
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIminium and enamine catalysis in enantioselective photochemical reactions
You-Quan
Zou
 *,
Fabian M.
Hörmann
and
Thorsten
Bach
*
*,
Fabian M.
Hörmann
and
Thorsten
Bach
*
Department Chemie and Catalysis Research Center (CRC), Technische Universität München, Lichtenbergstraße 4, 85747 Garching, Germany. E-mail: youquan.zou@gmail.com; thorsten.bach@ch.tum.de; Web: http://www.oc1.ch.tum.de/home_en/
First published on 20th November 2017
Abstract
Although enantioselective catalysis under thermal conditions has been well established over the last few decades, the enantioselective catalysis of photochemical reactions is still a challenging task resulting from the complex enantiotopic face differentiation in the photoexcited state. Recently, remarkable achievements have been reported by a synergistic combination of organocatalysis and photocatalysis, which have led to the expedient construction of a diverse range of enantioenriched molecules which are generally not easily accessible under thermal conditions. In this tutorial review, we summarize and highlight the most significant advances in iminium and enamine catalysis of enantioselective photochemical reactions, with an emphasis on catalytic modes and reaction types.
 You-Quan Zou | You-Quan Zou completed his PhD studies in 2014 under the supervision of Professor Wen-Jing Xiao at Central China Normal University. He has worked as an Alexander von Humboldt postdoctoral fellow with Professor Thorsten Bach at the Technische Universität München. In November 2017, he will take up a SAERI postdoctoral position with Professor David Milstein at the Weizmann Institute of Science. |
 Fabian M. Hörmann | Fabian M. Hörmann studied chemistry at the Technische Universität München, where he received his MSc degree in 2016. Currently, he is carrying out his PhD work in the group of Professor Thorsten Bach on asymmetric photo-catalysis. |
 Thorsten Bach | Thorsten Bach obtained his education at the University of Heidelberg and at the University of Southern California, where he conducted his Diploma thesis with G. A. Olah. He received his PhD in 1991 from the University of Marburg with M. T. Reetz and did post-doctoral work as a NATO fellow with D. A. Evans at Harvard University. He completed his Habilitation at the University of Münster in 1996, moved to the University of Marburg as an associate professor in 1997 and was appointed to the Chair of Organic Chemistry I at the Technische Universität München (TUM) in 2000. He is an elected member of the German Academy of Sciences (Leopoldina) since 2006 and of the Bavarian Academy of Sciences since 2009. |
Key learning points(1) The concept of synergistic combination of iminium and enamine catalysis with photocatalysis.(2) The photogeneration of ortho-quinodimethanes, open-shell free radicals and iminium cations. (3) The single-electron-transfer (SET) mechanism in enantioselective photochemical reactions. (4) The direct excitation of iminium ions and enamines in enantioselective photochemical reactions. (5) The electron donor–acceptor (EDA) complex mechanism in enantioselective photochemical reactions. |
1. Introduction
Chiral molecules, which can be found in diverse families of naturally occurring alkaloids and biologically active substances, play an important role in the field of synthetic chemistry, medicinal chemistry, material science, and industrial production. A key feature for the advancement of modern organic chemistry is the development of novel and efficient methods that allow for rapid construction of enantiomerically pure compounds from readily available starting materials.1 In this regard, enantioselective catalysis, also referred to as asymmetric catalysis,2 has emerged as a powerful and alluring strategy to advance this goal and it has fascinated chemists for the last decades. A plethora of catalytic transformations based on enantioselective catalysis has been established, and research in this discipline continues to expand vibrantly. Nevertheless, the enantioselective catalysis of photochemical reactions (enantioselective photocatalysis) is still a formidably challenging task resulting from the intrinsic factors of photochemical processes.3At the dawn of enantioselective photochemistry, circularly polarized light (CPL) was frequently used as the source of chirality.3 However, the use of CPL was limited to specific starting materials and the relatively low reaction efficiency largely stymied its application as a universal approach in enantioselective photochemistry. Additionally, asymmetric organocatalysis, a relatively large branch of enantioselective catalysis, started to blossom in the beginning of the 21st century, and it has developed into a flourishing research field. A large number of fine and useful enantioenriched chiral compounds with a well defined three-dimensional spatial arrangement have been constructed by means of organocatalytic protocols.4 The success of asymmetric organocatalysis is attributed to privileged enantiopure structures such as proline, cinchona alkaloids, BINOL (1,1′-bi-2-naphthol), and related compounds. These small molecules could be easily elaborated into different bespoke organocatalysts which have found a myriad of applications in asymmetric organocatalysis. Meanwhile, asymmetric organocatalysis also opens new avenues for chemists to approach the enantioselective catalysis of photochemical reactions. For instance, iminium and enamine catalysis using chiral amines, Brønsted acid catalysis employing chiral phosphoric acids, hydrogen bonding catalysis with chiral ureas and thioureas, N-heterocyclic carbene catalysis using chiral heteroazolium salts as well as phase-transfer catalysis with chiral ammonium salts have been successfully introduced into enantioselective photochemical reactions. In this context, iminium and enamine catalysis have recently grown in interest due to their high efficiency to activate aldehydes and ketones with excellent enantioselectivity, thus enabling the synthesis of diversely functionalized chiral carbonyl compounds.
Recently, some reviews have been written by us5 and other research groups6,7 in the field of enantioselective catalysis of photochemical reactions. However, to the best of our knowledge, there is still no specific and comprehensive review devoted to iminium and enamine catalysis in enantioselective photochemical reactions. Consequently, we herein provide a tutorial review to give a solid introduction to the field and highlight the recent breakthroughs in this area. Reactions which occur in heterogeneous conditions will be given less attention in this review. Organization of the data follows a subdivision according to catalytic modes (iminium or enamine catalysis) and reaction types. The emission wavelength or wavelength range of the irradiation is given in the schemes. The reaction temperatures are only specified if the reactions were not carried out at room temperature.
2. Iminium catalysis
Iminium catalysis is a frequently used strategy to activate α,β-unsaturated carbonyl compounds.8 Typically, the condensation of chiral amines with α,β-unsaturated aldehydes or ketones provides a reversible formation of iminium ions. The lowest unoccupied molecular orbitals (LUMO) of these iminium ions are lower in energy, which results in an enhanced electrophilicity and therefore can be more readily intercepted by nucleophiles. Various chiral amines were developed as iminium catalysts over the last decades to induce stereoselective reactions. They exhibit remarkable efficiencies and were largely applied in conventional thermal reactions. Recently, the application of selected chiral amines has also been extended into enantioselective photochemistry. Fig. 1 shows the structures of representative amines such as secondary amines 1–3 and primary amines 4–6 used in iminium catalysis of enantioselective photochemical reactions.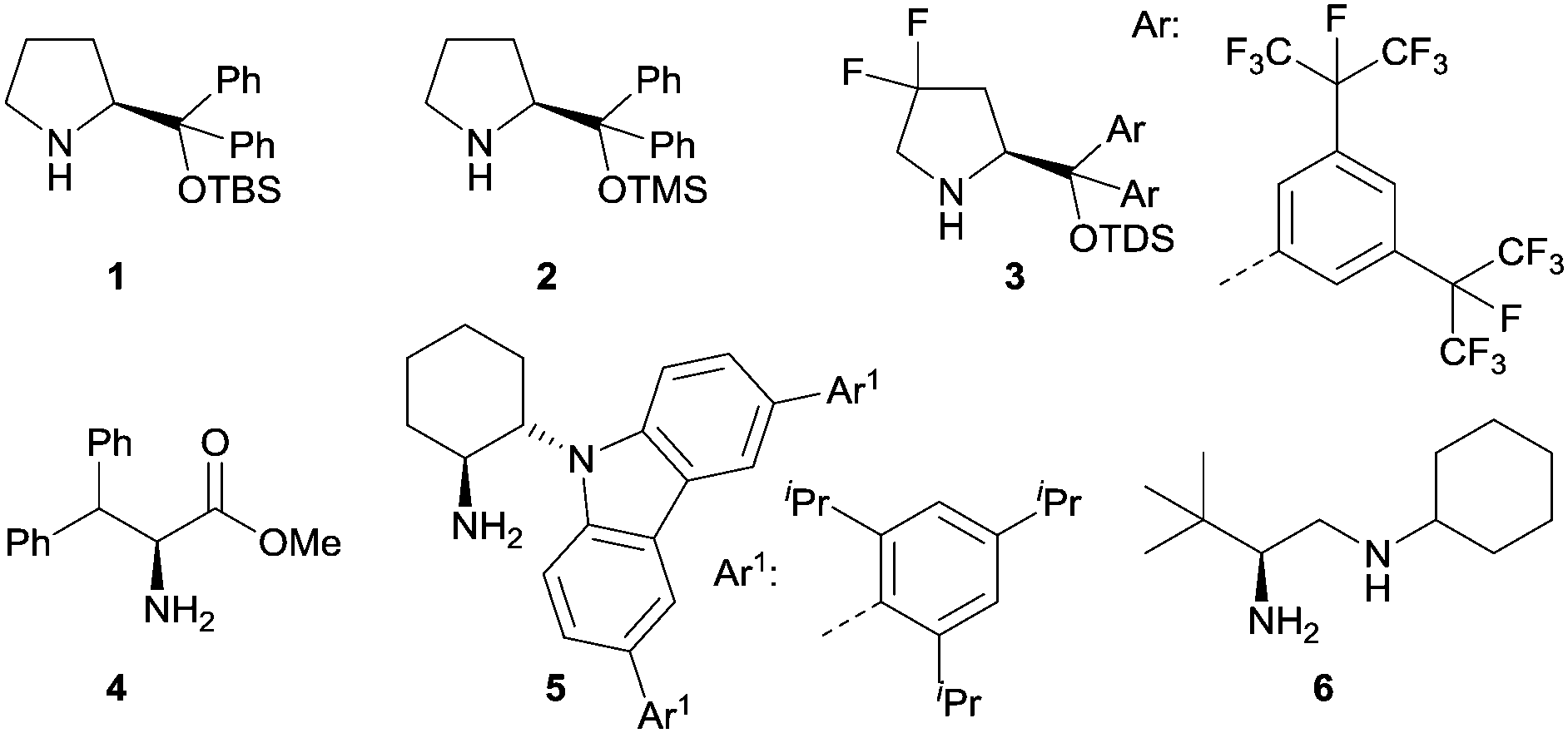 | ||
| Fig. 1 Structures of representative chiral amines 1–6 for iminium catalysis in enantioselective photochemical reactions. TBS = tert-butyldimethylsilyl, TMS = trimethylsilyl, TDS = tert-hexyldimethylsilyl. | ||
2.1 [2+2] Photocycloaddition reaction of iminium salts
One of the first examples involving chiral iminium ions in photochemical reactions was reported by the Mariano group in 2001.9 As part of their research program on iminium salt photochemistry, they synthesized the alkene-tethered, chiral iminium perchlorate 7 from a C2-symmetric chiral amine and tested its reactivity upon irradiation. The [2+2] photocycloaddition10 of iminium salt 7 did indeed occur and after hydrolysis ketone 8 was obtained with 82% ee at 40% conversion (Scheme 1). The enantioselectivity was found to decrease at higher conversions. Substituents at positions C2 and C5 of the pyrrolidine were crucial, and the corresponding 2,5-diphenyl iminium ion showed no reactivity which might result from the quenching of the iminium singlet excited state by the phenyl groups via single electron transfer (SET). Mechanistically, the reaction takes place at the singlet hypersurface via a concerted pathway resulting from the strong ππ* absorption (ε = 2–4 × 104 M−1 cm−1) of the iminium salt at λ ≅ 280 nm. Although the diastereoselectivity is good, the reaction capitalizes on a chiral auxiliary and is not catalytic.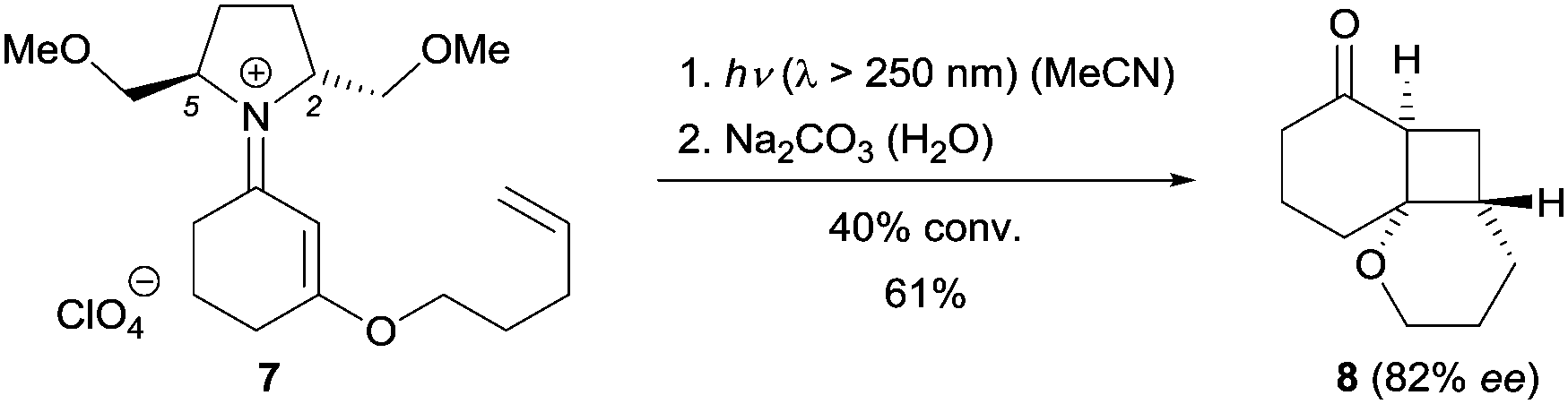 | ||
| Scheme 1 Diastereoselective intramolecular [2+2] photocycloaddition reaction of iminium salt 7. | ||
2.2 β-Benzylation of enals and enones
The photoexcitation of ortho-alkyl substituted benzaldehydes and benzophenones generates ortho-quinodimethanes as highly reactive species via an intramolecular H-abstraction.11 The resulting intermediates serve as dienes in Diels–Alder reactions and an enantioselective variant of this reaction was disclosed in 2003.12 In 2016, Melchiorre and co-workers reported a catalytic enantioselective version of this transformation by using a cinchona alkaloid-derived bifunctional tertiary amine–thiourea catalyst.13 More recently, Melchiorre, Maseras and co-workers discovered an enantioselective Michael-type addition reaction of photogenerated ortho-quinodimethanes to enals in the presence of a chiral secondary amine.14 Using 20 mol% of diphenylprolinol tert-butyldimethylsilylether (1) and 20 mol% of diphenylphosphoric acid (DPP), benzophenones 9 reacted with aliphatic enals (e.g., 10a–10f) smoothly in 1,2-dichlorobenzene upon irradiation at λ = 365 nm with a 15 W black light bulb to afford solely the β-benzylated products 11a–11f in moderate yields with good to excellent enantioselectivity (Scheme 2). By simply switching the catalyst to diphenylprolinol trimethylsilylether (2) (E)-cinnamaldehyde 10g, heteroaryl containing enals such as (E)-3-(furan-2-yl)acrylaldehyde (10h) were coupled with 2-methylbenzophenone in toluene to furnish the desired products with excellent enantioselectivity (94% and 90% ee, respectively).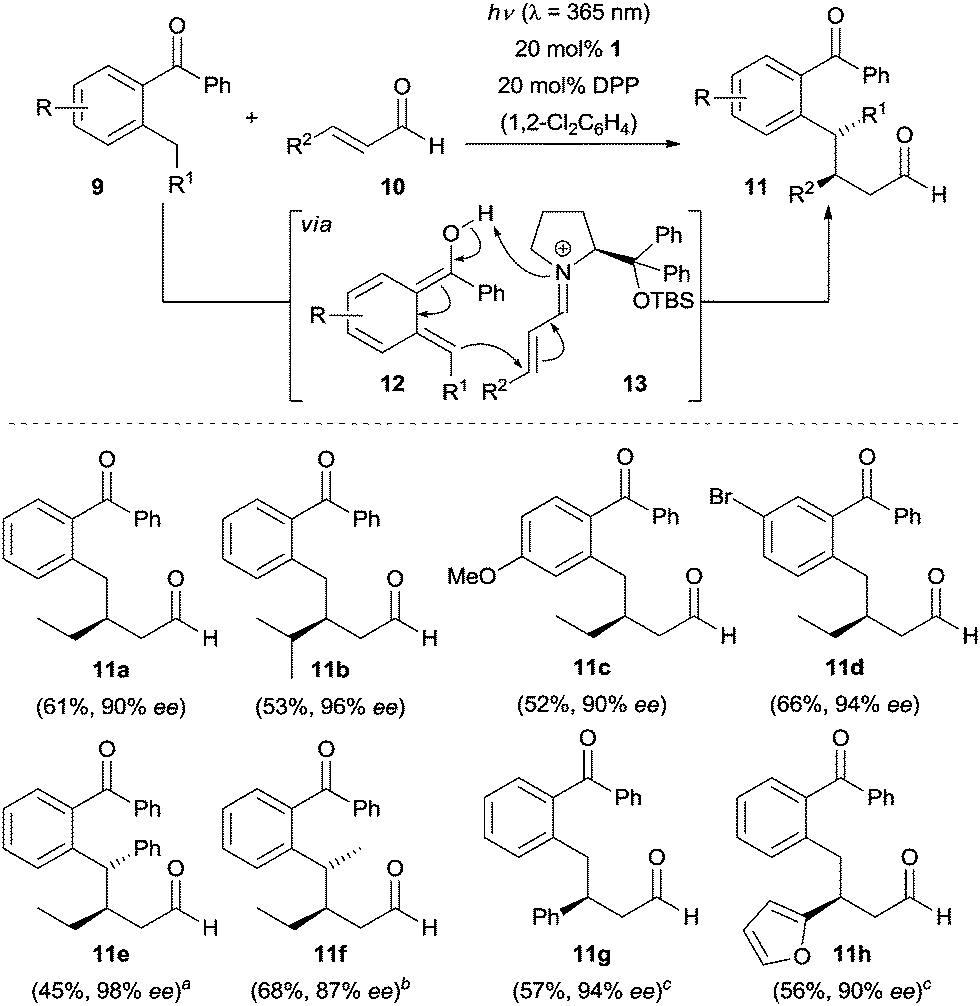 | ||
Scheme 2 Enantioselective β-benzylation of enals 10. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d.r. > 95/5. bd.r. = 56/44, the ee refers to the major diastereoisomer. cToluene employed as the solvent using 2 as the catalyst. d.r. > 95/5. bd.r. = 56/44, the ee refers to the major diastereoisomer. cToluene employed as the solvent using 2 as the catalyst. | ||
In this transformation, chiral secondary amine 1 is condensed with enals 10 to iminium salts 13. Additionally, a light induced enolization of ortho-alkyl substituted benzophenones to (E)-enol 12 occurs. The latter undergoes nucleophilic addition to the chiral iminium salt 13 to deliver the Michael-type addition products rather than [4+2] cycloadducts. A density functional theory (DFT) computational study was carried out by the authors to shed some light on this unusual reactivity and the results indicated that this transformation proceeds through a water assisted proton shuttle mechanism.
Recently, the Ye group15 reported an enantioselective β-benzylation reaction of enones 14via a similar strategy. As shown in Scheme 3, 2-cyclohexenone reacts – upon UV irradiation (λ = 365 nm) – with a variety of 2-alkyl benzophenones 9 in the presence of 20 mol% of chiral amino acid ester 4 furnishing the β-benzylation products in moderate to good yields with excellent enantioselectivity (e.g., 15a–15d). Electron donating and electron withdrawing groups on the enolizable aromatic ring of benzophenones did not have a significant effect on the reaction efficiency. The substrate scope included a β-substituted cyclohexenone (product 15e), an acyclic α,β-unsaturated ketone (product 15f), and a seven-membered cyclic enone (product 15g). None of the desired product was observed, when 2-cyclopentenone was used as the substrate. Interestingly, the reaction of 2-cyclopentenone did proceed to give the addition product 15h in 99% ee when chiral amine 6 was employed as the catalyst.
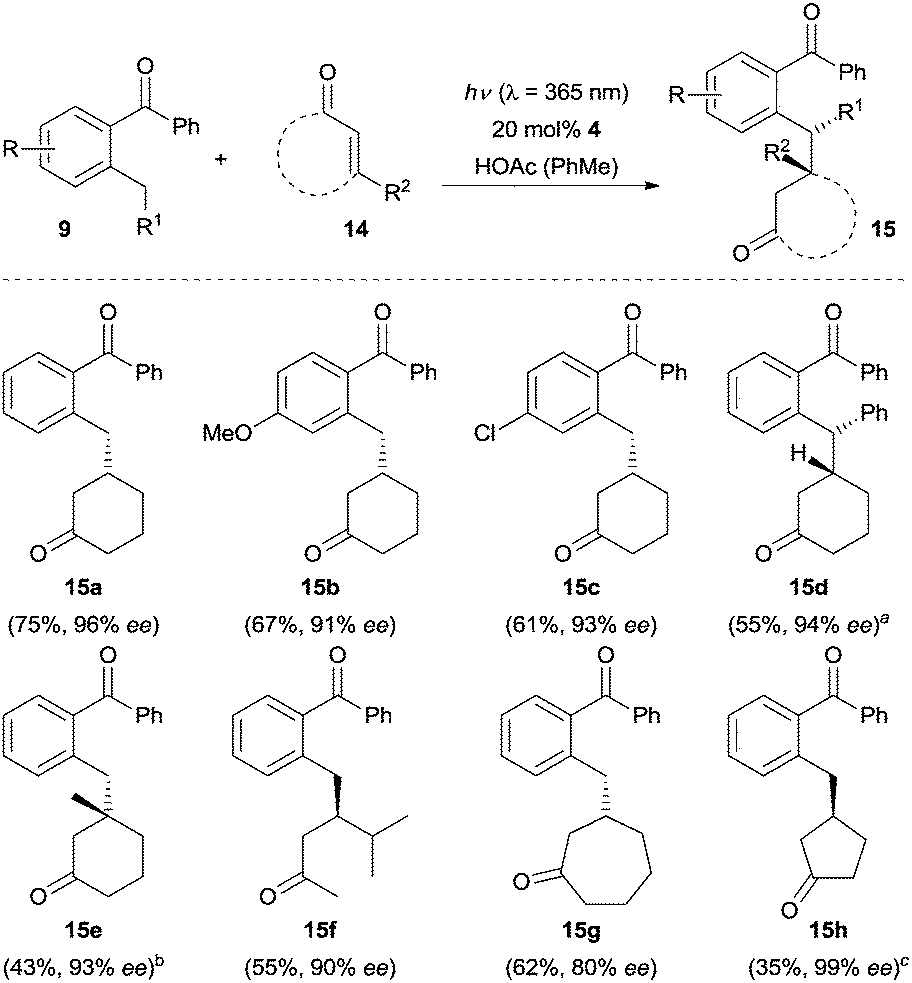 | ||
| Scheme 3 Enantioselective β-benzylation of enones 14. ad.r. > 95/5. b50 mol% benzoic acid was used instead of acetic acid. c6 was used as the chiral amine catalyst. HOAc = acetic acid. | ||
In biological systems, iminium ions have been found to absorb visible light and induce primary photochemical events. The reactivity of these ions in the excited state16 is dominated by electron transfer processes and has been extensively studied by the group of Mariano.17 Very recently, Melchiorre and co-workers disclosed the first asymmetric approach based on this reactivity enabling the enantioselective synthesis of β-alkylated aldehydes (Scheme 4).18 Crucial for the success was (a) the high redox potential of iminium ion 19 in the excited state and (b) the bathochromic shift in absorption which reaches to the visible light region when (E)-cinnamaldehyde (16) is condensed with chiral secondary amine 3 to iminium ion 19. Upon irradiation with visible light (λ = 420 nm), the iminium ion 19 populates the excited state, which is able to react with trimethylsilane 17 generating β-enaminyl radical 20 and silyl radical cation via a SET process. After solvent-assisted desilylation an intermolecular radical–radical coupling occurs and subsequent hydrolysis gives product 18 and amine 3 is regenerated. It is worth mentioning that no photocatalyst was needed in this reaction.
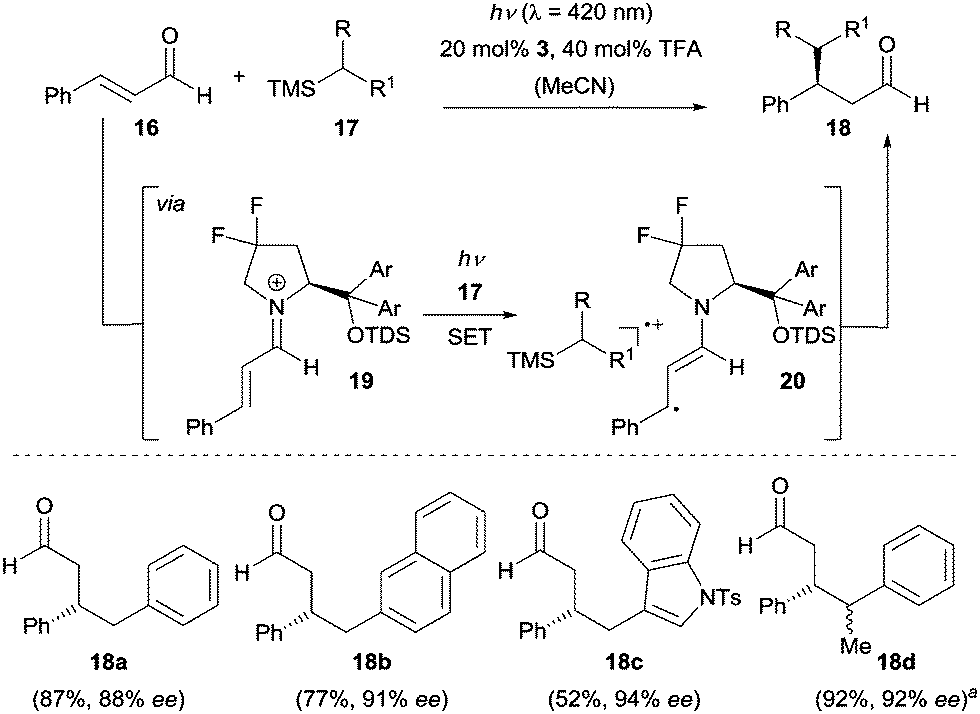 | ||
| Scheme 4 Enantioselective β-benzylation of enals via visible-light excitation of iminium ions catalyzed by chiral secondary amine 3. ad.r. = 55/45, the ee refers to the major diastereoisomer. TFA = trifluoroacetic acid. | ||
2.3 β-Alkylation of enals and enones
The Melchiorre group also achieved the first enantioselective radical conjugate addition (RCA) to β,β-disubstituted cyclic enones by a combination of photoredox catalysis and iminium catalysis (Scheme 5).19 The key feature for the success of this dual catalysis was the rationally designed organocatalyst 5 bearing a redox-active carbazole moiety (Fig. 1). In this reaction, the primary amine moiety first condenses with the cyclic enone to give the corresponding chiral iminium ion. In parallel, the benzodioxole is – upon irradiation by a single ultraviolet (UV) light-emitting diode (LED) in the presence of tetrabutylammonium decatungstate (TBADT) – converted to the corresponding nucleophilic carbon-centered radical. Subsequently, this radical adds to the chiral iminium ion generating the short-lived and highly reactive α-iminyl radical cation 22, which is rapidly reduced to enamine 23 by the carbazole moiety of the amine catalyst through an intramolecular SET process. After the enamine–imine tautomerization, regeneration of the photocatalyst TBADT, followed by a hydrolysis step, the terminal products 21 were furnished with good to excellent enantioselectivity with the release of the chiral amine 5.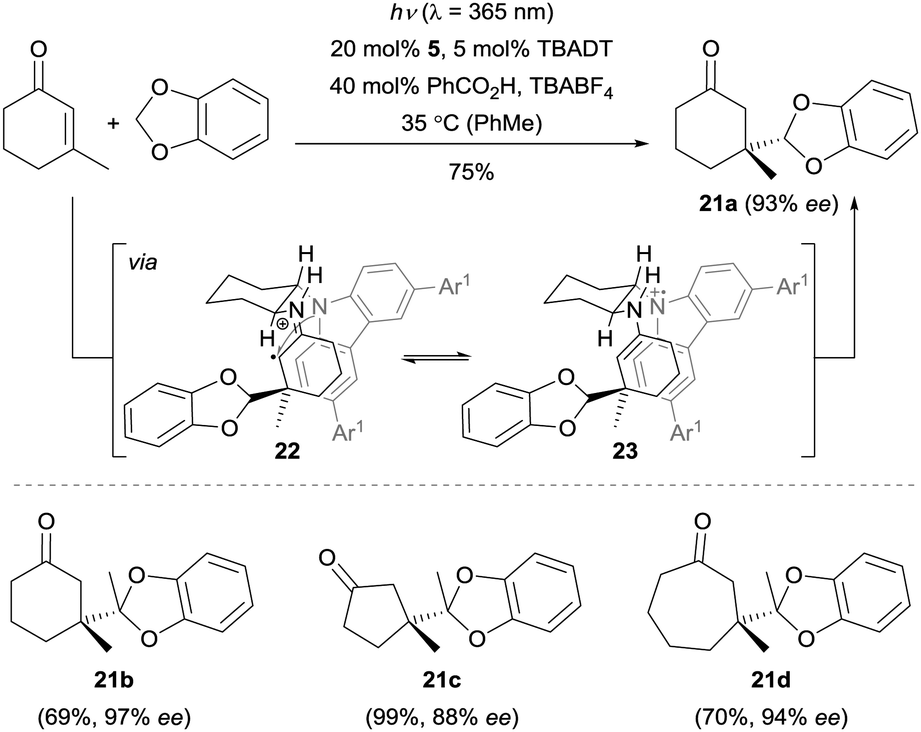 | ||
| Scheme 5 Formation of products 21 by enantioselective trapping of benzodioxole-derived radicals with enones catalyzed by amine 5. TBABF4 = tetrabutylammonium tetrafluoroborate. | ||
This methodology represents a major breakthrough in the field of asymmetric radical chemistry, and it also highlights the application of iminium catalysis in enantioselective photochemical reactions. The carefully designed catalyst not only served as a chiral amine to control the enantioselectivity, but also functioned as an efficient electron-rich center to prevent the radical elimination (β-scission) of radical cation 22 to its iminum ion precursor and to reduce 22 to the key intermediate enamine 23. This strategy could also be extended to a visible-light-induced enantioselective trapping of α-amino radicals.20 By using 1 mol% Ir[dF(CF3)ppy]2(dtbbpy)PF6 (dF(CF3)ppy = 3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl]phenyl, dtbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine) as the photocatalyst and ent-5 as the organocatalyst, a wide range of cyclic enones could be coupled with various tertiary amines with excellent stereocontrol (Scheme 6). Recently, detailed mechanistic studies regarding this radical conjugated addition reaction were carried out by the same group.21
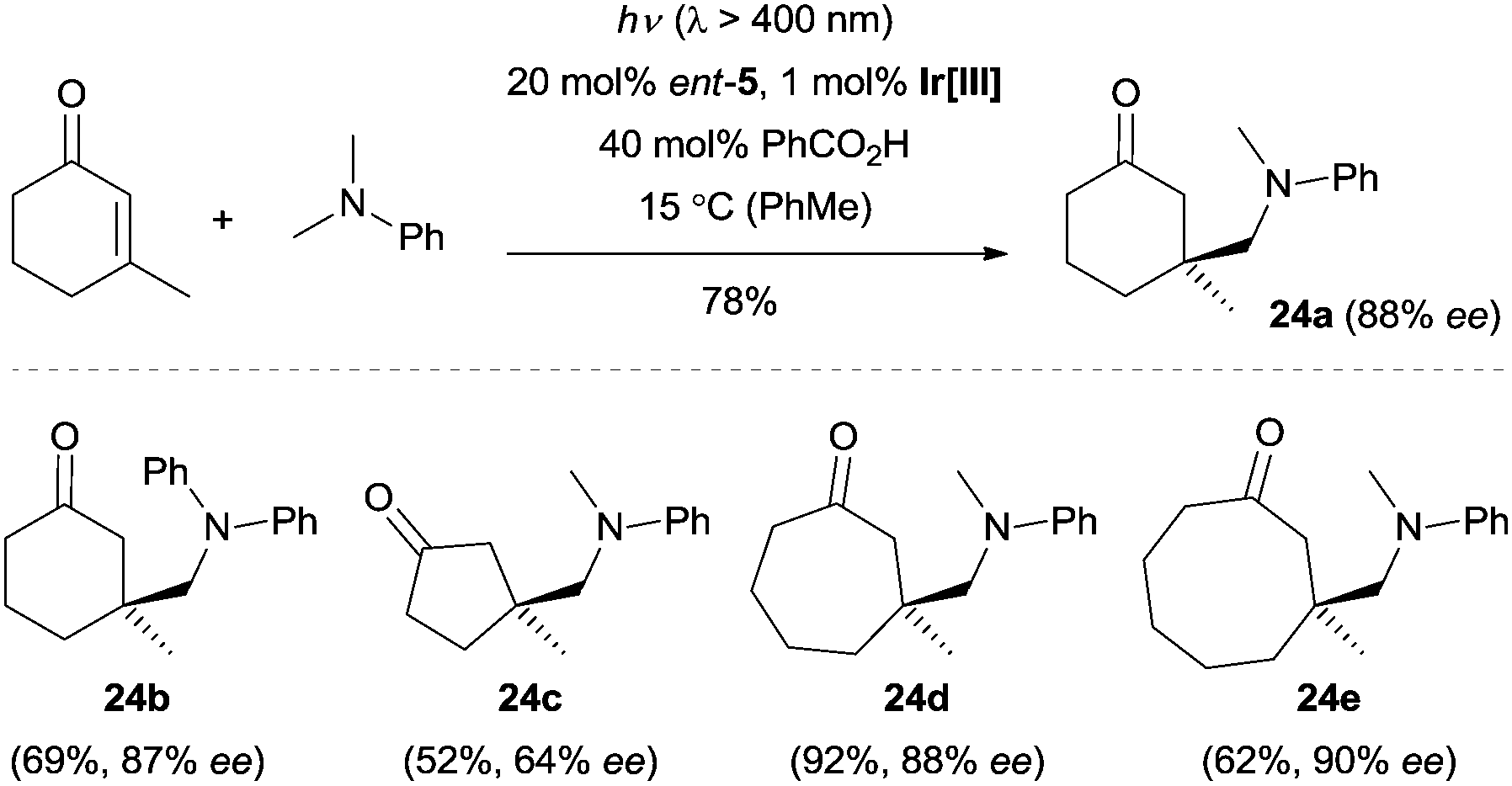 | ||
| Scheme 6 Enantioselective trapping of α-amino radicals with enones catalyzed by ent-5. Ir[III] = Ir[dF(CF3)ppy]2(dtbbpy)PF6. | ||
Additionally, an enantioselective photocatalytic Michael addition/oxyamination cascade of enals was discovered by Jang and co-workers.22 By using chiral secondary amine 2 along with N719/TiO2, a variety of α-oxyaminated β-alkylated aldehydes were produced in moderate to good yields with excellent enantio- and diastereoselectivities.
3. Enamine catalysis
In contrast to iminium catalysis, enamine catalysis23 implies that the carbonyl compound initially reacts with a primary or secondary amine to generate the corresponding enamine via dehydration. The highest occupied molecular orbital (HOMO) of the enamine is higher in energy than the HOMO of the carbonyl compound and therefore can more easily interact with the LUMO or SOMO (singly occupied molecular orbital) of an electrophile. Typical reaction pathways for an enamine are therefore reactions with electrophiles or with electrophilic radicals. Additionally, the enamine can be readily oxidized and the resulting radical cation can react with suitable substrates, e.g., olefins.The benefit of enamine catalysis is the high enantiocontrol which can be achieved by employing chiral amines as catalysts. In this context, a vast majority of amines have been developed and successfully employed in enamine catalysis, rendering enantioenriched functionalized carbonyl compounds under thermal reaction conditions. In recent years, the enamine catalytic mode has also been extended to enantioselective photochemistry, and Fig. 2 depicts the structures of selected chiral amines used in photochemistry, such as chiral secondary amines 25–31 and primary amines 32–37.
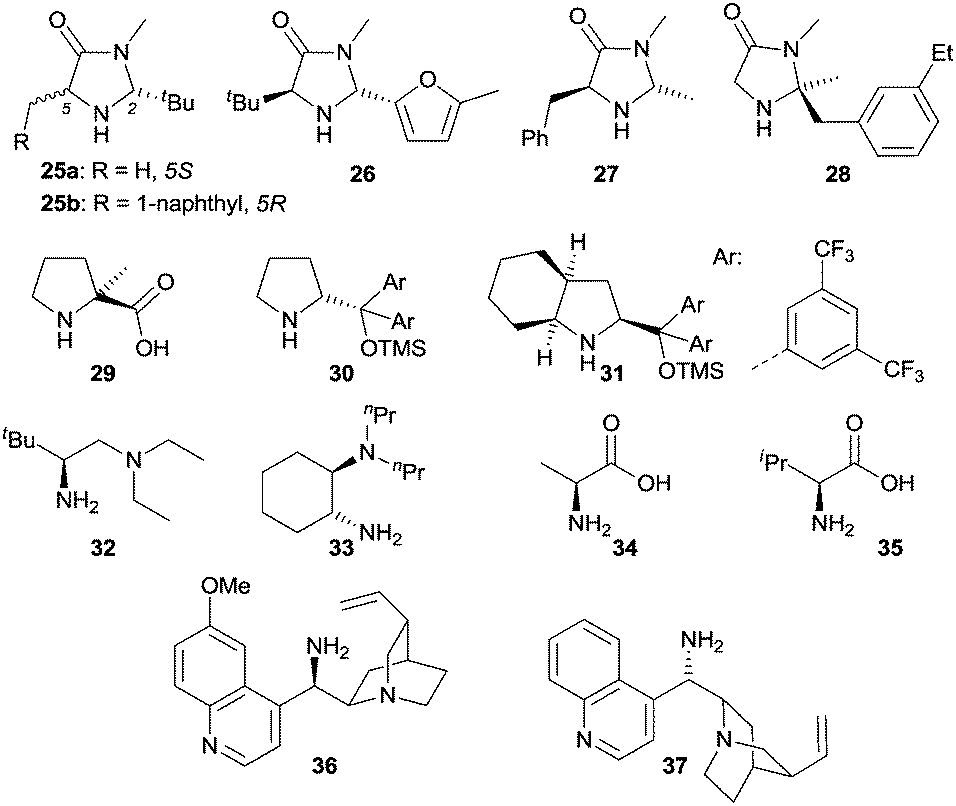 | ||
| Fig. 2 Structures of representative chiral amines 25–37 for enamine catalysis in enantioselective photochemical reactions. | ||
3.1 α-Alkylation of aldehydes, ketones and β-ketocarbonyls
In 2008, seminal work was reported by the group of MacMillan in the field of enantioselective α-alkylation of aldehydes by merging photoredox catalysis24 and organocatalysis. By using 0.5 mol% of Ru(bpy)3Cl2 (bpy = 2,2′-bipyridine) as the photocatalyst and 20 mol% of chiral imidazolidinone 25a as the organocatalyst, a number of aldehydes reacted effectively with various electron-deficient α-bromo carbonyls and produced the α-alkylated aldehydes in good yields with excellent enantioselectivity (Scheme 7).25 Mechanistically, the reactant aldehyde was suggested to be transiently converted to the more nucleophilic chiral enamine intermediate 39 in the organocatalytic cycle. Meanwhile, in the photoredox catalytic cycle, the photocatalyst is initially populated into its excited state *Ru[II] upon irradiation by visible light. The photoexcited metal complex accepts a single electron from a sacrificial quantity of enamine 39 and generates the strong reductant Ru[I] in the first catalytic cycle. Subsequently, the Ru[I] species reduces the α-bromo carbonyl compound to an electrophilic free radical 40, while regenerating the photocatalyst Ru[II]. An intermolecular radical–radical coupling was proposed to rationalize the formation of the electron-rich α-amino radical 41, which acts as a reductive quencher to reduce the excited *Ru[II] to Ru[I] in the following catalytic cycle with release of the iminium ion. Rapid hydrolysis of the iminium ion gives the optically enriched α-alkylated aldehyde 38 and the chiral organocatalyst 25a is regenerated.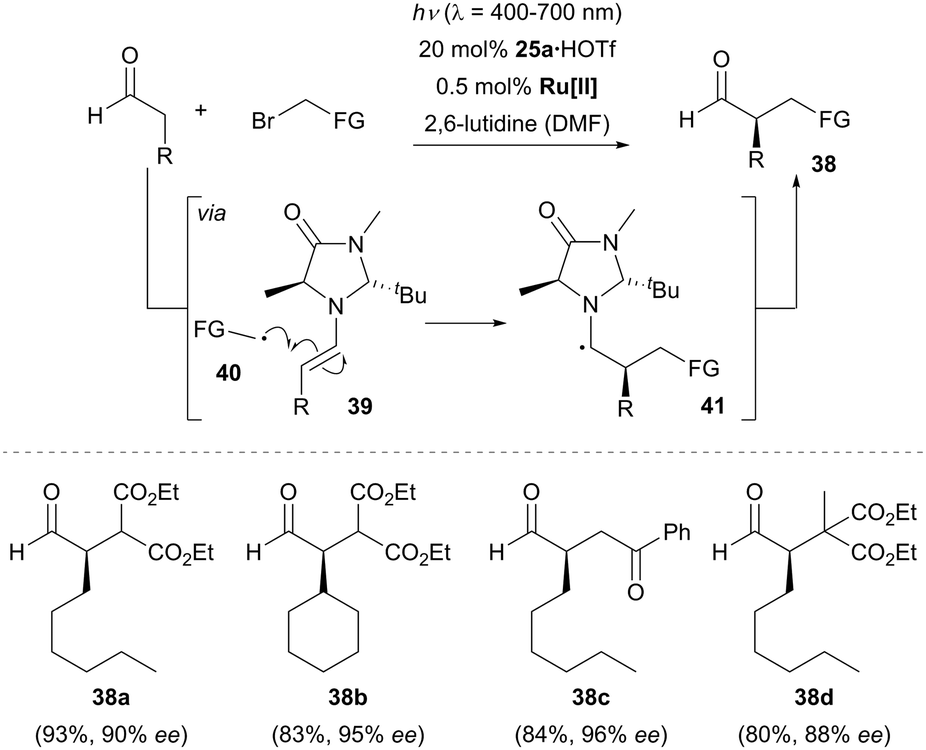 | ||
| Scheme 7 Enantioselective α-alkylation of aldehydes catalyzed by chiral imidazolidinone 25a. Ru[II] = Ru(bpy)3Cl2, FG = functional group, HOTf = trifluoromethanesulfonic acid, DMF = N,N-dimethylformamide. | ||
Recent work from the Yoon group shed some doubt on the proposed action of two synergistic catalytic cycles.26 It was found that the quantum yield for the formation of product 38a was Φ = 18, which indicates that the enantioselective α-alkylation reaction likely proceeds through a radical chain propagation mechanism. Chain propagation can occur if α-amino radical 41 is oxidized by the α-bromo carbonyl substrate but not by the excited photocatalyst. Ceroni, Cozzi and co-workers postulated a similar chain process when using Fe(bpy)3Br2 as the photocatalyst.27
Organic dyes, a class of environmentally friendly, cheap, readily available and easy to handle photocatalysts, have been successfully applied in photocatalysis.28 In 2011, Zeitler and co-workers realized an enantioselective α-alkylation reaction of aldehydes by using eosin Y as the photocatalyst.29 As exemplarily shown in Scheme 8, the reaction of octanal and diethyl 2-bromomalonate in the presence of 20 mol% of imidazolidinone 25a and 0.5 mol% of eosin Y gave the desired product in 85% yield with 88% ee. From a mechanistic point of view, the authors proposed that the photocatalyst eosin Y initially undergoes a rapid intersystem crossing (ISC) upon irradiation. Subsequently, the excited triplet state of eosin Y follows the same pathway as the excited state *Ru[II] to mediate the following steps. The Ferroud group found Rose Bengal was also suitable for this transformation.30 Both metal free methods gave comparable results to the ruthenium catalyzed process.
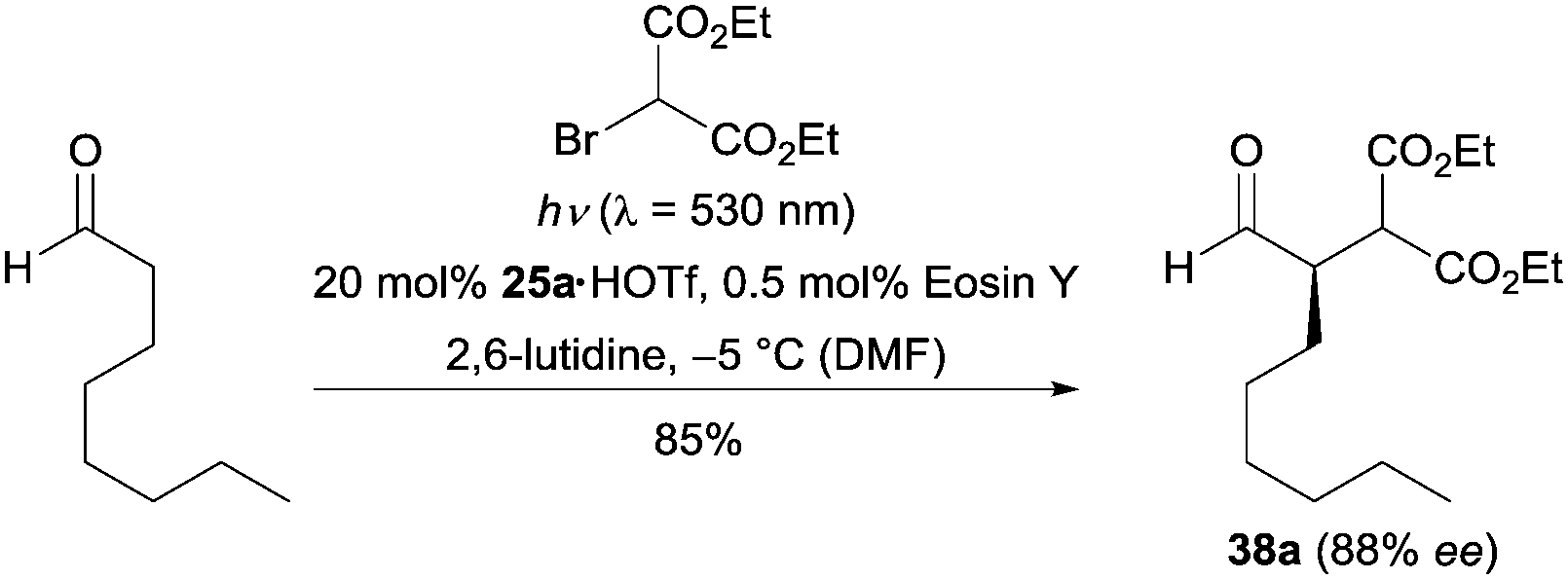 | ||
| Scheme 8 Enantioselective α-alkylation of aldehydes catalyzed by chiral imidazolidinone 25a using eosin Y as the photocatalyst. | ||
Heterogeneous catalysts are often highly efficient and recyclable, and therefore would provide an alternative to the use of noble metal photocatalysts and organic dyes in photochemistry. In this regard, several heterogeneous photocatalysts such as PbBiO2Br, Bi2O3 as well as NbSe2 nanosheet supported PbBiO2Br have been used in the visible light-driven asymmetric α-alkylation of aldehydes by the König group,31 the Pericàs and Palomares groups,32 and the Fan group,33 respectively. Duan and co-workers demonstrated that chiral metal–organic frameworks (MOFs) of Zn-PYI1 and Zn-PYI1 also showed remarkable catalytic activity in the same transformation.34
The MacMillan group successfully expanded their dual catalysis platform to target enantioenriched α-trifluoromethylated and α-perfluoromethylated aldehydes.35 Using a combination of Ir(ppy)2(dtbbpy)PF6 and chiral secondary amine 25a, a wide range of aldehydes could be coupled with trifluoromethyl iodide furnishing the α-trifluoromethylation products in moderate to good yields with excellent enantioselectivity (Scheme 9). Functional groups such as ethers, esters, amines, carbamates (e.g., 42b) and aromatic rings (e.g., 42c) were tolerant under the optimized reaction conditions. Perfluoroalkyl iodides were also suitable in this enantioselective alkylation reaction, and various chiral α-perfluoromethylated aldehydes were synthesized under the same reaction conditions (e.g., 42d and 42e). To demonstrate the potential synthetic applications of this methodology, α-trifluoromethyl 3-phenylpropanal (42c) was readily converted to the corresponding α-trifluoromethyl-substituted acid, the respective β-trifluoromethyl-substituted amine, as well as to the β-trifluoromethyl-substituted alcohol without significant loss of enantiopurity. Notably, high enantioselectivities could only be achieved at −20 °C but not at ambient temperature.
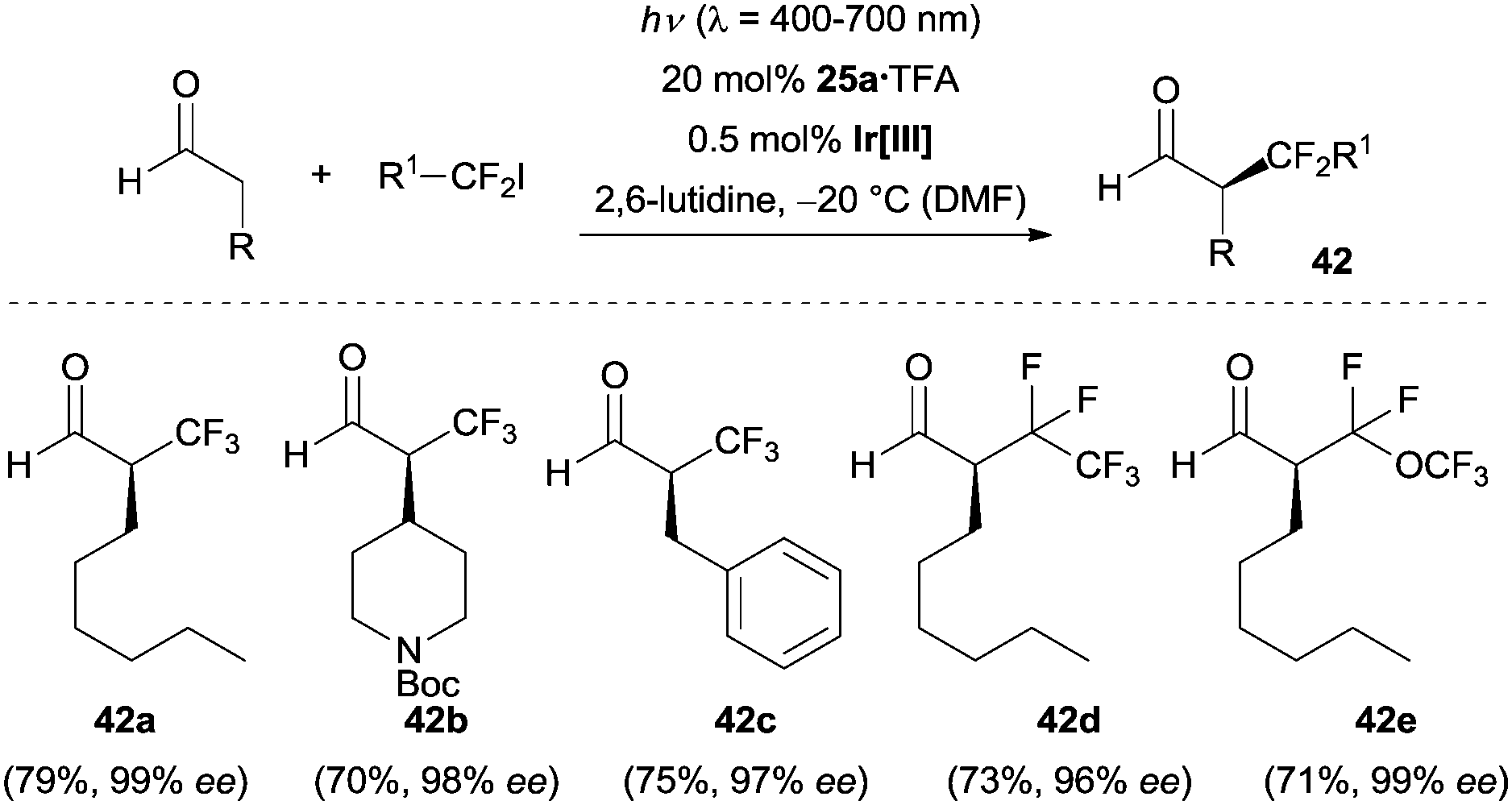 | ||
| Scheme 9 Enantioselective α-trifluoromethylation and α-perfluoromethylation of aldehydes catalyzed by chiral imidazolidinone 25a. Ir[III] = Ir(ppy)2(dtbbpy)PF6, Boc = tert-butyloxycarbonyl. | ||
Nitrile group containing compounds are a class of versatile building blocks in organic synthesis, which can be readily transformed into carbonyl, amine, imidate motifs, etc. Similarly, β-cyanoaldehydes can be readily converted into their corresponding lactone, pyrrolidine, lactam and cyanoalcohol derivatives. In light of the synthetic potential of nitrile compounds, the enantioselective synthesis of β-cyanoaldehydes is highly desirable. In 2015, MacMillan and co-workers reported the first example of enantioselective α-cyanoalkylation of aldehydes by means of their synergistic combination of photoredox catalysis and organocatalysis, providing rapid access to chiral β-cyanoaldehydes 43.36 As depicted in Scheme 10, under the optimal conditions, a large array of α-bromoacetonitriles reacted with aldehydes to give the desired products (e.g., 43a–43d) with excellent enantioselectivity. Interestingly, the product derived from 3-phenylpropanal could be smoothly converted to the corresponding alcohol, ether, lactone, aldehyde, ketone, amide, amine and lactam without erosion of stereochemical integrity. A four-step synthesis of the natural product (−)-bursehernin was also carried out using the aforementioned strategy.
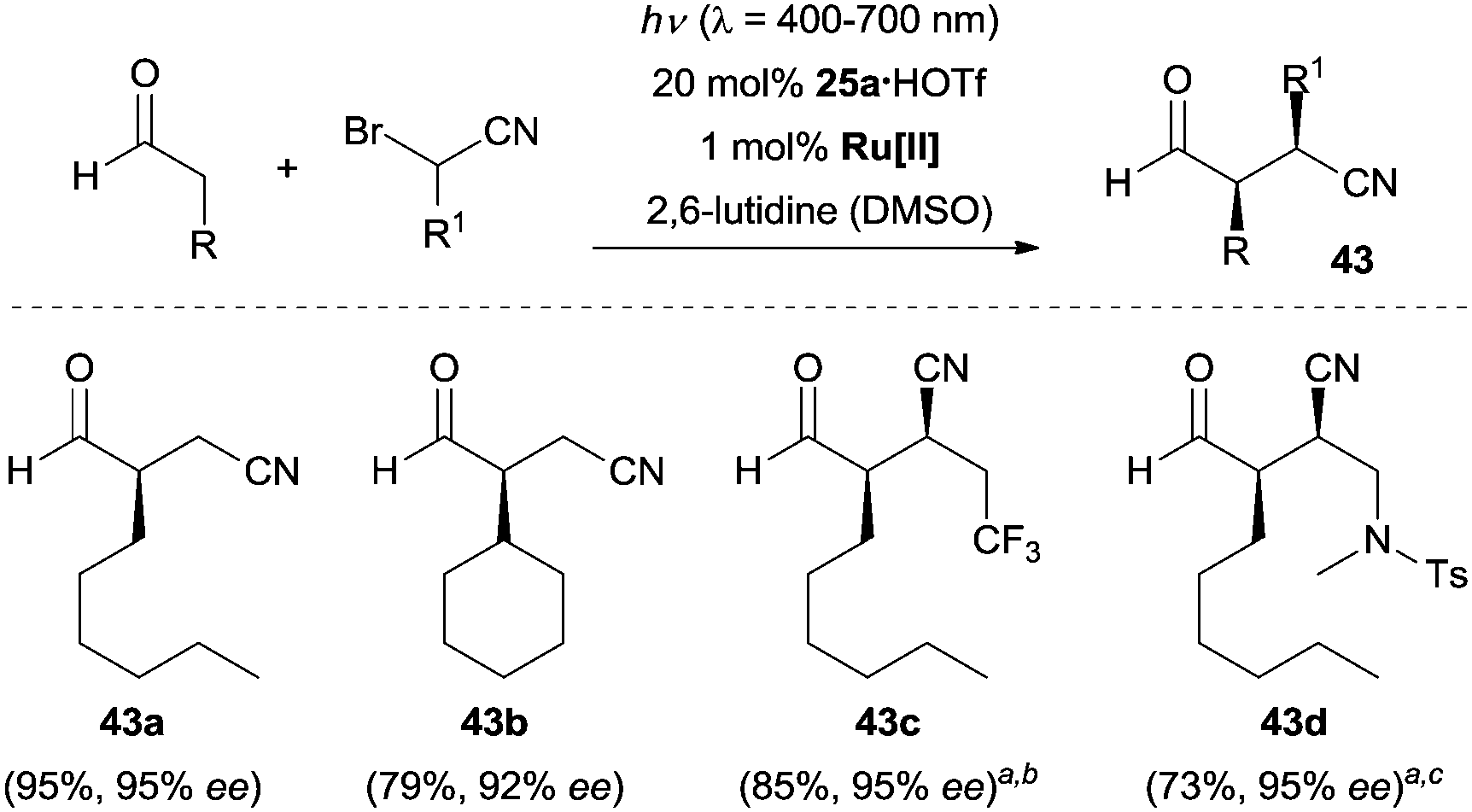 | ||
| Scheme 10 Enantioselective synthesis of β-cyanoaldehydes 43 by photoredox organocatalysis. Ru[II] = Ru(bpy)3Cl2. aChiral secondary amine 26 was used as the organocatalyst. bd.r. = 53/47. cd.r. = 55/45. The ee refers to major diastereoisomer. DMSO = dimethyl sulfoxide, Ts = para-toluenesulfonyl. | ||
Since all of the above-mentioned dual catalysis examples require the use of photocatalyst, the Melchiorre group developed a conceptually new approach for the enantioselective α-alkylation of aldehydes via an electron donor–acceptor (EDA) complex37 mechanism. As illustrated in Scheme 11, an enamine intermediate was first generated by condensing butyraldehyde (44) with the chiral secondary amine 31.38 Upon association of enamine with the electron-deficient 2-bromo-1-phenylethan-1-one (45), a transient photo-absorbing chiral EDA complex 47 was formed via an n → π* interaction in the ground state. Visible light irradiation of the colored EDA complex 47 initiated a SET process to give the contact radical ion pair 48, which is stabilized by coulombic attractive forces to preserve the original orientation. Subsequently, a positively charged intermediate pair was generated with the release of the bromide anion. Following radical–radical coupling and hydrolysis, the enantioenriched α-alkylated aldehyde was produced in the absence of any external photocatalyst.
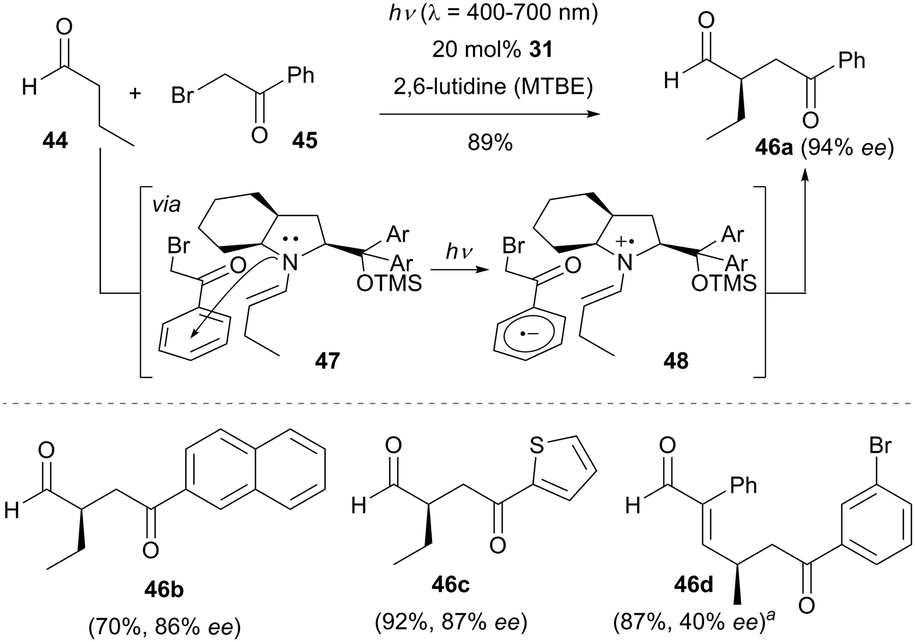 | ||
| Scheme 11 Enantioselective α-alkylation of aldehydes catalyzed by 31via an electron donor–acceptor (EDA) complex mechanism. aChiral amine 2 was used as the organocatalyst. MTBE = methyl tert-butyl ether. | ||
It should be noted that a radical-chain propagation pathway cannot entirely ruled out at this stage. The success of this reaction is attributed to the formation of the colored EDA complex 47 which absorbs the visible light. This methodology tolerates a wide range of aldehydes and phenacyl bromides, even a thiophene containing bromide reacted with butanal to give the product 46c in 92% isolated yield with 87% ee. γ-Site selective alkylation of enals was also achieved by using chiral amine 2 as the organocatalyst (e.g., 46d).
The EDA complex strategy could also be applied to the enantioselective α-alkylation of cyclohexanone (49).39 Switching the light source to a 300 W xenon lamp (λ = 300–600 nm), cyclohexanone (49) was coupled with a broad array of phenacyl bromides in the presence of quinidine-derived chiral primary amine 36, which led to the α-alkylated cyclohexanones 50 with excellent enantioselectivity (Scheme 12). The work described here expanded the substrate scope of enantioselective amine-catalyzed α-alkylation reactions to cyclic ketones.
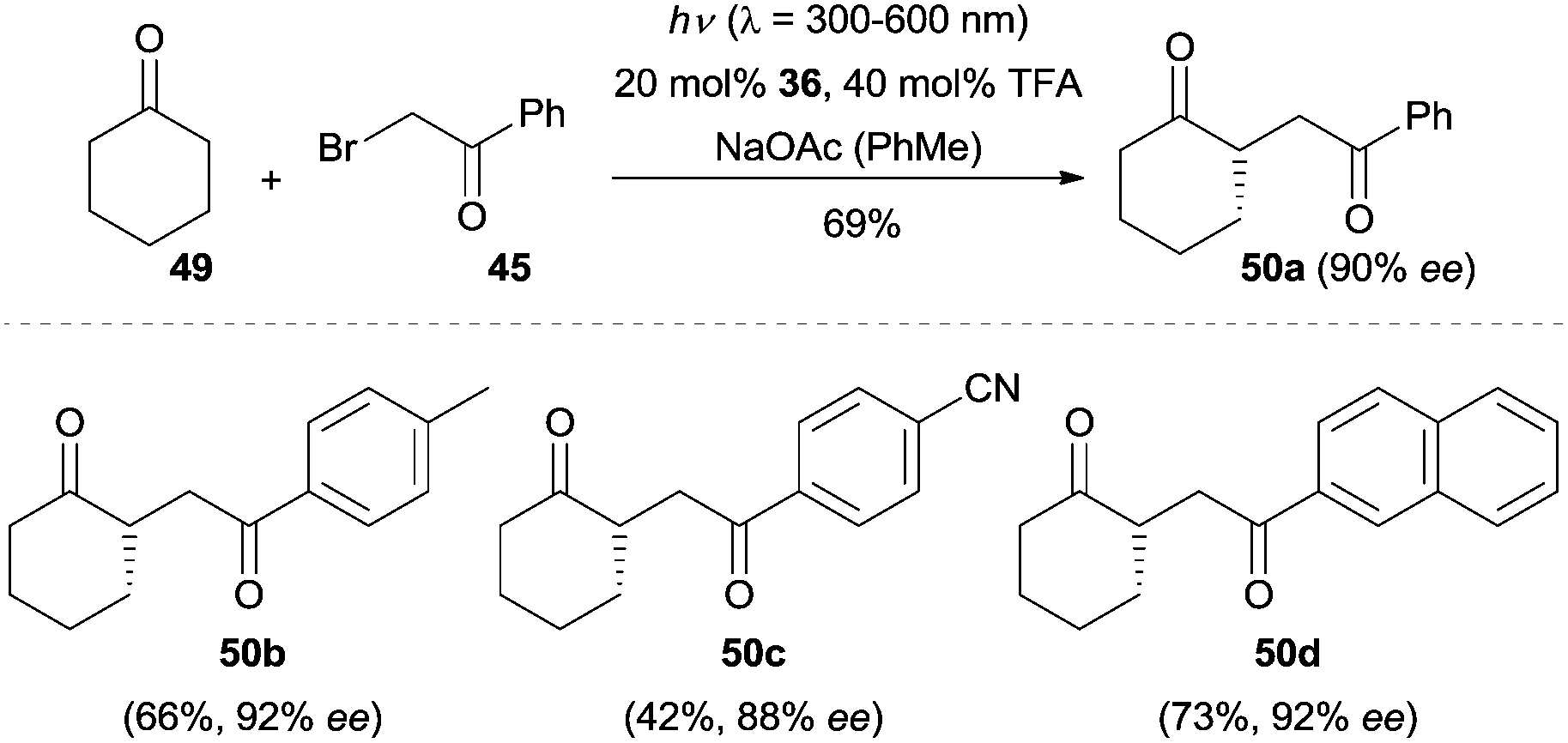 | ||
| Scheme 12 Enantioselective α-alkylation of cyclohexanone (49) via an electron donor–acceptor (EDA) complex mechanism. | ||
At a later stage, Melchiorre and co-workers disclosed a direct photoexcitation strategy to achieve the enantioselective α-alkylation of aldehydes.40 By using bromomalonates as the reaction partners, no color change was observed when exposed to the enamine intermediate 52. This observation led the authors to propose a unique direct excitation pathway, where upon irradiation the enamine 52 populates its excited state 52* (Scheme 13). Then 52* acts as a photosensitizer to reduce the bromomalonate to the open-shell radical species, which in turn couples with the ground state enamine 52 followed by an oxidation via another bromomalonate and following hydrolysis the final products are generated. In the entire catalytic cycle, the excited 52* was sacrificed and a chain propagation mechanism was involved. To gain more insight into the mechanism of this transformation, detailed photophysical studies, nuclear magnetic resonance (NMR) spectroscopy as well as kinetic studies were recently conducted by the same group.41
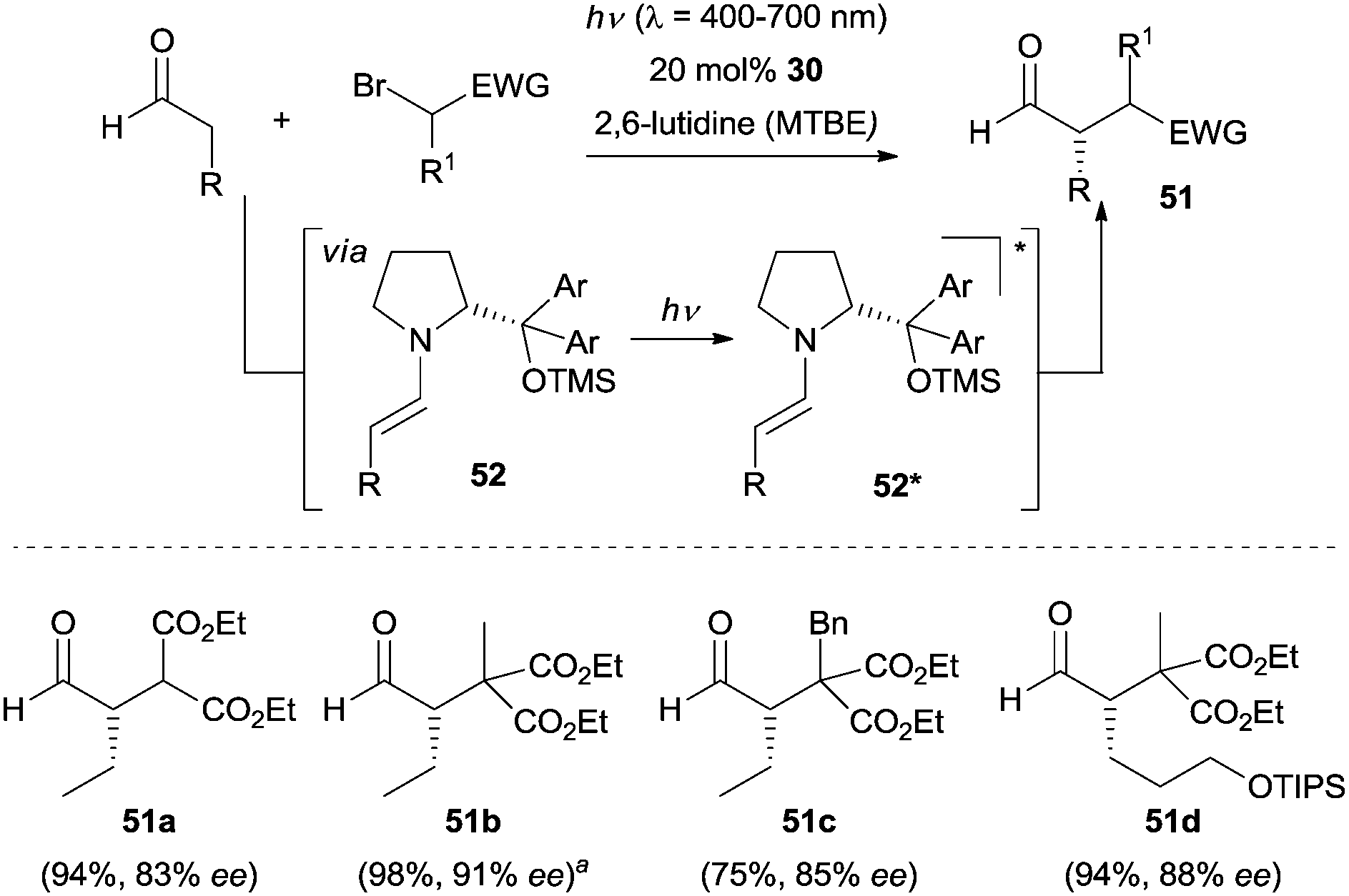 | ||
| Scheme 13 Enantioselective α-alkylation of aldehydes via the direct excitation of enamines 52. aReaction performed upon sunlight irradiation. TIPS = triisopropylsilyl. | ||
Furthermore, the above direct excitation protocol could be extended to the enantioselective α-(phenylsulfonyl)methylation of aldehydes by using ent-30 as the organocatalyst.42 Na2S2O3 was added to remove possible trace amounts of iodine in the reaction mixture and therefore improved the reaction efficiency. A (phenylsulfonyl)methyl or (methylsulfonyl)methyl group could readily be incorporated into various aldehyde components, and the chiral α-alkylated products were isolated as the corresponding alcohols after in situ reduction with NaBH4 in moderate to good yields with good enantioselectivity (Scheme 14). The reaction proceeds via an atom-transfer radical addition (ATRA) mechanism and the phenylsulfonyl moiety from the iodide substrates acted as a redox auxiliary group to facilitate the formation of the radical key intermediate.
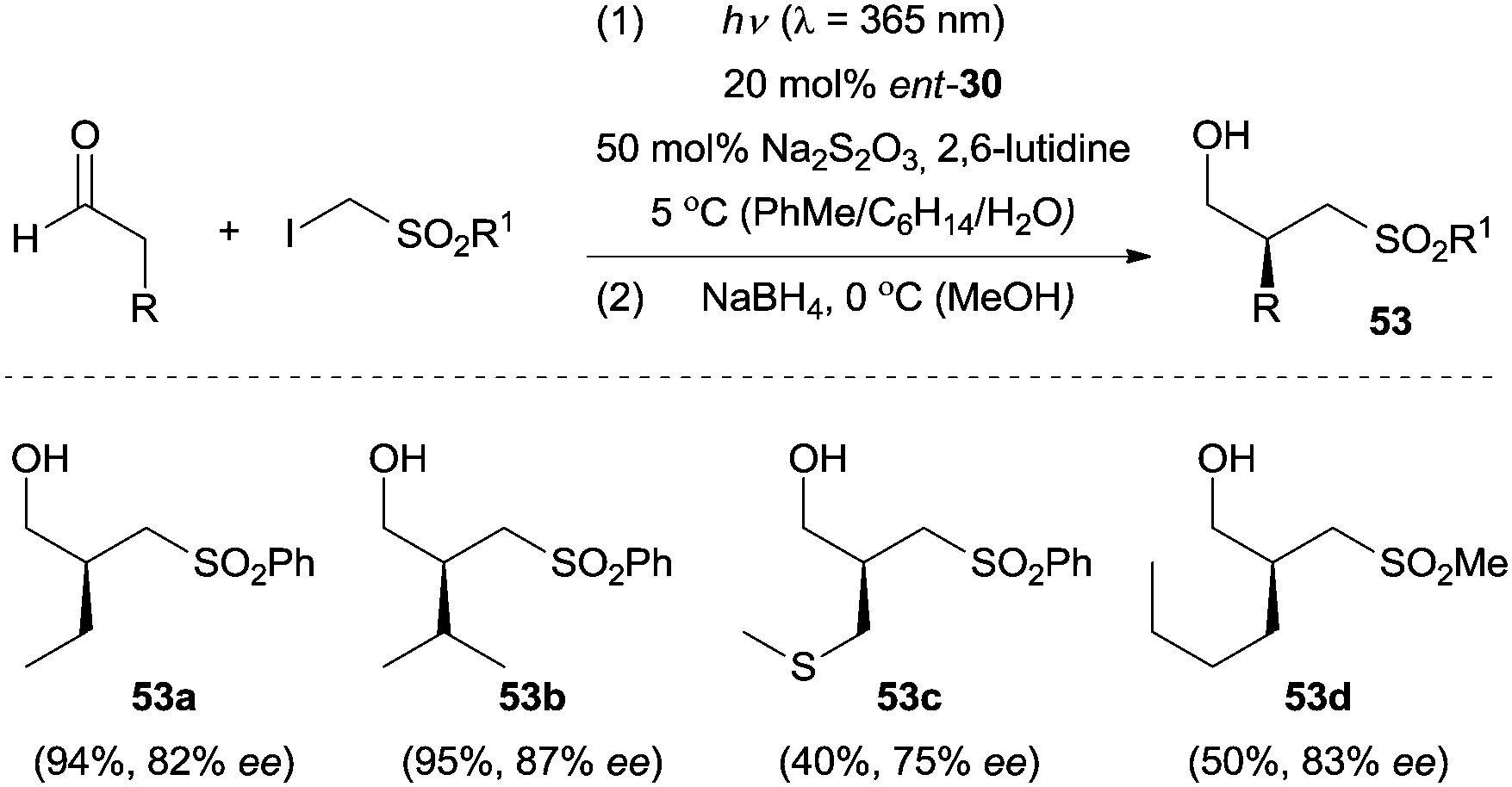 | ||
| Scheme 14 Enantioselective α-(phenylsulfonyl)- and α-(methylsulfonyl)methylation of aldehydes catalyzed by ent-30. | ||
As shown above, the alkylation reagents used in the enantioselective synthesis of α-alkylated carbonyl compounds are always pre-activated halides, which do not meet the atom economy ideals of modern organic synthesis. Consequently, the direct enantioselective α-alkylation of carbonyl compounds from simple and abundant synthetic building blocks is highly desirable. Recently, MacMillan and co-workers reported an elegant enantioselective α-alkylation of aldehydes with simple olefins by synergistically merging photoredox, enamine and hydrogen-atom transfer (HAT) catalysis.43 As described in Scheme 15, the N-tethered aldehydic olefin 54 is initially converted to the chiral enamine by condensation with organocatalyst 25b, which in turn is oxidized to the enaminyl radical 56 by the excited photocatalyst *Ir[III]. The enaminyl radical 56 rapidly adds to the olefin moiety to generate a nucleophilic radical 57 through an intramolecular radical–radical coupling. Subsequently, radical 57 participates in a HAT catalytic cycle mediated by 2,4,6-triisopropylbenzenethiol affording the key iminium ion following a hydrolysis to the desired product 55a. Concurrently, the thiyl radical arising from the HAT catalytic cycle reacts with the reduced photocatalyst Ir[II] to regenerate the ground state photocatalyst Ir[III] and the HAT thiol catalyst. N-Tethered, O-tethered and even alkyl tethered aldehydic olefins were all compatible with the optimised conditions, giving rise to chiral hetero- or carbocyclic ring systems (e.g., 55a–55e) in moderate to good yields with high enantiocontrol.
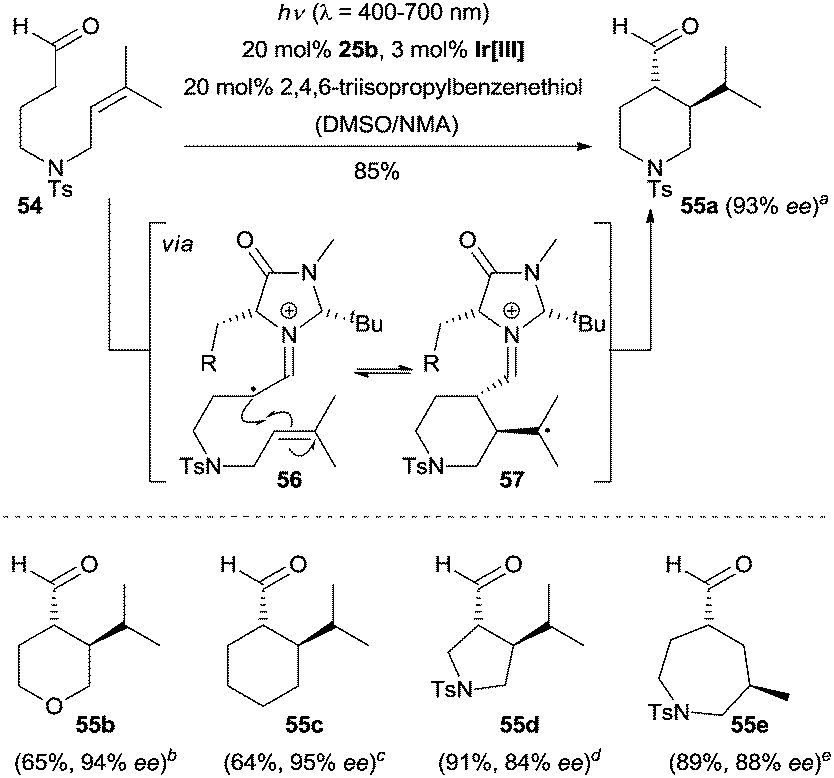 | ||
| Scheme 15 Enantioselective α-alkylation of aldehydes catalyzed by 25b. Ir[III] = Ir(Fmppy)2(dtbbpy)PF6 [Fmppy = 2-(4-fluorophenyl)-4-(methylpyridine)]. ad.r. = 91/9. bd.r. = 91/9. cd.r. > 95/5. dd.r. > 95/5. ed.r. = 87.5/12.5. The ee refers to the major diastereoisomer. NMA = N-methyl acetamide. | ||
The authors found this tricatalytic strategy was also suitable for an intermolecular variant. Using 20 mol% of chiral amine ent-30 as the organocatalyst, 1 mol% of Ir(dmppy)2(dtbbpy)PF6 [dmppy = 2-(4-methylphenyl)-4-(methylpyridine)] as the photocatalyst and 10 mol% of 2,4,6-tri-tert-butyl benzenethiol as the HAT catalyst, a large number of aldehydes reacted with different styrenes, providing the α-alkylated aldehydes with excellent enantioselectivity (e.g., 58a–58d). Additionally, methylenecyclopentane was also subjected to these conditions outlined in Scheme 16, and product 58e was obtained in 90% ee.
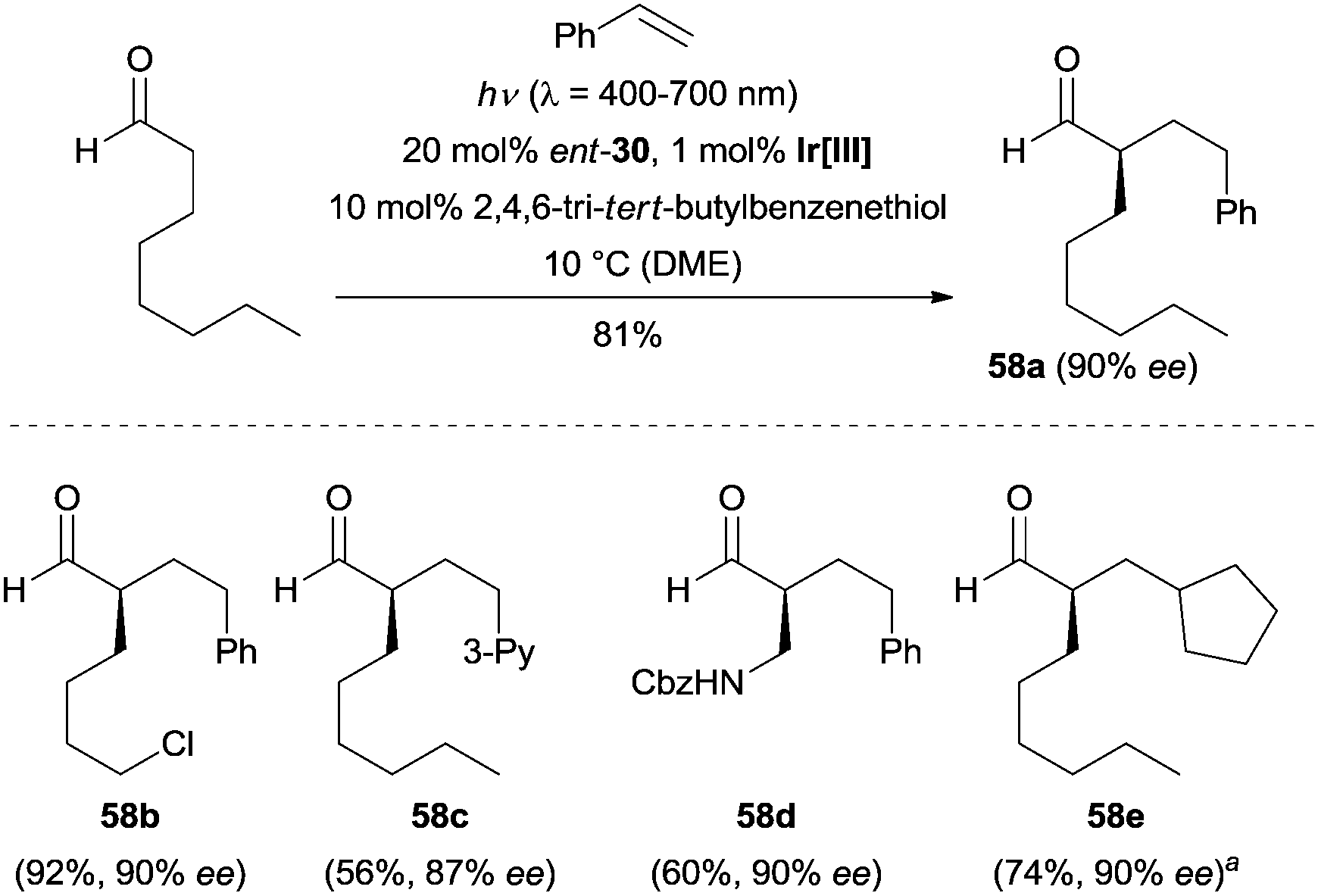 | ||
| Scheme 16 Intermolecular enantioselective α-alkylation of aldehydes catalyzed by ent-30. Ir[III] = Ir(dmppy)2(dtbbpy)PF6. aReaction was performed at −65 °C. DME = dimethoxyethane, 3-Py = 3-pyridine, Cbz = carboxybenzyl. | ||
The Luo group recognized that the chiral primary amine 32 was a robust catalyst for the enantioselective α-alkylation of β-ketocarbonyl compounds by merging photoredox catalysis and enamine catalysis.44 Under their optimized reaction conditions, a large array of acyclic ketoesters, cyclic ketoesters, and aliphatic 1,3-diketones were coupled to various phenylacyl bromides, and the enantioenriched compounds 59a–59d bearing an all-carbon quaternary stereogenic center were constructed in a highly concise fashion (Scheme 17). Surprisingly, when N-aryl substituted β-ketoamides were employed as the reactants, spiro-γ-lactams (e.g., 59e–59g) with two nonadjacent quaternary stereogenic centers were isolated as products of an α-alkylation/intramolecular ketalization cascade with excellent enantioselectivity and diastereoselectivity. The hydrogen bonding between the protonated chiral tertiary amine moiety and the keto moiety of β-ketocarbonyl compounds was crucial for the stereocontrol, which guided the well-defined orientation of the chiral complex 60 formed from enamine intermediate and the open-shell radical species.
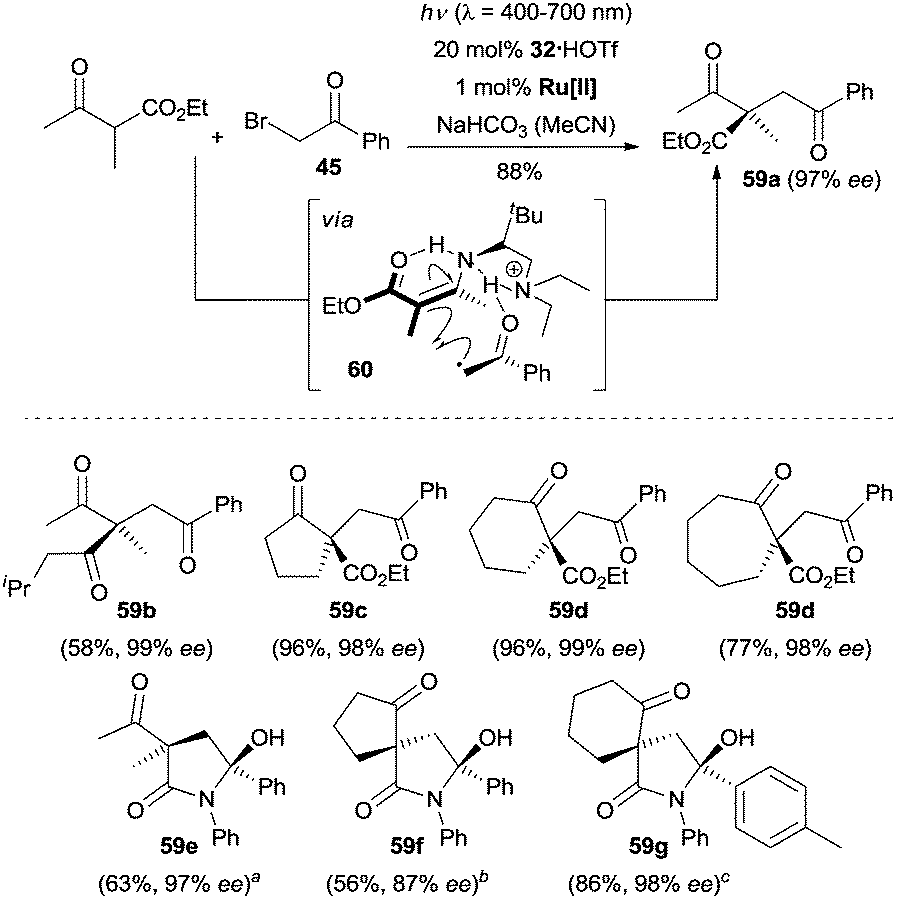 | ||
| Scheme 17 Enantioselective α-alkylation of β-ketocarbonyl compounds catalyzed by 32. Ru[II] = Ru(bpy)3Cl2·6H2O. ad.r. = 96/4. bd.r. = 94/6. cd.r. > 99/1. | ||
3.2 α-Benzylation of aldehydes and ketones
Although α-benzylation represents a subdivision of α-alkylation of carbonyl compounds, a subchapter is included to highlight the recent advances in the enantioselective photocatalytic α-benzylation of carbonyl compounds via enamine catalysis. The synergistic strategy combining photoredox catalysis and organocatalysis pioneered by the MacMillan group also proved to be effective for the enantioselective α-benzylation of aldehydes.45Scheme 18 shows that various electron-deficient aryl and heteroaryl methylene bromides can serve as viable benzylation reagents. The redox conditions were compatible with a wide range of aldehydes bearing different functional groups, all coupling reactions worked efficiently and enabled the facile access to α-benzylated aldehydes (e.g., 61a–61c) with excellent enantioselectivity.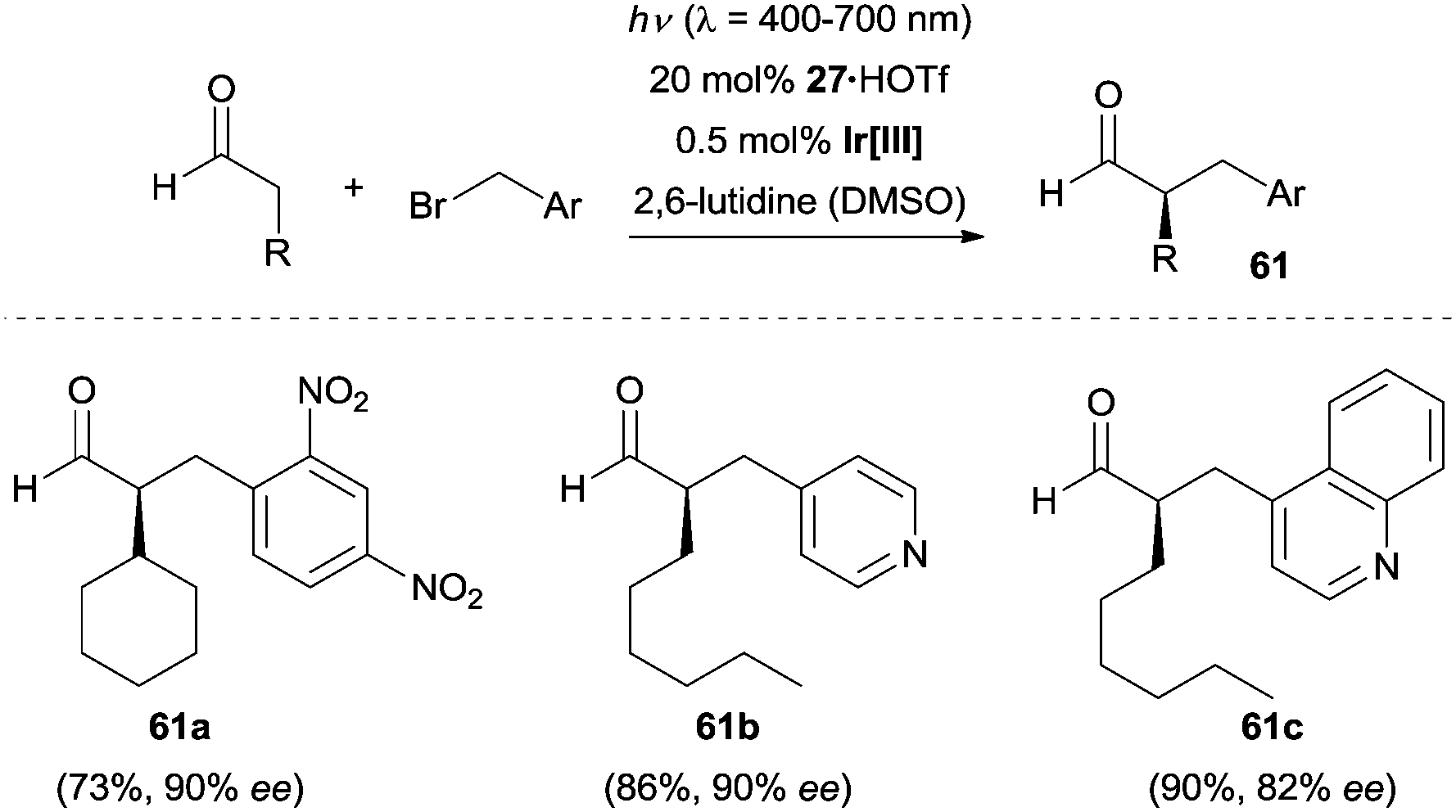 | ||
| Scheme 18 Enantioselective α-benzylation of aldehydes catalyzed by 27. Ir[III] = fac-Ir(ppy)3. | ||
In contrast to MacMillan's dual catalysis, the EDA complex mechanism can operate without the need of any external photocatalyst and this reaction has found wide applications in the enantioselective α-benzylation of aldehydes.38 The coloured EDA complex 64 was observed by Melchiorre and co-workers when treating electron-deficient benzyl bromides such as 1-(bromomethyl)-2,4-dinitrobenzene (62) with the chiral enamine intermediate derived from the corresponding aldehydes and the secondary amine 31. The EDA complex 64 undergoes an intermolecular SET process to generate a chiral radical ion pair 65 which in turn triggers the following radical–radical coupling step. As shown in Scheme 19, several electron-deficient benzyl bromides reacted with a range of aldehydes to achieve the α-benzylation of aldehydes with good enantiocontrol. Interestingly, 2-phenylpropanal could also be employed in the reaction when using chiral amine 2 as the organocatalyst and the transformation forged an all-carbon quaternary stereogenic centre product 63d in 86% ee.
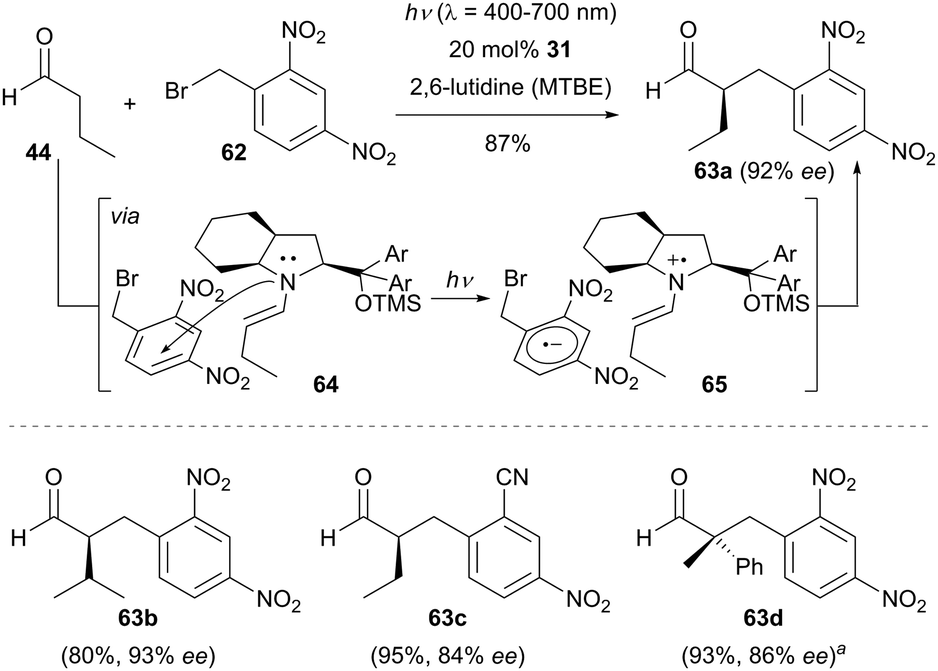 | ||
| Scheme 19 Enantioselective α-benzylation of aldehydes catalyzed by 31via an electron donor–acceptor (EDA) complex mechanism. aChiral amine 2 was used as the organocatalyst. | ||
The majority of α-alkylations and α-benzylations of carbonyl compounds are largely based on aldehyde precursors, and the α-alkylation or benzylation of ketones is still largely unexplored. In 2014, Melchiorre and co-workers realized the direct enantioselective α-alkylation of cyclohexanone via an EDA complex mechanism.39 They demonstrated that this protocol was also suitable for the α-benzylation of cyclic ketones by simply switching the light source to a 23 W compact fluorescent light (CFL) bulb (Scheme 20). Various cyclic ketones such as cyclopentanone, cyclohexanone, cycloheptanone as well as N-Boc-piperidin-4-one could be benzylated with 1-(bromomethyl)-2,4-dinitrobenzene (62) as the reaction partner, generating the desired products (e.g.66a–66d) with moderate to good enantioselectivity.
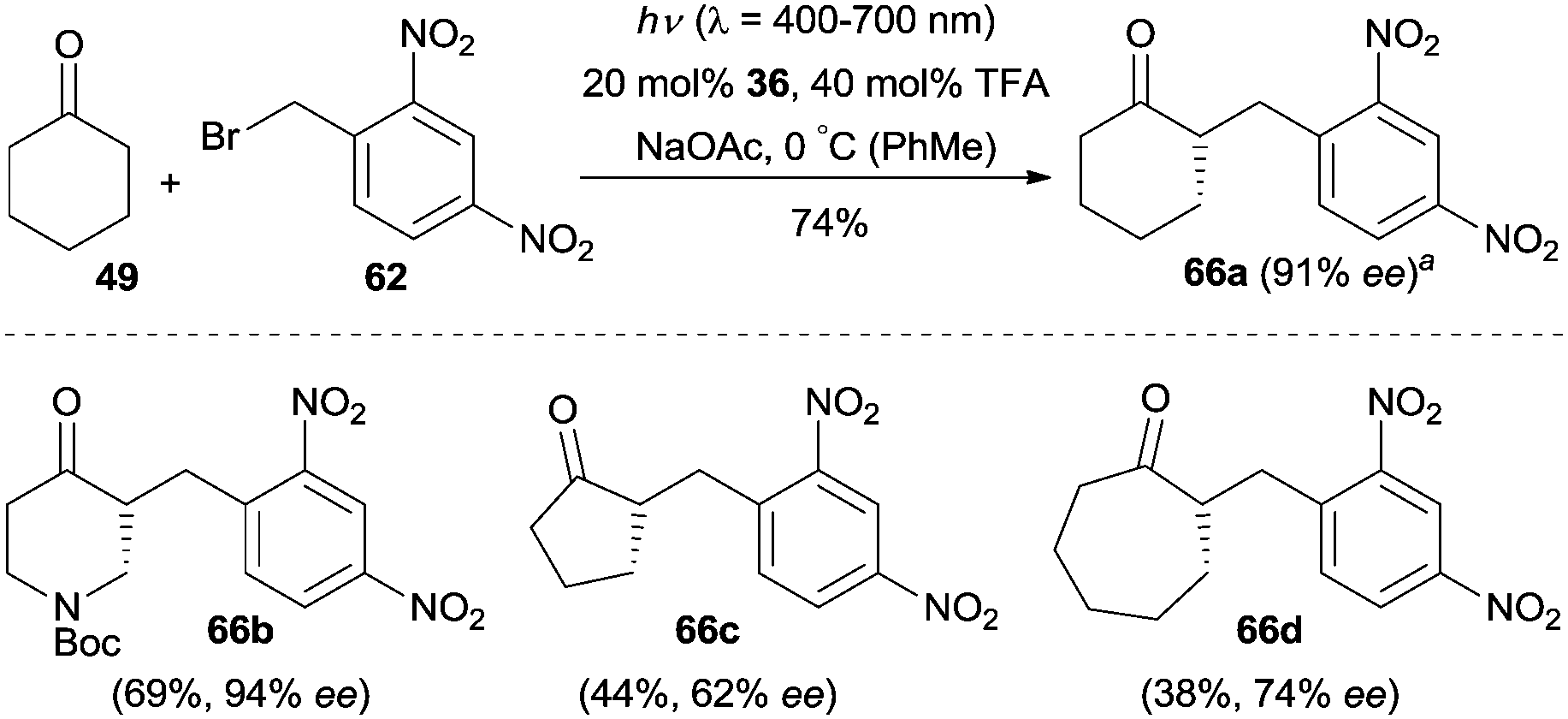 | ||
| Scheme 20 Enantioselective α-benzylation of cyclic ketones catalyzed by 36via an electron donor–acceptor (EDA) complex mechanism. aReaction performed on a 1 mmol scale. | ||
More recently, Wu, Luo and co-workers reported a unique tricatalytic system involving enamine catalysis, photocatalysis and transition metal catalysis to realize the enantioselective cross-dehydrogenative coupling (CDC) reaction between tetrahydroisoquinolines and ketones.46 By using 3 mol% of Ru(bpy)3Cl2·6H2O and 8 mol% of Co(dmgH)2Cl2 (dmgH = dimethylglyoximate), numerous cyclic and acyclic ketones were coupled with N-aryl tetrahydroisoquinolines and an enantioselective α-benzylation of ketones could be achieved by irradiation with visible light in the presence of 20 mol% of chiral amine 33 and 40 mol% of 3-nitrobenzoic acid (Scheme 21). The authors proposed an oxidative quenching mechanism in the photocatalytic cycle, where the excited photocatalyst Ru[II]* is first oxidized to Ru[III] by Co[III]. Subsequently, Ru[III] is suggested to oxidize the N-aryl tetrahydroisoquinoline to the iminium cation by SET and the release of an additional electron and a proton. By the aid of 3-nitrobenzoic acid, the reduced Co[II] captures the electron and the proton to regenerate Co[III]. In the enamine catalytic cycle, the ketone condenses with amine 33 to form a chiral enamine, which then intercepts the iminium cation via the transition state 68 to give the corresponding products. It is worth mentioning that 3-nitrobenzoic acid not only acts as an acid additive to promote the formation of the chiral enamine intermediate, but also plays an important role as a hydrogen acceptor undergoing an in situ hydrogenation step to 3-aminobenzoic acid.
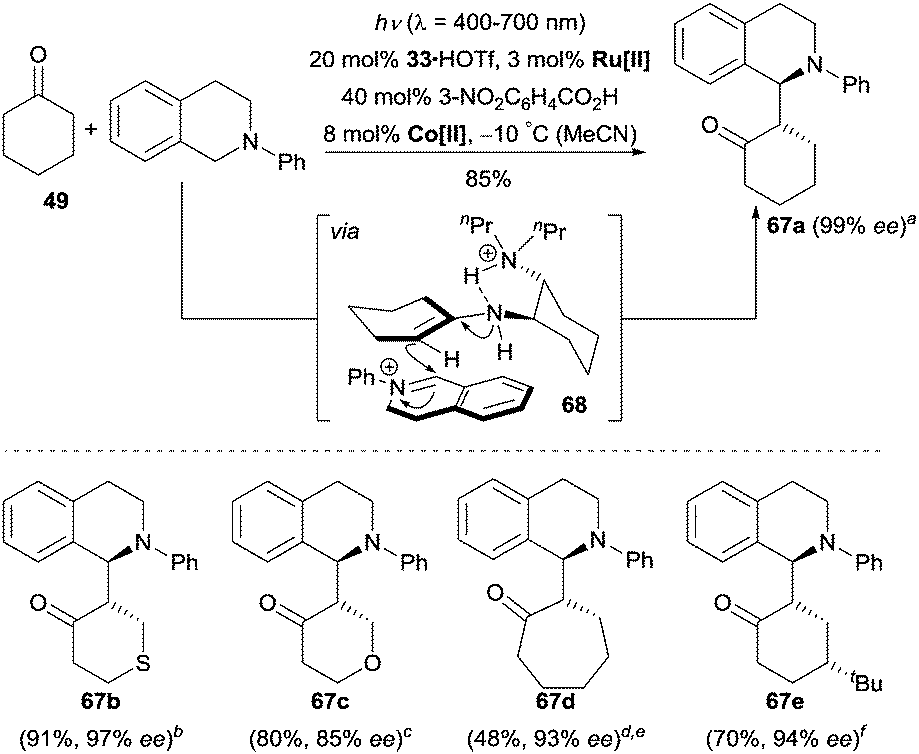 | ||
| Scheme 21 Enantioselective α-benzylation of cyclic ketones catalyzed by 33. Ru[II] = Ru(bpy)3Cl2·6H2O, Co[II] = Co(dmgH)2Cl2. ad.r. = 90/10. bd.r. = 89/11. cd.r. = 67/33. dd.r. = 92/8. eReaction performed at −5 °C. fd.r. = 89/11. The ee refers to the major diastereoisomer. | ||
3.3 α-Hydroxylation of aldehydes and ketones
Molecular oxygen is regarded as a green oxidant and has been widely used in organic synthesis. The direct incorporation of molecular oxygen into organic molecules is a straightforward and effective method for the oxidation of a desired target molecule. Given the fact that excited singlet oxygen (1O2) is more reactive than the ground state triplet oxygen (3O2), Córdova and co-workers reported the first example of an amine-catalyzed enantioselective α-hydroxylation of aldehydes under photochemical conditions.47 Optimization of the reaction conditions revealed (L)-α-Me proline (29) as the most effective organocatalyst. As depicted in Scheme 22, the 1,2-diols were isolated with moderate enantioselectivity after in situ reduction by NaBH4. The authors postulated a possible mechanism which was related to the enamine catalysis under thermal reactions. The chiral amino acid converts the aldehyde to the more electron-rich enamine 70, which exhibits increased nucleophilicity. Simultaneously, the photosensitizer tetraphenylporphyrin (TPP) sensitizes 3O2 to 1O2 upon irradiation with visible light. An ene-type reaction was suggested to operate linking 1O2 and enamines 70 and leading to the formation of α-hydroperoxides 71 followed by a reduction to afford diols 69. Later work from the same group showed that chiral amine 2 proved to be a more effective catalyst and the ee could be improved to 98% in some cases.48 Mechanistic studies of this transformation were carried out by the group of Gryko.49 They proposed the formation of a zwitterionic enamine peroxide intermediate and elucidated the enantioface preference for the individual amine catalysts in this reaction.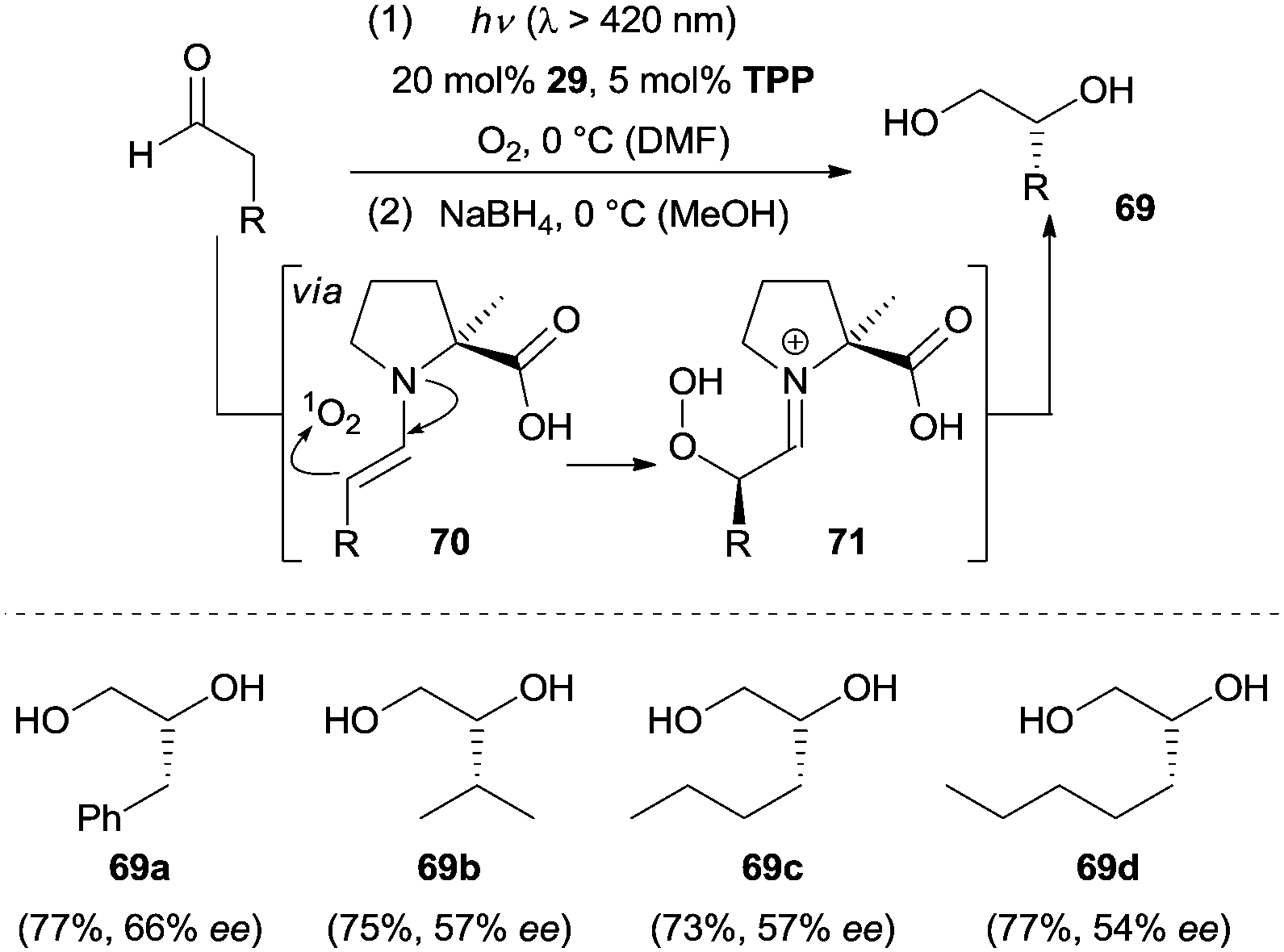 | ||
| Scheme 22 Enantioselective α-hydroxylation of aldehydes catalyzed by 29. | ||
A further application of the oxygenation methodology was achieved with ketones as the substrates and chiral amine 34 as the catalyst (Scheme 23).50 Cyclohexanone and C4-substituted cyclohexanones were readily oxidized, allowing for the synthesis of chiral α-hydroxylated cyclic ketones (e.g., 72a–72c). Furthermore, acyclic ketones such as octan-2-one could also be favourably employed for the enantioselective α-hydroxylation, albeit with lower ee (72d, 28% ee) by using 35 as the organocatalyst.
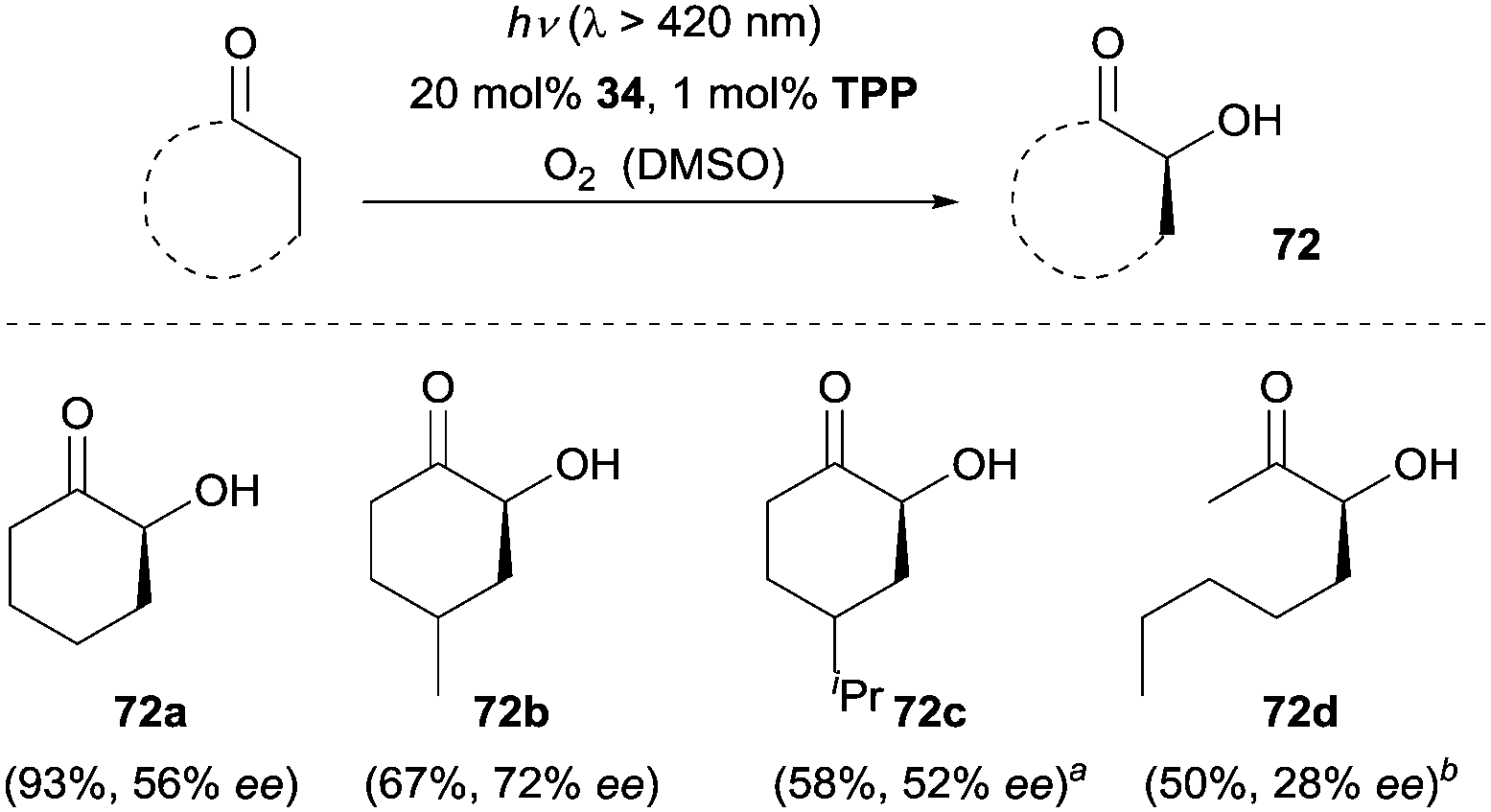 | ||
| Scheme 23 Enantioselective α-hydroxylation of ketones catalyzed by 34. aReaction was performed in N-methylpyrrolidinone (NMP). b35 was used as the catalyst. | ||
Jang and co-workers developed an enantioselective photocatalytic α-oxyamination of aldehydes under heterogeneous conditions.51 In this case, commercially available TiO2 was introduced as the photocatalyst, and chiral amines 2 or ent-30 were employed as the chiral amine catalysts. Remarkably, a wide range of aldehydes were coupled with (2,2,6,6-tetramethylpiperidine-1-yl)oxyl (TEMPO) to deliver the desired α-oxyaminated aldehydes with moderate to good enantioselectivity (up to 78% ee).
3.4 α-Amination of aldehydes
The photocatalytic generation of N-centered radicals has recently attracted increasing attention, resulting in new methods for the synthesis of nitrogen-containing compounds.52 In 2013, a breakthrough in this field was achieved by the group of MacMillan. They accomplished the direct enantioselective α-amination of aldehydes via a combination of photoredox catalysis and enamine catalysis.53 High enantioselectivities were observed when using chiral secondary amine 28 as the catalyst. Some representative products 74a–74d are shown in Scheme 24. Under optimal conditions the corresponding α-aminated aldehydes were produced in good yields with high enantioselectivity.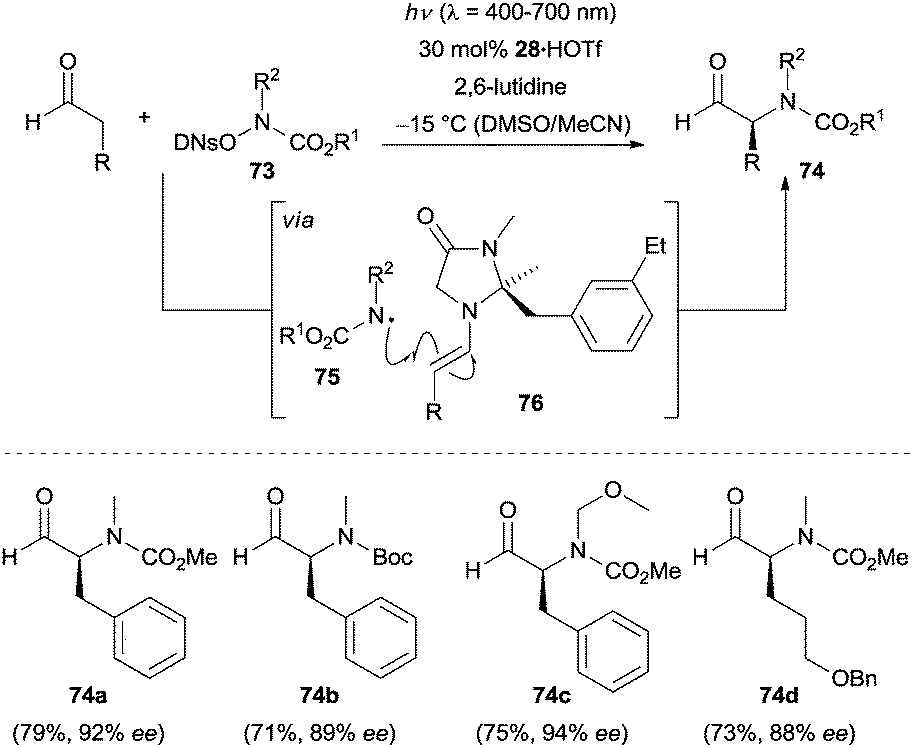 | ||
| Scheme 24 Enantioselective α-amination of aldehydes catalyzed by 28. | ||
An external photocatalyst was not employed in this amination reaction. The success of the transformation strongly depends on the leaving group 2,4-dinitrophenylsulfonyl (DNs) in the amination reagent 73. Mechanistically, the open-shell N-centered radical 75 is initially generated upon irradiation with visible light, which then rapidly adds to the electron-rich enamine 76 generated from the condensation of aldehyde and chiral amine 28. Oxidation of the resulting α-amino radical intermediate by the photoexcited amination reagent 73 forms the second equivalent of N-centered radical 75 accompanying the formation of the iminium ion. Hydrolysis of the iminium ion furnishes the final product and releases the chiral amine 28 to re-enter the catalytic cycle.
3.5 β-Arylation of ketones
Owing to the low reactivity of the β-methylene position of saturated aldehydes and ketones, the direct β-functionalization of this class of substrates is a challenging task. Previous methods used for β-carbonyl activation were exclusively based on the addition of soft nucleophiles to pre-oxidized α,β-unsaturated aldehydes and ketones. By means of their synergistic photoredox and organocatalysis, MacMillan and his group illustrated that a variety of saturated aldehydes and ketones react efficiently with a large array of cyanobenzenes and cyanoheteroarenes to deliver the β-arylated carbonyl compounds without the formation of any α-arylated products (Scheme 25).54 As an asymmetric variant of this reaction, a promising level of enantioselectivity was reported in the reaction of cyclohexanone (49) and 1,4-dicyanobenzene (77) using the cinchona-derived catalyst 37 (50% ee).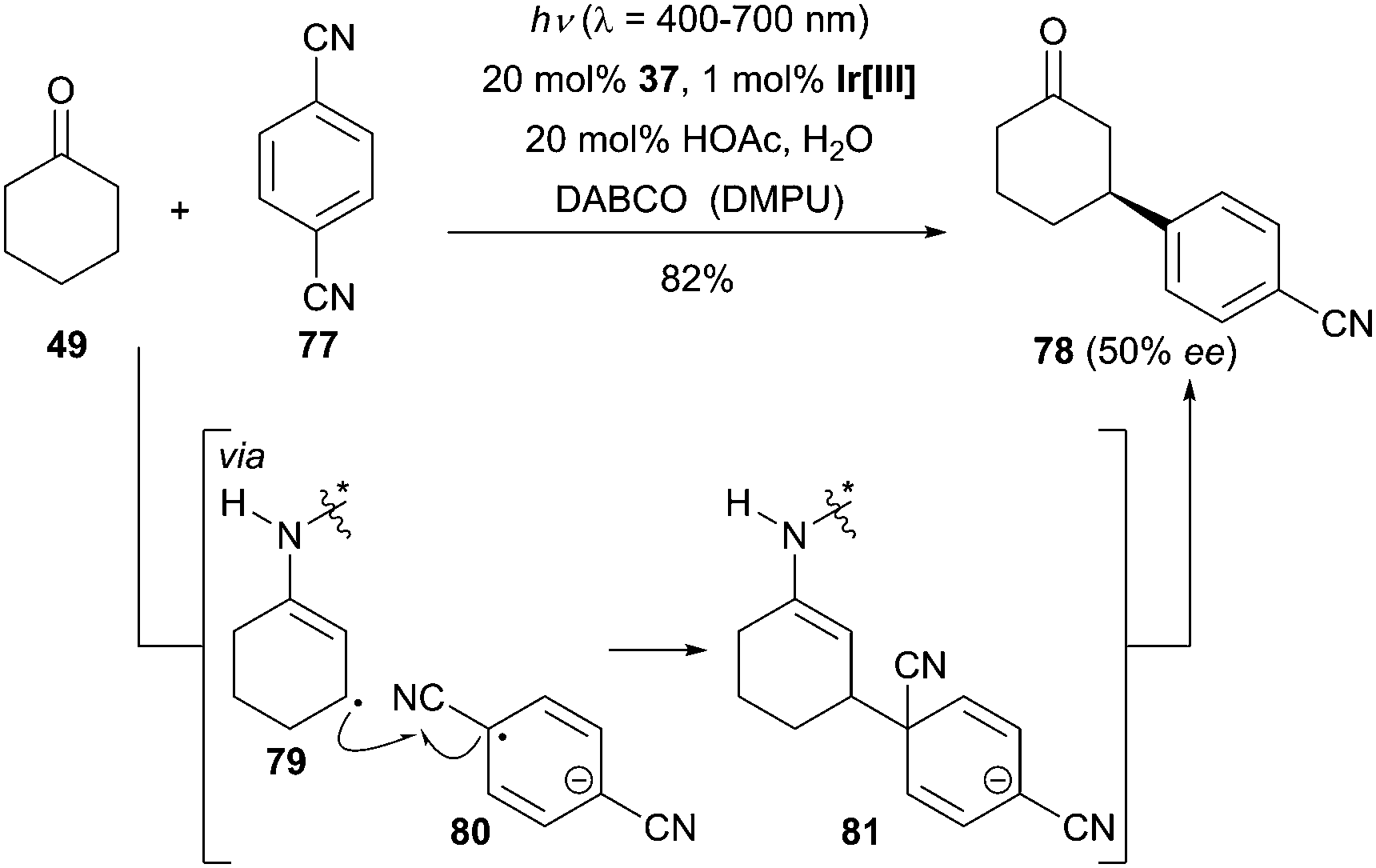 | ||
| Scheme 25 Enantioselective β-arylation of cyclohexanone (49) catalyzed by 37. Ir[III] = Ir(ppy)3, DABCO = 1,4-diazabicyclo[2.2.2]octane, DMPU = 1,3-dimethyltetrahydropyrimidin-2(1H)-one. | ||
The authors postulated a possible mechanism to explain the formation of the β-arylated products. The initial step entailed an oxidative quenching of the photoexcited catalyst Ir[III] by 1,4-dicyanobenzene (77) giving rise to the arene radical anion 80. The authors hypothesized that the resulting Ir[IV] would oxidize the enamine intermediate following deprotonation to yield the β-enamine radical 79. Subsequently, intermolecular radical–radical coupling occurs allowing the formation of intermediate 81, which then undergoes rapid elimination of cyanide and hydrolysis to the desired products. Later, the above-mentioned strategy was also expanded to the β-alkylation of aldehydes.55
4. Conclusions and outlook
The continuously growing interest in enantioselective catalysis has resulted in new developments in the field of enantioselective photocatalysis especially using visible light. This research area promises use of light as an abundant and sustainable source of energy. The development of enantioselective organocatalysis has inspired chemists to develop novel concepts in the pursuit of enantioselective photochemistry, and numerous elegant transformations have been designed and implemented by synergistic enantioselective organocatalysis and photocatalysis. In this tutorial review, we have described the recent achievements in iminium and enamine catalysis in enantioselective photochemical reactions. Two different chiral active species, iminium ions or enamines derived from the condensation of carbonyl groups and chiral amines are generated in the organocatalytic cycle, which are intercepted by the photogenerated reactive species to furnish enantioenriched useful molecules.Despite the advances in the field, several challenges and research topics remain: (1) the enantioselective photochemical reactions illustrated above are mainly based on the α-functionalization of aldehydes and ketones and the β-functionalization of enals and enones. It would be desirable to extend these protocols to other reactions types. (2) The chiral amine toolbox is not yet as versatile as the toolbox for thermal iminium and enamine catalysis and the development of novel and robust chiral amines is highly required. (3) Although some experimental data have been obtained, further theoretical and spectroscopic studies are required, which will help to understand the detailed mechanisms and guide the design of novel methods. (4) Applying the concepts described in this review to the total synthesis of some biologically important natural products and pharmaceuticals would be attractive and further work is expected along these lines.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y.-Q. Zou acknowledges the Alexander von Humboldt foundation and the Carl Friedrich von Siemens foundation for a research fellowship. Funding of our own work in the field is provided by the European Research Council under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 665951 – ELICOS). We thank Dr T. S. Chung and Dr J. D. Jolliffe for proof-reading the final version of this manuscript.References
- K. Ding and L.-X. Dai, Organic Chemistry – Breakthroughs and Perspectives, Wiley-VCH, Weinheim, 2012 Search PubMed.
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer-Verlag, Heidelberg, 1999, vol. I–III Search PubMed.
- Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS PubMed.
- T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed.
- E. Meggers, Chem. Commun., 2015, 51, 3290–3301 RSC.
- A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416–5470 CrossRef PubMed.
- C. Chen, V. Chang, X. Cai, E. Duesler and P. S. Mariano, J. Am. Chem. Soc., 2001, 123, 6433–6434 CrossRef CAS PubMed.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- N. C. Yang and C. Rivas, J. Am. Chem. Soc., 1961, 83, 2213 CrossRef CAS.
- B. Grosch, C. N. Orlebar, E. Herdtweck, W. Massa and T. Bach, Angew. Chem., Int. Ed., 2003, 42, 3693–3696 CrossRef CAS PubMed.
- L. Dell’Amico, A. Vega-Peñaloza, S. Cuadros and P. Melchiorre, Angew. Chem., Int. Ed., 2016, 55, 3313–3317 CrossRef PubMed.
- L. Dell’Amico, V. M. Fernández-Alvarez, F. Maseras and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 3304–3308 CrossRef PubMed.
- X. Yuan, S. Dong, Z. Liu, G. Wu, C. Zou and J. Ye, Org. Lett., 2017, 19, 2322–2325 CrossRef CAS PubMed.
- D. A. Nagib, Chem, 2017, 2, 616–618 CAS.
- P. S. Mariano, Tetrahedron, 1983, 39, 3845–3879 CrossRef CAS.
- M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868–873 CrossRef CAS PubMed.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946–1956 CrossRef CAS PubMed.
- A. Bahamonde, J. J. Murphy, M. Savarese, É. Brémond, A. Cavalli and P. Melchiorre, J. Am. Chem. Soc., 2017, 139, 4559–4567 CrossRef CAS PubMed.
- H.-S. Yoon, X.-H. Ho, J. Jang, H.-J. Lee, S.-J. Kim and H.-Y. Jang, Org. Lett., 2012, 14, 3272–3275 CrossRef CAS PubMed.
- S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471–5569 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927–5931 CrossRef CAS.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- K. Fidaly, C. Ceballos, A. Falguières, M. S.-I. Veitia, A. Guy and C. Ferroud, Green Chem., 2012, 14, 1293–1297 RSC.
- M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062–4066 CrossRef CAS PubMed.
- P. Riente, A. M. Adams, J. Albero, E. Palomares and M. A. Pericàs, Angew. Chem., Int. Ed., 2014, 53, 9613–9616 CrossRef CAS PubMed.
- X. Li, J. Wang, D. Xu, Z. Sun, Q. Zhao, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, ACS Sustainable Chem. Eng., 2015, 3, 1017–1022 CrossRef CAS.
- P. Wu, C. He, J. Wang, X. Peng, X. Li, Y. An and C. Duan, J. Am. Chem. Soc., 2012, 134, 14991–14999 CrossRef CAS PubMed.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- E. R. Welin, A. A. Warkentin, J. C. Conrad and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 9668–9672 CrossRef CAS PubMed.
- C. G. S. Lima, T. de, M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- E. Arceo, A. Bahamonde, G. Bergonzini and P. Melchiorre, Chem. Sci., 2014, 5, 2438–2442 RSC.
- M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed.
- A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed.
- G. Filippini, M. Silvi and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 4447–4451 CrossRef CAS PubMed.
- A. G. Capacci, J. T. Malinowski, N. J. McAlpine, J. Kuhne and D. W. C. MacMillan, Nat. Chem., 2017 DOI:10.1038/nchem.2797.
- Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2014, 136, 14642–14645 CrossRef CAS PubMed.
- H.-W. Shih, M. N. Vander Wal, R. L. Grange and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 13600–13603 CrossRef CAS PubMed.
- Q. Yang, L. Zhang, C. Ye, S. Luo, L.-Z. Wu and C.-H. Tung, Angew. Chem., Int. Ed., 2017, 56, 3694–3698 CrossRef CAS PubMed.
- A. Córdova, H. Sundén, M. Engqvist, I. Ibrahem and J. Casas, J. Am. Chem. Soc., 2004, 126, 8914–8915 CrossRef PubMed.
- I. Ibrahem, G.-L. Zhao, H. Sundén and A. Córdova, Tetrahedron Lett., 2006, 47, 4659–4663 CrossRef CAS.
- D. J. Walaszek, K. Rybicka-Jasińska, S. Smoleń, M. Karczewski and D. Gryko, Adv. Synth. Catal., 2015, 357, 2061–2070 CrossRef CAS.
- H. Sundén, M. Engqvist, J. Casas, I. Ibrahem and A. Córdova, Angew. Chem., Int. Ed., 2004, 43, 6532–6535 CrossRef PubMed.
- X.-H. Ho, M.-J. Kang, S.-J. Kim, E. D. Park and H.-Y. Jang, Catal. Sci. Technol., 2011, 1, 923–926 CAS.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- G. Cecere, C. M. König, J. L. Alleva and D. W. C. MacMillan, J. Am. Chem. Soc., 2013, 135, 11521–11524 CrossRef CAS PubMed.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef CAS PubMed.
- J. A. Terrett, M. D. Clift and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 6858–6861 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |