investigated the NAD(P)H:quinone oxidoreductase isozyme 1 (NQO1). As detailed in the following section, NQO1 is a critical enzyme in cancer progression, being overexpressed in several types of cancer cells. QD probes containing Mn:ZnS reporters and trimethylquinone-propionic acids as reactive sites were prepared. The activation mechanism of these conjugates relied on an intramolecular cyclisation reaction facilitated by the reduction of the trimethylquinone core, which led to the activation of the QDs and the observation of increased fluorescence emission in human A549 cells.
3. Trimethyl lock quinones to monitor enzymatic processes
Taming the reactivity of quinone-based chemical probes within biological systems is a remarkable challenge due to the complexity of the intracellular environment. Chemical control over the reactivity of biomolecules in a temporal and spatial manner can be attained with the trimethyl lock system, an o-hydroxydihydrocinnamic acid derivative that undergoes rapid lactonization under physiological conditions to yield a dihydrocoumarin derivative. Its name arises from the three interlocking methyl groups that are responsible for its fast reaction rate. The lactonization can be triggered by enzymatic, chemical or photolytic processes. This key reactivity was first identified by Cohen et al. and it is the hallmark of this family of compounds.19Nowadays, the use of the trimethyl lock system represents an efficient and elegant chemical strategy to trigger the release of fluorescent compounds in well-defined biological environments and to enable the monitoring of specific intracellular events. As previously described, quinones have also the ability to quench fluorescence emission, and fluorescent trimethyl lock quinone-based probes have been developed to exploit this property for optical imaging. Despite the complexity of the biological mechanisms involved in the activation of these molecules, the simple underlying concept is the release of a fluorescent species from a non-fluorescent precursor after enzyme activation, such as NQO1 (Fig. 2A).
 | ||
| Fig. 2 (A) General overview of enzymatic prossesses involving trimethyl lock quinones. (B) Schematic representation of the release of fluorescent reporters after reaction with NQO1. (C) Chemical probes including different fluorophores with diverse spectral properties. | ||
NQO1, also known as DT diaphorase, is a detoxifying enzyme present in low levels in most healthy tissues, but it is often over-expressed after exposure to quinones producing reactive oxygen species (e.g., quinones found in some foods, pollutants and cigarette smoke). NQO1 expression in normal cells after exposure to specific quinones is increased by c-fos/AP1, c-jun and the NF-E2-related factor 2 (NRF2) transcription factors, which regulate the NQO1 gene at its hARE (human Antioxidant Response Element). NQO1 is over-expressed in many human solid cancers, including prostate, breast, colon, pancreatic and head and neck cancers. Whereas the mechanisms behind NQO1 over-expression in cancer formation, growth and metastasis have not been entirely elucidated, there is evidence that NQO1 stabilizes the protein p53, increases the efficacy of hypoxia-inducible factor 1 alpha (HIF1-α), and increases the metastatic capacity of cancer cells.20 Studies in melanoma cells also suggest that NQO1 oxidoreductase could protect cancer cells from oxidative stress and promote their proliferation by increasing expression of cyclins A2, B1 and D1. Moreover, NQO1 can enhance the activity of hypoxia-inducible factors (HIFs), which adapt cellular functions to low oxygen concentration conditions.21 Further investigation of the role(s) of increased NQO1 expression in carcinogenic induction and progression are currently under investigation, and NQO1 is a promising target for the development of anticancer therapies based on bioactivatable prodrug quinones. In this context, McCarley et al. have investigated the use of the trimethyl lock quinone systems for the interrogation of enzymatic processes involving NQO1 and delivered important contributions to the field.22–25
Trimethyl lock quinones have been used to construct activatable reporter molecules for selective and sensitive detection of NQO1 activity. One of the first examples was a rhodamine-morpholino urea derivative, which was conjugated to a trimethyl lock quinone in order to generate activatable fluorescence emission.26 The probe was readily prepared by coupling a trimethyl lock quinone to a rhodamine derivative in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) using a mixture of DMF/pyridine as the solvent. Notwithstanding the ease of the synthesis, only a moderate 34% yield was attained. The activation mechanism relied on the reduction of the quinoidal moiety by NQO1 to afford the hydroquinone analogue, which underwent lactonization to release the fluorescent rhodamine derivative (Fig. 2B). Compound 4 exhibited poor fluorescence with a quantum yield lower than 1%, whereas the final released molecule 6 displayed around 100-fold increase and bright fluorescence emission (e.g. quantum yields around 50%).
Optimal trimethyl lock quinone-based probes should display (a) rapid cell uptake, (b) selective enzymatic activation, (c) low fluorescence quantum yields for the unactivated probes and high fluorescence emission upon release of the reporter, (d) good retention of the reporter inside the target cells and (e) significant differences in energies of absorption maxima (λabs. probe vs. λabs. reporter) and emission maxima (λem. probe vs. λem. reporter) to maximize signal-to-background ratios.25Fig. 2C features several probes which release fluorescent reporters upon specific stimuli and display the properties listed above, being suitable candidates for direct imaging of enzymatic biological processes. The naphthalimide derivative 7 was designed for the detection and visualization of intracellular enzymatic processes involving NQO1 with high sensitivity and minimal cytotoxicity, even at higher concentrations than those used for biological experiments. The enzymatic reduction of the quinoidal system – which quenched the naphthalimide core by PeT in the intact probe – released bright green fluorescence emission, enabling the detection of NQO1 in cancer cells.22
Despite rather basic chemistry is mostly sufficient for the preparation of the probes in this research area, the intrinsic complexity of biological systems poses challenges to the identification of good reporter-quinone pairs. In addition to naphthalimides, the rhodamine scaffold has been also successfully used for NQO1 detection. The bright fluorescence emission of rhodamine – with fluorescence quantum yields over 80% – enabled the acquisition of microscope images of NQO1-expressing tumor-derived colorectal and ovarian cancer cells with unprecedented signal-to-background ratios.22–25
Imaging agents emitting fluorescence in the near-infrared region (i.e., 650–900 nm) enable deeper tissue penetration with low phototoxicity and high signal-to-background ratios due to minimal tissue autofluorescence, which are necessary for in vivo imaging studies.27,28 In this context, near-infrared probes including trimethyl lock quinones have been reported by the groups of McCarley, Zhang and Yi (Fig. 3).29,30 These compounds have remarkable translational potential for the visualization of solid tumors and the determination of tissue resection borders during intraoperative imaging in cancer surgery. The probes were prepared through synthetic routes coupling trimethyl lock quinones and selected near-infrared reporters so that the latter would be released into the media upon enzymatic recognition. Initially, the heterocyclic reporter tricarbocyanine 16 was prepared in two steps from 1-ethyl-2,3,3-trimethyl-3H-indol-1-ium iodide. Subsequently, the trimethyl lock derivative 15 was chlorinated and then linked to the cyanine unit via a N-alkylation (Fig. 3A).29 Notable features of the resulting probes were their near-infrared emission wavelengths, long Stokes shifts, and reasonable photostability. The electron-deficient nature of the trimethyl lock quinone was important to exert effective PeT quenching on the tricarbocyanine scaffold and to maintain the reporter unit intact before its reaction with the NQO1 enzyme. Notably, very large differences in the spectral properties before and after enzymatic reaction were observed (e.g. λabs. (16): 606 nm vs. λabs. (17): 786 nm; λem. (16): 755 nm vs. λem. (17): 798 nm). Furthermore, the release of the fluorophore upon interaction with the enzyme resulted in a 14-fold increase in the fluorescence quantum yields. The low background fluorescence of the fluorogenic precursor together with the significant ‘turn-on’ effect resulted critical to obtain high signal-to-background ratios in cell imaging studies. Bioimaging assays were performed in colorectal carcinoma cell lines (HT-29) and ovarian cancer cell lines (OVCAR-3 and SHIN3), which express high levels of the enzyme NQO1. H596 non-small lung carcinoma cells were used as negative controls expressing low levels of NQO1. Microscope cell images confirmed brighter fluorescence emission in HT-29 (NQO1+), OVCAR-3 (NQO1+), SHIN3 (NQO1+) whereas H596 cells (NQO1−) presented very low fluorescence intensity. Compound 17 was recently used for in vivo imaging of ovarian cancer in a mouse model upon activation by hNQO1, enabling the identification of cancerous lesions with minimal interference from tissue autofluorescence.31
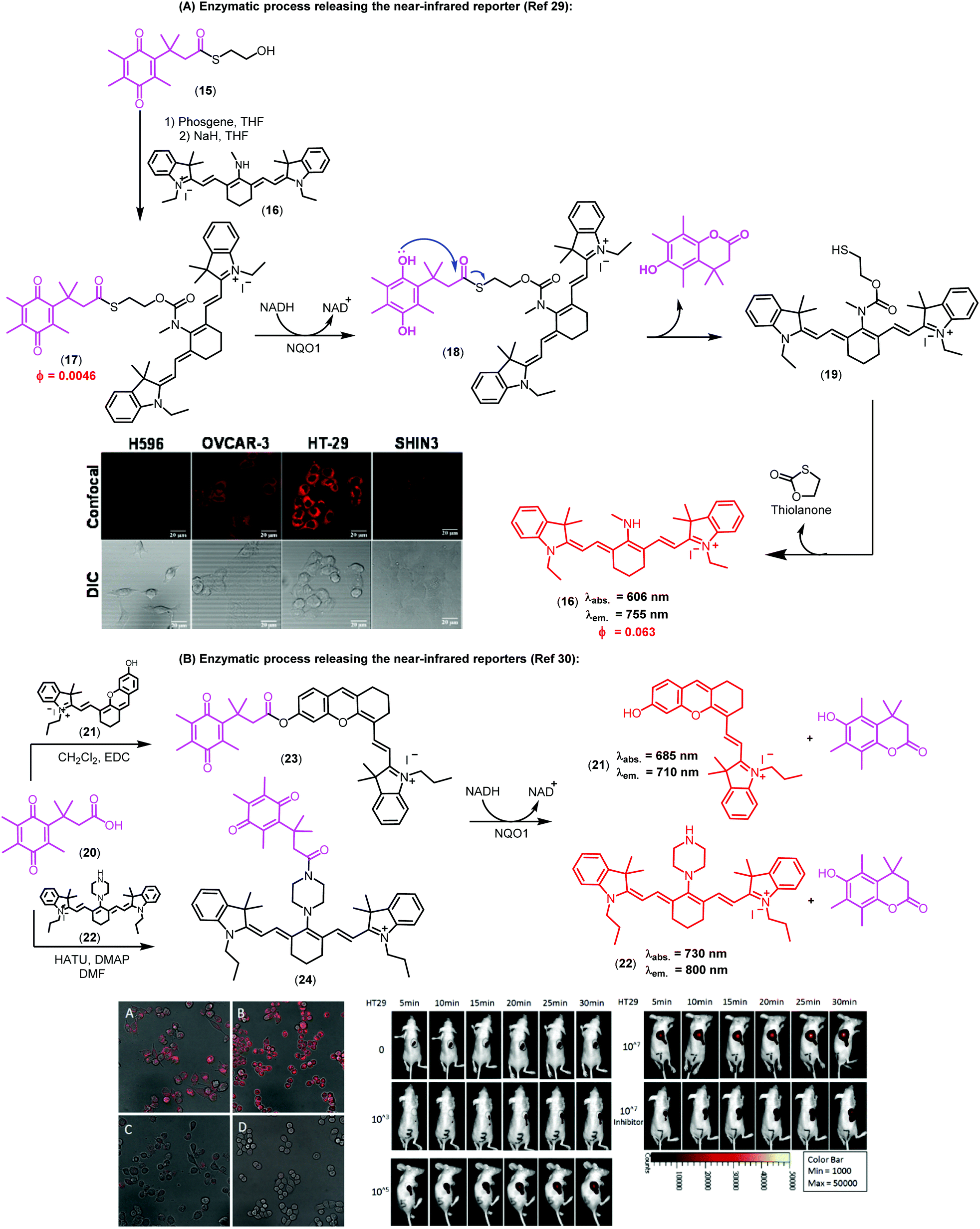 | ||
| Fig. 3 Trimethyl lock quinone fluorescent probes. (A) Mechanism of activation for NQO1-sensitive tricarbocyanine probes and their use in confocal and differential interference contrast images of H596 (negative), OVCAR-3 (positive), HT-29 (positive), and SHIN3 (positive) cells. Fluorescence images were obtained using 633 nm excitation and collecting fluorescence emission in the 660–820 nm range. Scale bar: 20 μm. (B) Microscopy images of NQO1-positive MB231-1640 cells (A and C) and HT29 cells (B and D) in the absence (A and B) or in the presence (C and D) of the inhibitor dicoumarol together with probe 24. Cancer cell detection limit and sensitivity of the probe (24) in living mice. Fluorescence images of mice after injection of 107, 105, 103 and no HT29 cells at different time points. Mice were also pre-treated with dicoumarol, in the case where they were administered 107 HT29 cells. Images reproduced with permission from American Chemical Society (ref. 29) and Elsevier (ref. 30). | ||
In the context of near-infrared fluorophores, Zhang and Yi recently described two novel probes (23 and 24, Fig. 3B) for the detection of NQO1 activity in vitro and in mice. These near-infrared ‘turn-on’ probes relied on activation by intramolecular charge transfer (ICT) and PeT mechanisms using merocyanine and tricarbocyanine structures.30 The synthesis was accomplished in a single step and designed to release the near-infrared reporters after enzymatic reduction (Fig. 3B). Compound 24 showed a 33-fold fluorescence enhancement after NQO1 treatment and marginal cytotoxicity, being further applicable to in vitro imaging of human colorectal and breast cancer cells as well as in vivo whole-body imaging in mouse models.
In the last decade, in vivo imaging studies have been revolutionised by the emergence of two-photon fluorescence microscopy, which allows deep imaging of biological specimens with low phototoxicity. Recently, the groups of Cui, Kim and Yoon have described a two-photon fluorescent probe for NAD(P)H:quinone oxidoreductase detection in which an aminoacetyl-naphthalene derivative was conjugated to a trimethyl lock quinone.32 In this case, the interaction of the probe with hNQO1 led to the release of a two-photon compatible reporter with an emission wavelength around 540 nm, as demostrated in assays for the detection of NQO1 in living cells. The authors also corroborated the specificity in cells expressing high and low levels of NQO1. Specifically, HT29 colorectal cancer cells displayed a strong fluorescence ‘turn-on’ effect, whereas NQO1-negative breast cancer cell lines (i.e., MDA-MB-231 and MDA-MB-468) showed negligible fluorescence response (Fig. 4A).
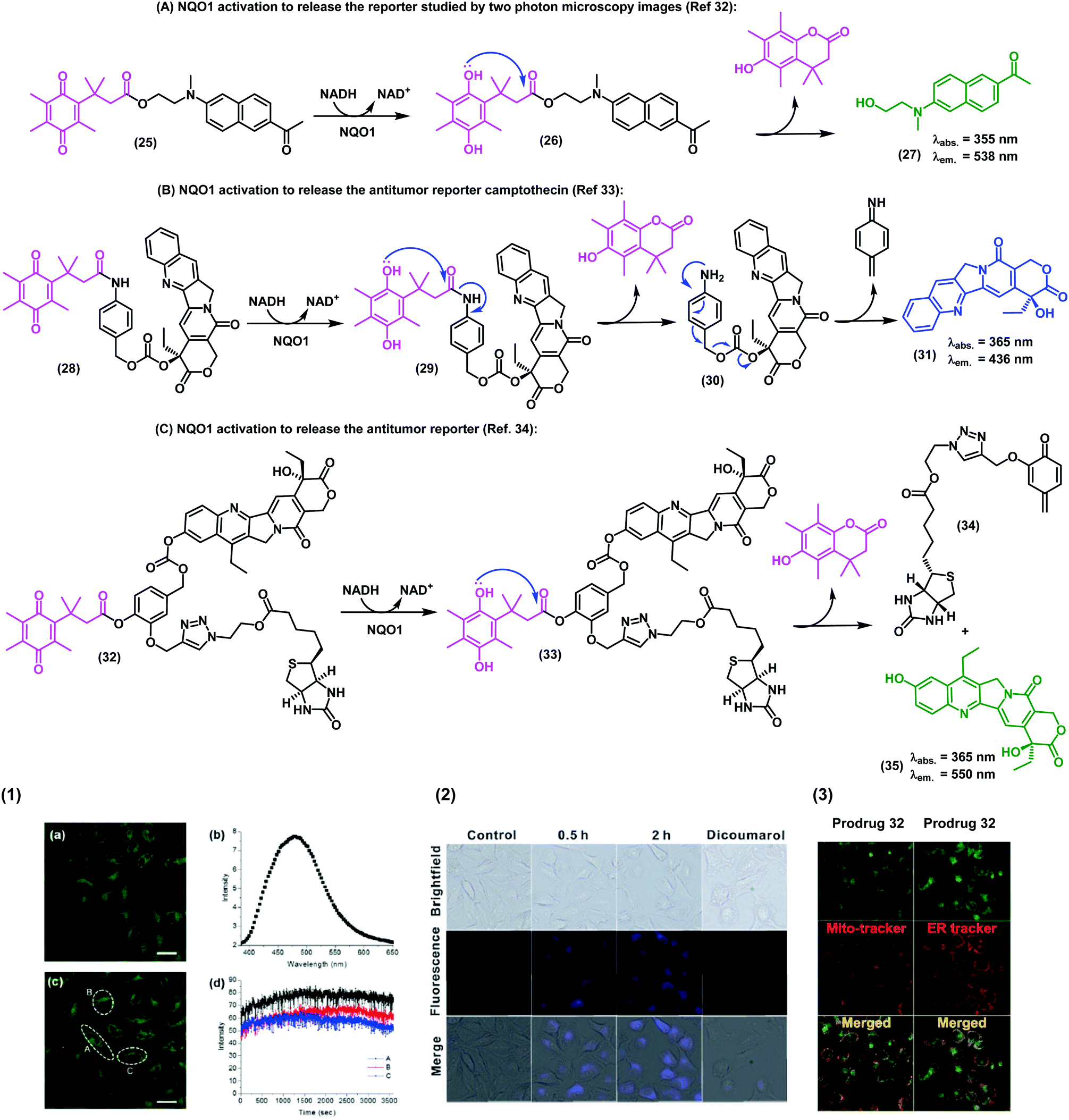 | ||
| Fig. 4 Synthesis of the trimethyl lock imaging and theranostic probes targeting NQO1 and respective characterisation studies. (1) Two-photon microscope images of NQO1-positive HeLa cells (a and c), fluorescence spectrum (b), and time-course fluorescence (d) after incubation with the probe 25 (1 μM) for 30 min. (2) Fluorescence images of A549 cells in the absence (control) or in the presence of the prodrug 28 (10 μM) at different incubation times. For the inhibition experiments, A549 cells were pre-treated with dicoumarol (20 μM), then the prodrug 28 (10 μM) was added and incubated for another 2 h. (3) Subcellular localisation of compound 32. Fluorescence co-localisation studies with Mito-tracker Red FM and ER-tracker Red in A549 cells, where HeLa cells were treated with the prodrug 35 (10 μM) for 24 h. Images reproduced with permission from Royal Society of Chemistry (ref. 32 and 33) and American Chemical Society (ref. 34). | ||
In addition to the visualization of disease biomarkers, fluorescent probes can also be used for real-time quantitative monitoring of drug localisation (e.g. anticancer therapy) in targeted cells. Some anticancer drugs do contain inherent chromophore groups that enable direct fluorescence imaging in cells, and their conjugation to trimethyl lock quinones has been used to generate prodrugs and monitor NQO1-dependent release in cells. The groups of Zeng and Wu reported a NQO1-activatable theranostic prodrug (28) containing the anticancer drug camptothecin, which is an inhibitor of the enzyme topoisomerase I (Fig. 4B).33 In this case, the emission of compound 28 was quenched by PeT, and fluorescence was only observed when the prodrug was cleaved and camptothecin was released to the intracellular space. Cell viability assays indicated that camptothecin on its own was not able to exert enough cytotoxicity, highlighting the crucial role of the quinone propionic acid moiety. This chemical strategy opens multiple avenues for the use of NQO1-activatable theranostic prodrugs to generate enzyme-dependent antitumor drugs that effectively target cancer cells overexpressing NQO1 with reduced side effects and toxicity in other cell types.
Shin et al.34 developed an efficient chemical strategy for enzyme-triggered targeted therapy (Fig. 4C) by making use of trimethyl lock quinone prodrugs. In addition to the NQO1-activatable component, the prodrugs included a biotin molecule as a cancer-targeting unit to enhance the selective uptake of the prodrug in cancer cells expressing a biotin receptor in their surface. Once internalised, the trimethyl lock system was activated by NQO1 to release the cytotoxic drug camptothecin only in target cancer cells. The synthesis of these complex architectures was achieved in 9 steps, including a copper-catalyzed alkyne–azide cycloaddition to form a triazole bond between the ligand (i.e., biotin) and the drug (i.e., camptothecin). Finally, the trimethyl lock quinone was incorporated via an aromatic linker to assemble these sophisticated theranostic quinone-based prodrugs.
The latest addition from McCarley et al. in trimethyl lock based turn-on probes for the detection of NQO1 in cancer cells involved a rhodamine fluorophore (λem.: 520 nm) that was activated via hydrolysis of the spyrocycle moiety. The compound displayed 200-fold fluorescence increase after interaction with the enzyme target, which facilitated fluorescence microscopy experiments of colorectal and ovarian cancer cell lines with excellent signal-to-background ratios.35
Despite the fast-growing development in this area of research, the generation of probes based on the trimethyl lock system is still incipient and often requires multidisciplinary approaches in chemical biology. In particular, the discovery of probes that release stable and efficient near-infrared reporters for in vivo imaging remains challenging. The challenges of developing activatable near-infrared probes which can be obtained through simple synthetic methods and possess optimal spectral properties are reflected in the paucity of reported examples in the literature.
4. Turning non-fluorescent quinone scaffolds into fluorescent therapeutic probes
Nature is an endless source of inspiration for chemists due to the broad complex array of chemical structures found in natural products. Natural products have been used in therapy since ancient times, and are still currently an important chemical resource in the development of drugs for numerous diseases. Lapachones (Fig. 5A) and their derivatives are quinone-based natural substrates widely used as therapeutics for different applications.36–38 Because these quinones are typically non-fluorescent and therefore unsuitable for direct visualization of therapeutic targets and/or interrogation of mechanisms of action or gene expression,39 a number of approaches have been pursued to incorporate appropriate chemical modifications to quinoidal structures. The straightforward generation of fluorescent heterocycles such as phenazines, oxazoles and imidazoles from lapachone-type quinoidal derivatives has emerged as an important strategy to produce new disease biomarkers.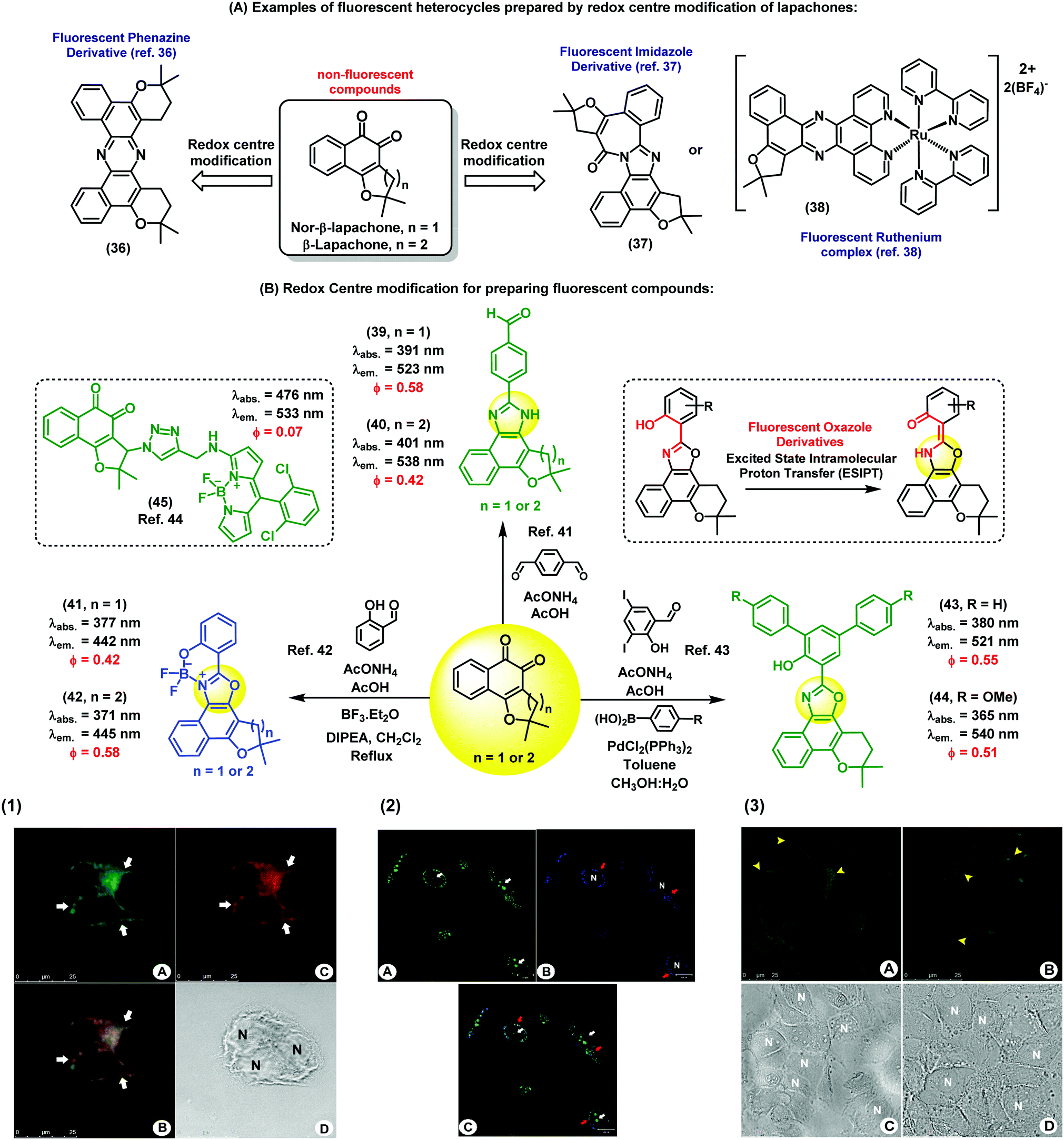 | ||
| Fig. 5 Fluorescent probes obtained from the C-ring modification of lapachones. (1) Fluorescence microscope images of MCF-7 cells incubated with compound 40 and Mitotracker. Image A shows the staining profile of compound 40 and image C shows the staining profile of Mitotracker. The white arrows indicate the perinuclear staining accumulation for all markers used. Image B displays the overlay of both green and red fluorescence channels, showing co-localisation in yellow. Cell nuclei are identified in the brightfield image D with the letter N. Scale bar: 25 μm. (2) Fluorescence microscope images of live PC3 cancer cells upon staining with compound 41 (blue). As a positive control, cells were stained with acridine orange (green). Compound 41 was localised within subcellular organelles corresponding to the endocytic pathway. Red arrows point at the staining of lysosomes, and white arrows point at the nucleus stained with acridine orange (non-specific staining). Scale bar: 20 μm. (3) Fixed Caco-2 cells stained with compound 44 and a commercial available BODIPY dye. (A) Cells stained with compound 44 showing its accumulation in the lipid droplets (yellow arrowheads) and in some areas of the cytoplasm. (B) Lipid droplets are also stained with BODIPY dyes. (C) and (D) correspond to the brightfield images. N: nucleus, scale bar: 25 μm. Images reproduced with permission from Wiley-VCH (ref. 41) and Royal Society of Chemistry (ref. 42 and 43). | ||
Brinn et al. have used β-lapachones as the starting materials in the synthesis of symmetric phenazines (Fig. 5A).36 This fluorescent nitrogen-containing heterocycle was prepared in a single step by reacting the lapachone precursor with an ammonia source. Advantageously, compound 36 displayed bright fluorescence with a quantum yield around 40%, being an optimal fluorophore for bioimaging. Similarly, Pinto et al. have reported the synthesis of complex fluorescent diazaazulenones37 from the reaction of lapachones with ammonium acetate in acidic conditions. More recently, our group has reported cyclometallated ruthenium complexes from naturally-occurring lapachones. The investigation of their photophysical properties revealed emission maxima in the red region of the visible spectra (λem.: 620 nm), being promising candidates for subsequent applications in fluorescence imaging.38 In these examples, the common synthetic approach is based on the modification of the redox centre of the lapachone core to prepare fluorescent heterocycles. Alternatively, aldehyde-containing dyes can be also synthesized from non-fluorescent lapachones. Some of these compounds possess multiple emission bands, which is a remarkable feature for the preparation of fluorescent ratiometric probes or the design of fluorescent dye cocktails.40 As shown in Fig. 5B, a number of derivatives displayed fluorescence emission in the blue (e.g. between 420 and 450 nm) and green (e.g. between 510 nm and 560 nm) regions of the visible spectra. Fluorescence microscope images of both live and fixed cells upon incubation with compounds 39 and 40 showed intracellular patterns resembling mitochondrial staining, with preferential accumulation in areas around the nucleus and slight diffusion throughout the cytoplasm.41 Co-localisation experiments with the commercially available dye Mitotracker corroborated the fluorescence localisation of compounds 39 and 40 in the mitochondria of human MCF7 breast cancer cells. This work represents the first chemical synthesis of lapachone-derived imidazoles and their bioimaging application as selective fluorescent markers of mitochondria in live cells.
Another important example of lapachone-derivatisation using the same redox centre modification strategy has resulted in the preparation of fluorescent naphthoxazoles and their boron derivatives. These compounds are useful markers to monitor endocytic pathways in live PC3 cancer cells by fluorescence microscopy.42 Furthermore, comparative assays with acridine orange suggest that these new fluorophores present better selectivity for the internalisation of endocytic processes. The boron-containing analogues showed enhanced spectral properties as efficient π-conjugated systems, including large Stokes shifts (i.e., around 70 nm) and high fluorescence quantum yields. Compounds 41 and 42 were capable of selectively staining different organelle structures in the canonical endocytic pathway (i.e., early endosomes, late endosomes and lysosomes) without causing cell toxicity. These probes enabled dynamic studies of the endocytic trafficking in live cells and direct visualisation of endocytosis through caveolar vesicles and the canonical pathway. As a result, the simple and efficient synthesis of these nature-inspired lapachone derivatives represents a valuable chemical strategy to produce new tools to study complex cellular uptake processes by means of fluorescence imaging.
Other examples have been described towards chemical probes targeting specific intracellular microenvironments. For instance, oxazole-based compounds have been designed to target lipophilic environments.43 Initially, long hydrophobic alkyl chains were conjugated to the furan ring through triazole linkers, but the resulting products showed low efficacy for staining intracellular lipid structures. On the other hand, phenyl-substituted oxazoles (compounds 43 and 44) showed significant accumulation in lipid droplets. In order to confirm their intracellular localisation, compound 44 was compared to lipid-staining BODIPY dyes to analyse their intracellular staining patterns. The oxazole-lapachone displayed little photobleaching or degradation, indicating high chemical stability and photostability inside cells. These oxazole derivatives constitute an interesting family of fluorophores because of their high molar extinction coefficients and their ability to undergo ESIPT (excited-state intramolecular proton transfer), which often results in extraordinarily long Stokes shifts (i.e., over 140 nm).
The C-ring modification of lapachones has been also exploited for the preparation of theranostic fluorescent probes. For instance, a lapachone-based BODIPY hybrid (45) was recently reported as a novel theranostic construct with green fluorescence emission and potent antitumor activity.44 Confocal microscopy studies revealed the ability of this hybrid compound to stain mitochondria. Furthermore, studies to elucidate the cytotoxic activity and the mechanism of action of this hybrid were described. This adduct represents the first example of a fluorescent quinoidal system with both capabilities as a fluorophore and as a potent antitumor drug. Importantly, this report demonstrates that the intrinsic lack of fluorescence of the quinoidal structure can be overcome by conjugation to appropriately bright fluorescent scaffolds to generate highly fluorescent derivatives with therapeutic activity.
In summary, these examples prove the adaptability of lapachones to different chemical approaches introducing modifications at either the redox centre and/or the C-ring to generate fluorescent derivatives for imaging assays. The versatility of these approaches allows for the extension of π-conjugated planar systems, thus opening multiple avenues for the design of fluorophores with bespoke photophysical properties for different bioimaging studies.
5. Switchable quinone/hydroquinone pairs for bioimaging applications
Fluorophores endowed with quinone/hydroquinone (Q/HQ) pairs constitute another remarkable family of quinone-based molecules.45 These compounds are prototypical redox species with important roles in cells, particularly in electron transfer and energy conservation systems. In general, probes with Q/HQ pairs work by means of PeT activation/deactivation between a quinoid redox center (PeT acceptor) and a fluorescent reporter (PeT donor), which modulates the fluorescence emission. Different reporters have been employed so far, including BODIPY and cyanines among others, yielding probes for the imaging of redox processes with high sensitivity. The groups of Cosa and Jiang have described interesting examples of quinoidal systems based on the BODIPY scaffold (Fig. 6A).46,47 While Cosa et al. synthesized a para-quinone containing BODIPY (46), Jiang et al. prepared an ortho-quinone coupled to aza-BODIPY (48). In both cases, the reduction of the quinoidal moiety, generating HQs (47) and (49), respectively led to a significant increase in fluorescence. Additional examples using quinones found in natural metabolites have been also reported. An activatable fluorescent analogue of vitamin K was synthesized via coupling of a BODIPY core to menadione to render a fluorogenic probe showing 70-fold fluorescence increase upon reduction of the quinone moiety.48 The probe functioned as an electron shuttle in the presence of a NAD(P)H:quinone oxidoreductase, either NADP or NADPH and cytochrome C. Other protein complexes, such as intracellular complex I, can be also detected by monitoring the redox state with Q/HQ pairs. Long et al. described the synthesis and application of bioconjugates containing Q/HQ pairs attached to QDs for the monitoring of complex I levels in human neuroblastoma SH-SY5Y cells (Fig. 6B).17 Therein, the authors also demonstrated the potential use of these compounds for early-stage diagnosis of Parkinson disease, thus highlighting the potential of these nanosensors for future clinical applications.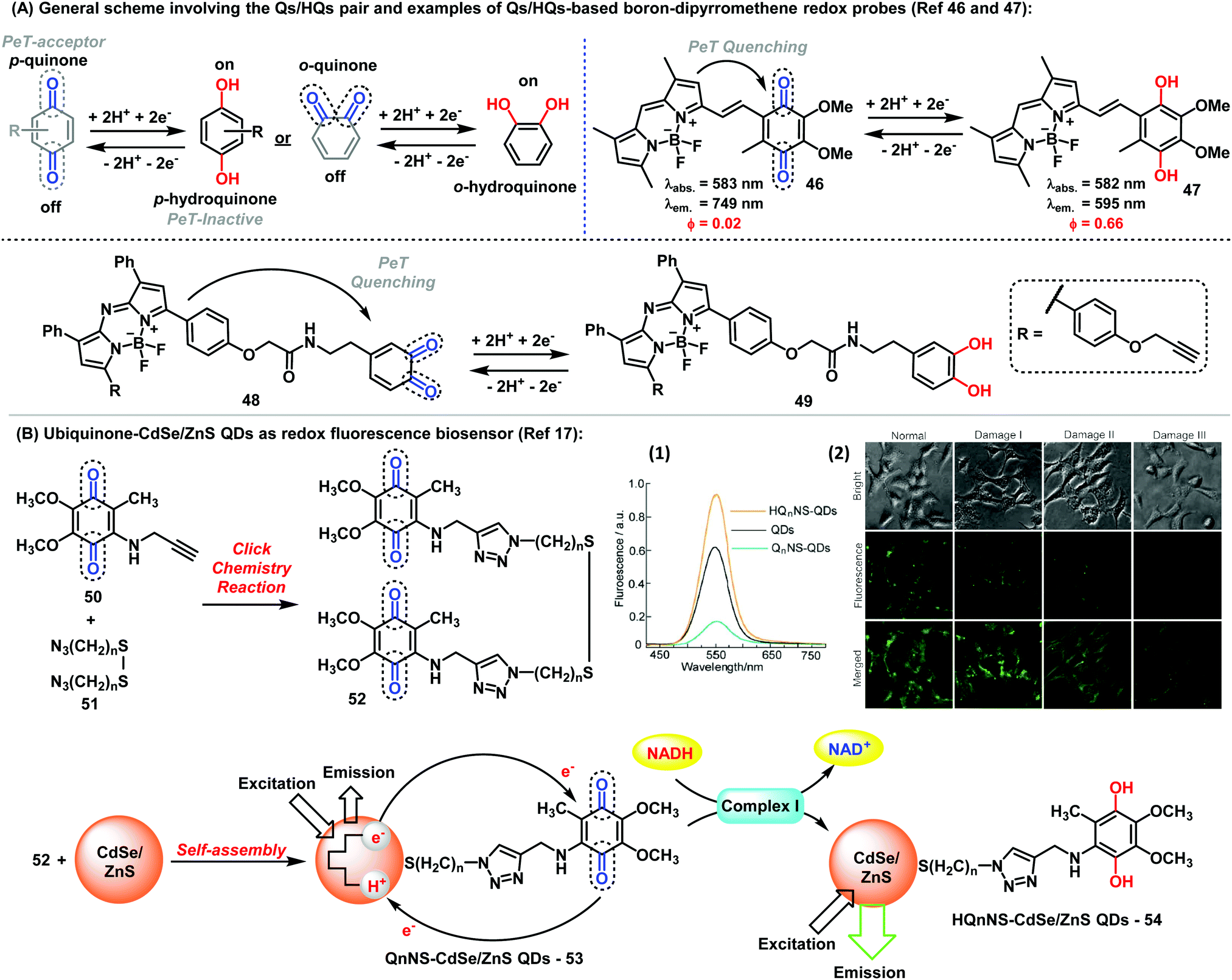 | ||
| Fig. 6 (A) General representation of the Q/HQ pairs and examples of BODIPY-based redox probes. (B) Preparation of ubiquinone-CdSe/ZnS QDs used as fluorescent redox nanoprobes. (1) Fluorescence spectra of ubiquinone/ubiquinol-functionalised CdSe/ZnS QDs. (2) Fluorescence microscopy images showing the intracellular complex I level using Q2NS-QDs. Brightfield and fluorescent images were collected from human neuroblastoma SH-SY5Y cells with Q2NS-functionalised QDs. The expression levels of complex I increased in human SH-SY5Y cells after 24 h or upon exposure to rotenone at 100 nM (Damage I), 500 nM (Damage II) or 1 μM (Damage III). Images reproduced with permission from Springer Nature (ref. 17). | ||
Recently, Chao et al. have described Ir(III)-anthraquinone complexes as two-photon probes (Fig. 7A). The authors demonstrated the ability of compounds 55–58 to act as mitochondrial markers.49 The phosphorescence of Ir(III) complexes was quenched by the quinoidal moiety, and restored under hypoxic conditions after a two-electron bioreduction process. The probes were activated by reductases in the presence of NADPH, which triggered the conversion of the anthraquinone moiety into the hydroquinone, rendering 26-fold increase in emission. These complexes also enabled imaging mitochondrial morphological changes in hypoxic environments. Since conventional mitochondrial imaging agents (e.g. MitoTracker dyes) show poor efficacy under hypoxic conditions, these molecules are a significant addition to the field. In solid tumors, several key processes take place under stress conditions involving low levels of nutrients and oxygen, including diffusion-limited hypoxia.
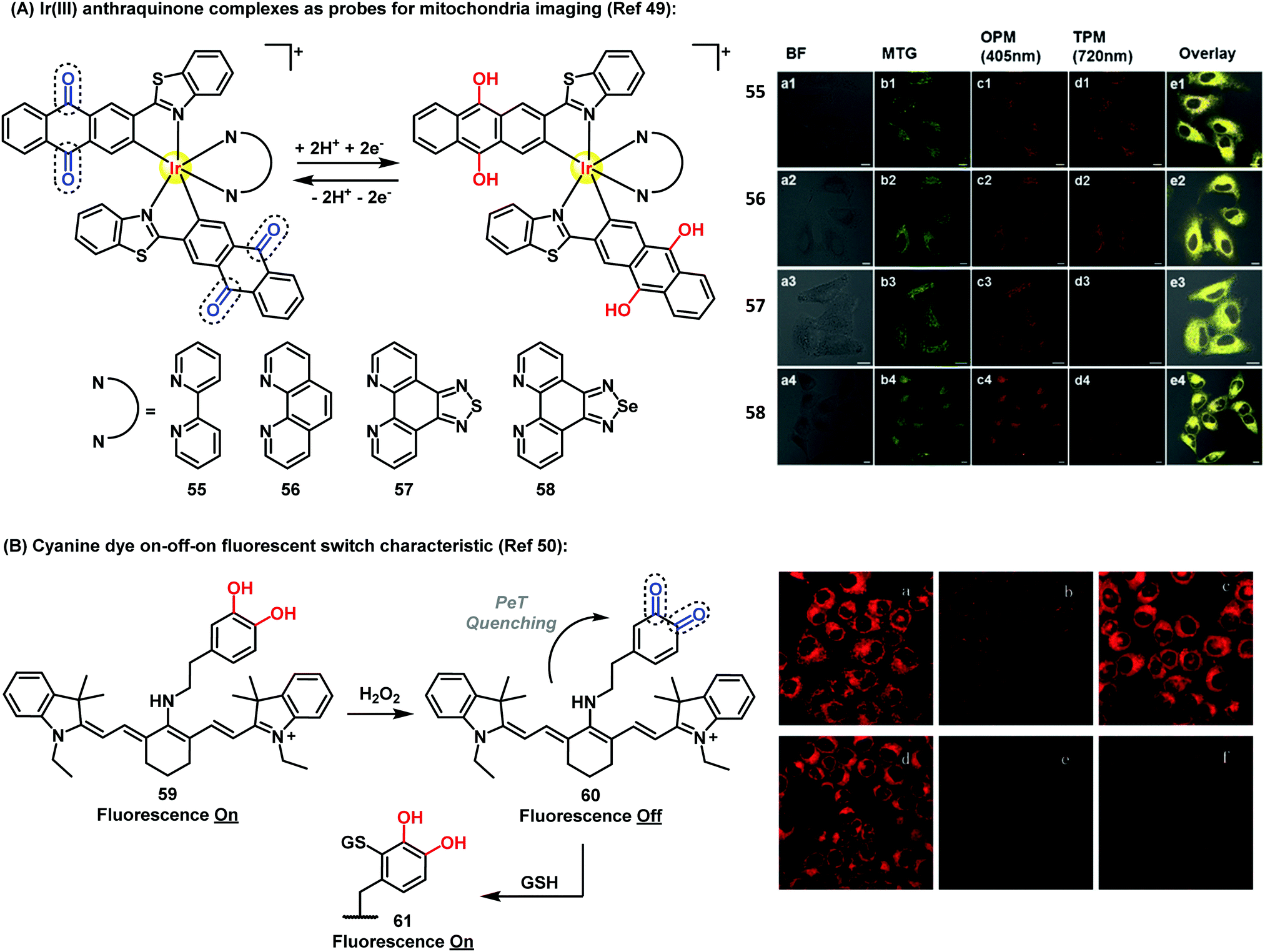 | ||
| Fig. 7 (A) Fluorescence microscopy images of A549 cells incubated with compounds 55–58 (5 μM) under hypoxic (1% O2) conditions for 30 min at 37 °C, followed by addition of 50 nM Mitotracker Green. Column a: brightfield images; column b: cells stained with Mitotracker Green; column c: OPM confocal phosphorescence images; column d: TPM confocal phosphorescence images; column e: overlay images of columns a-d; scale bars: 10 μm. (B) Fluorescence microscopy images of HL-7702 cells under H2O2 oxidative stress/thiol-induced repair: (a) HL-7702 cells loaded with 5 μM 59 for 3 min; (b) 59-loaded cells treated with 25 μM H2O2 for 9 min; (c) cells treated as described in (b) after culture for 3 h; (d) cells incubated for 30 min with N-ethylmaleimide (100 μM), then for 3 min with 59 (5 μM); (e) cells described in (d) after exposure to H2O2 (25 μM); (f) cells treated as described in (e) after additional culture for 3 h. Images reproduced with permission from Wiley-VCH (ref. 49) and Royal Society of Chemistry (ref. 50). | ||
Therefore, the use of efficient ‘turn-off’/‘turn-on’ compounds such as Q/HQ pairs represents an elegant strategy to develop probes for imaging intracellular events in live cells. Han et al. described a quinone-based cyanine derivative as an on–off–on fluorescent probe for monitoring oxidative stress and subsequent repair in live cells.50 In this example, the authors took advantage of the variations in fluorescence intensity of the cyanine (59) in oxidative and reductive environments. Compound 59 showed a fluorescence decrease and a color change from blue to purple in the presence of H2O2, then the fluorescence was gradually restored to upon reaction of quinone 60 with GSH via Michael addition (Fig. 7B).
6. Conclusions
The versatility of biological functions exhibited by the quinoidal compounds is well-documented and makes them privileged chemical structures for a wide variety of disorders, including cancer, malaria and leishmaniasis, among others. The main mechanisms by which quinones exert their action in cells involve the stimulation of oxidative stress and/or the alkylation of specific biomolecules. As a result, there has been growing interest in designing chemical strategies to synthesize novel quinone derivatives and to evaluate their properties in different biological systems. Quinones constitute a relatively simple chemical structure, and yet the development of new synthetic strategies presents remarkable challenges. The ability of quinones to participate in bioreduction and biooxidation processes enables their use for numerous applications in different fields, from cell biology to energy storage. Furthermore, the multiple features of quinones make them a ‘Swiss Army knife’-like chemical scaffold for the discovery of new materials, therapeutic agents and analytical probes. In this review, we covered the emerging rise of quinone-based fluorophores for imaging biological processes in multiple environments, from cells to in vivo animal models. Recent work describing the synthesis of fluorescent probes from naturally occurring quinones (e.g. lapachones) as well as their application to stain subcellular organelles or to monitor specific intracellular environments (e.g. hypoxia) exemplify the value of these novel fluorescent molecules. Over the years, the importance of the quinoidal framework in the preparation of chemical agents for medicinal purposes has been indisputable. Chemists are now facing the challenge of exploiting quinones to construct sophisticated structures (e.g. activatable fluorescent probes) that can bring new properties to this interesting family of chemical structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. N. da Silva Júnior acknowledges funding from CNPq (474797/2013-9, Chamada Universal 404466/2016-8, PVE 401193/2014-4 and PQ 305385/2014-3), FAPEMIG (Edital 01/2014 APQ-02478-14 and Programa Pesquisador Mineiro – PPM X), CAPES and INCT-Catálise. M. V. acknowledges funding from the Marie Curie Integration Grant (333847), the Biotechnology and Biological Sciences Research Council (BB/M025160/1), and The Royal Society (RG160289). The authors also acknowledge Prof. David Boothman (University of Texas) for helpful discussions and advice about NQO1.References
- F. L. Crane, Y. Hatefi, R. L. Lester and C. Widmer, Biochim. Biophys. Acta, 1957, 25, 220–221 CrossRef CAS
.
- P. Srinivas, G. Gopinath, A. Banerji, A. Dinakar and G. Srinivas, Mol. Carcinog., 2004, 40, 201–211 CrossRef CAS PubMed
- E. A. Blanche, L. Maskell, M. A. Colucci, J. L. Whatmore and C. J. Moody, Tetrahedron, 2009, 65, 4894–4903 CrossRef CAS
- P. I. T. Thomson, V. L. Camus, Y. Hu and C. J. Campbell, Anal. Chem., 2015, 87, 4719–4725 CrossRef CAS PubMed
- A. Fernández and M. Vendrell, Chem. Soc. Rev., 2016, 45, 1182–1196 RSC
- M. Vendrell, E. Angulo, V. Casadó, C. Lluís, R. Franco, F. Albericio and M. Royo, J. Med. Chem., 2007, 50, 3062–3069 CrossRef CAS PubMed
- F. Yraola, R. Ventura, M. Vendrell, A. Colombo, J. C. Fernández, N. de la Figuera, D. Fernández-Forner, M. Royo, P. Forns and F. Albericio, QSAR Comb. Sci., 2004, 23, 145–152 CAS
- F. de Moliner, N. Kielland, R. Lavilla and M. Vendrell, Angew. Chem., Int. Ed., 2017, 56, 3758–3769 CrossRef CAS PubMed
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed
- S. T. Huang, K. N. Ting and K. L. Wang, Anal. Chim. Acta, 2008, 620, 120–126 CrossRef CAS PubMed
- X. Li, X. Zhang, Z. Chi, X. Chao, X. Zhou, Y. Zhang, S. Liu and J. Xu, Anal. Methods, 2012, 4, 3338–3343 RSC
- R. R. Nawimanage, B. Prasai, S. U. Hettiarachchi and R. L. McCarley, Anal. Chem., 2014, 86, 12266–12271 CrossRef CAS PubMed
- X. Dai, Z. F. Du, L. H. Wang, J. Y. Miao and B. X. Zhao, Anal. Chim. Acta, 2016, 922, 64–70 CrossRef CAS PubMed
- H. Komatsu, Y. Shindo, K. Oka, J. P. Hill and K. Ariga, Angew. Chem., Int. Ed., 2014, 53, 3993–3995 CrossRef CAS PubMed
- K. Tanabe, N. Hirata, H. Harada, M. Hiraoka and S. I. Nishimoto, ChemBioChem, 2008, 9, 426–432 CrossRef CAS PubMed
- H. Komatsu, H. Harada, K. Tanabe, M. Hiraoka and S. Nishimoto, MedChemComm, 2010, 1, 50–53 RSC
- W. Ma, L.-X. Qin, F.-T. Liu, Z. Gu, J. Wang, Z. G. Pan, T. D. James and Y.-T. Long, Sci. Rep., 2013, 3, 1537–1544 CrossRef PubMed
- Y. M. Sung, S. R. Gayam, P. Y. Hsieh, H. Y. Hsu, E. W. G. Diau and S. P. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 25961–25969 CAS
- S. Milstien and L. A. Cohen, J. Am. Chem. Soc., 1972, 94, 9158–9165 CrossRef CAS PubMed
- B. Madajewski, M. A. Boatman, G. Chakrabarti, D. A. Boothman and E. A. Bey, Mol. Cancer Res., 2016, 14, 14–25 CrossRef CAS PubMed
-
(a) E.-T. Oh, J.-W. Kim, J. M. Kim, S. J. Kim, J.-S. Lee, S.-S. Hong, J. Goodwin, R. J. Ruthenborg, M. G. Jung, H.-J. Lee, C.-H. Lee, E. S. Park, C. Kim and H. J. Park, Nat. Commun., 2016, 7, 13593–13606 CrossRef CAS PubMed
- B. Prasai, W. C. Silvers and R. L. McCarley, Anal. Chem., 2015, 87, 6411–6418 CrossRef CAS PubMed
- S. U. Hettiarachchi, B. Prasai and R. L. McCarley, J. Am. Chem. Soc., 2014, 136, 7575–7578 CrossRef CAS PubMed
- Q. A. Best, B. Prasai, A. Rouillere, A. E. Johnson and R. L. McCarley, Chem. Commun., 2017, 53, 783–786 RSC
- Q. A. Best, A. E. Johnson, B. Prasai, A. Rouillere and R. L. McCarley, ACS Chem. Biol., 2016, 11, 231–240 CrossRef CAS PubMed
- W. C. Silvers, A. S. Payne and R. L. McCarley, Chem. Commun., 2011, 47, 11264–11266 RSC
- A. L. Vahrmeijer, M. Hutteman, J. R. van der Vorst, C. J. H. van de Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10, 507–518 CrossRef CAS PubMed
- M. Vendrell, A. Samanta, S.-W. Yun and Y.-T. Chang, Org. Biomol. Chem., 2011, 9, 4760–4762 CAS
- Z. Shen, B. Prasai, Y. Nakamura, H. Kobayashi, M. S. Jackson and R. L. McCarley, ACS Chem. Biol., 2017, 12, 1121–1132 CrossRef CAS PubMed
- C. Zhang, B.-B. Zhai, T. Peng, Z. Zhong, L. Xu, Q.-Z. Zhang, L.-Y. Li, L. Yi and Z. Xi, Dyes Pigm., 2017, 143, 245–251 CrossRef CAS
- Y. Nakamura, Z. Shen, T. Harada, T. Nagaya, K. Sato, S. Okuyama, F. Ogata, P. L. Choyke, R. L. McCarley and H. Kobayashi, OncoTargets Ther., 2017, 8, 61181–61192 Search PubMed
- N. Kwon, M. K. Cho, S. J. Park, D. Kim, S.-J. Nam, L. Cui, H. M. Kim and J. Yoon, Chem. Commun., 2017, 53, 525–528 RSC
- P. Liu, J. Xu, D. Yan, P. Zhang, F. Zeng, B. Lia and S. Wu, Chem. Commun., 2015, 51, 9567–9570 RSC
- W. S. Shin, J. Han, P. Verwilst, R. Kumar, J.-H. Kim and J. S. Kim, Bioconjugate Chem., 2016, 27, 1419–1426 CrossRef CAS PubMed
- Q. A. Best, B. Prasai, A. Rouillere, A. E. Johnson and R. L. McCarley, Chem. Commun., 2017, 53, 783–786 RSC
- C. E. M. Carvalho, N. C. de Lucas, J. O. M. Herrera, A. V. Pinto, M. C. F. R. Pinto and I. M. Brinn, J. Photochem. Photobiol., A, 2004, 167, 1–9 CrossRef CAS
- F. S. Emery, M. C. F. R. Pinto, C. A. de Simone, V. R. S. Malta, E. N. da Silva Júnior and A. V. Pinto, Synlett, 2010, 1931–1934 CAS
- C. M. de Souza, R. C. Silva, P. O. Fernandes, J. D. de Souza Filho, H. A. Duarte, M. H. Araujo, C. A. de Simone, S. L. de Castro, R. F. S. Menna-Barreto, C. P. Demicheli and E. N. da Silva Júnior, New J. Chem., 2017, 41, 3723–3731 RSC
- S. J. Park, H. C. Yeo, N. Y. Kang, H. Kim, J. Lin, H. H. Ha, M. Vendrell, J. S. Lee, Y. Chandran, D. Y. Lee, S.-W. Yun and Y.-T. Chang, Stem Cell Res., 2014, 12, 730–741 CrossRef CAS PubMed
- J. C. Er, M. Vendrell, M. K. Tang, D. Zhai and Y.-T. Chang, ACS Comb. Sci., 2013, 15, 452–457 CrossRef CAS PubMed
- F. S. dos Santos, G. G. Dias, R. P. de Freitas, L. S. Santos, G. F. de Lima, H. A. Duarte, C. A. de Simone, L. M. S. L. Rezende, M. J. X. Vianna, J. R. Correa, B. A. D. Neto and E. N. da Silva Júnior, Eur. J. Org. Chem., 2017, 3763–3773 CrossRef CAS
- G. G. Dias, B. L. Rodrigues, J. M. Resende, H. D. R. Calado, C. A. de Simone, V. H. C. Silva, B. A. D. Neto, M. O. F. Goulart, F. R. Ferreira, A. S. Meira, C. Pessoa, J. R. Correa and E. N. da Silva Júnior, Chem. Commun., 2015, 51, 9141–9144 RSC
- G. G. Dias, P. V. B. Pinho, H. A. Duarte, J. M. Resende, A. B. B. Rosa, J. R. Correa, B. A. D. Neto and E. N. da Silva Júnior, RSC Adv., 2016, 6, 76056–76063 RSC
- T. B. Gontijo, R. P. de Freitas, G. F. de Lima, L. C. D. de Rezende, L. F. Pedrosa, T. L. Silva, M. O. F. Goulart, B. C. Cavalcanti, C. Pessoa, M. P. Bruno, J. R. Corrêa, F. S. Emery and E. N. da Silva Júnior, Chem. Commun., 2016, 52, 13281–13284 RSC
- W. Ma and Y.-T. Long, Chem. Soc. Rev., 2014, 43, 30–41 RSC
- L. E. Greene, R. Godin and G. Cosa, J. Am. Chem. Soc., 2016, 138, 11327–11334 CrossRef CAS PubMed
- L. Han, M. Liu, D. Ye, N. Zhang, E. Lim, J. Lu and C. Jiang, Biomaterials, 2014, 35, 2952–2960 CrossRef CAS PubMed
- M. N. Belzile, R. Godin, A. M. Durantini and G. Cosa, J. Am. Chem. Soc., 2016, 138, 16388–16397 CrossRef CAS PubMed
- L. Sun, Y. Chen, S. Kuang, G. Li, R. Guan, J. Liu, L. Ji and H. Chao, Chem. – Eur. J., 2016, 22, 8955–8965 CrossRef CAS PubMed
- F. Yu, P. Li, P. Song, B. Wang, J. Zhao and K. Han, Chem. Commun., 2012, 48, 4980–4982 RSC
| This journal is © The Royal Society of Chemistry 2018 |