
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic (de)hydrogenation promoted by non-precious metals – Co, Fe and Mn: recent advances in an emerging field†
Georgy A.
Filonenko
 *ab,
Robbert
van Putten
ab,
Emiel J. M.
Hensen
a and
Evgeny A.
Pidko
*bc
*ab,
Robbert
van Putten
ab,
Emiel J. M.
Hensen
a and
Evgeny A.
Pidko
*bc
aInorganic Materials Chemistry Group, Schuit Institute of Catalysis, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
bDepartment of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: g.a.filonenko@tudelft.nl; e.a.pidko@tudelft.nl
cITMO University, Lomonosova 9, St. Petersburg, 191002, Russia
First published on 15th January 2018
Abstract
Catalytic hydrogenation and dehydrogenation reactions form the core of the modern chemical industry. This vast class of reactions is found in any part of chemical synthesis starting from the milligram-scale exploratory organic chemistry to the multi-ton base chemicals production. Noble metal catalysis has long been the key driving force in enabling these transformations with carbonyl substrates and their nitrogen-containing counterparts. This review is aimed at introducing the reader to the remarkable progress made in the last three years in the development of base metal catalysts for hydrogenations and dehydrogenative transformations.
 Georgy A. Filonenko | Georgy Filonenko (Elizovo, Russia, 1988) received his PhD from Eindhoven University of Technology in 2015 under the supervision of Prof. Emiel Hensen. After completing a two-year research study at Okinawa Institute of Science and Technology he joined the team of Evgeny Pidko in July 2017 as a postdoctoral researcher. His research interests include physical organometallic chemistry and mechanistic studies of catalytic hydrogenation reactions. |
 Robbert van Putten | Robbert van Putten (The Netherlands, 1992) received his MSc from Eindhoven University of Technology in 2017. During his work in the Inorganic Materials Chemistry group at TU/e, he contributed to the development of homogeneous catalysts for the hydrogenation of CO2 and carboxylic acid esters based on Ru and Mn metals. After completing industrial internships at Shell and ASML, he joined the PhD program at Delft University of Technology with the group of Evgeny Pidko in 2017. |
 Emiel J. M. Hensen | Emiel Hensen (Geleen, The Netherlands, 1971) obtained his PhD from Eindhoven University of Technology in 2000. After working as an assistant professor at the University of Amsterdam he returned to Eindhoven in 2001 and became a full professor in 2009. From 2006 to 2008, he was a visiting research scientist at Shell, Amsterdam. He is chairman of the Netherlands Institute for Catalysis Research (NIOK), board member of the European Research Institute on Catalysis (ERIC) and management team member of the Netherlands gravitation centre on Multiscale Catalytic Energy Conversion (MCEC). His research interests include mechanisms of heterogeneous catalysis for natural and syngas conversion, biomass conversion, solar fuels and topics related to synthesis of porous catalysts. |
 Evgeny A. Pidko | Evgeny Pidko (Moscow, Russia, 1982) received his PhD from Eindhoven University of Technology in 2008, wherein from 2011 to 2017 he was an Assistant Professor of Catalysis for Sustainability. In 2016 he became a part-time professor of theoretical chemistry at ITMO University, St. Petersburg. Since September 2017 he has been an Associate Professor and head of the Inorganic Systems Engineering group at the Chemical Engineering Department of Delft University of Technology. In his research he successfully combines experiments and theory to understand the molecular mechanisms underlying the behaviour of various chemical systems ranging from heterogeneous and homogeneous catalysis to inorganic functional materials. |
1. Introduction
Interconversions of organic substrates involving hydrogen transfer constitute a broad class of industrially relevant chemical reactions. Either in molecular form or in the form of protons and hydrides, hydrogen is added, abstracted or shuffled between organic compounds in reactions that are almost universally catalytic. Efficient catalysis can promote both addition of hydrogen in a reductive process and hydrogen abstraction in the oxidative process. Moreover, multistep reactions involving oxidative, reductive and bond-forming events are also possible given that the right catalyst and conditions are ensured. As a result, a vast number of interconnected reactions are accessible via (de)hydrogenative catalysis (Scheme 1). Obtaining control over these reactions would grant chemists access to a wide variety of useful synthons and building blocks. In this setting, homogeneous catalysis becomes a powerful tool that allows desired products to be obtained via multiple catalytic pathways.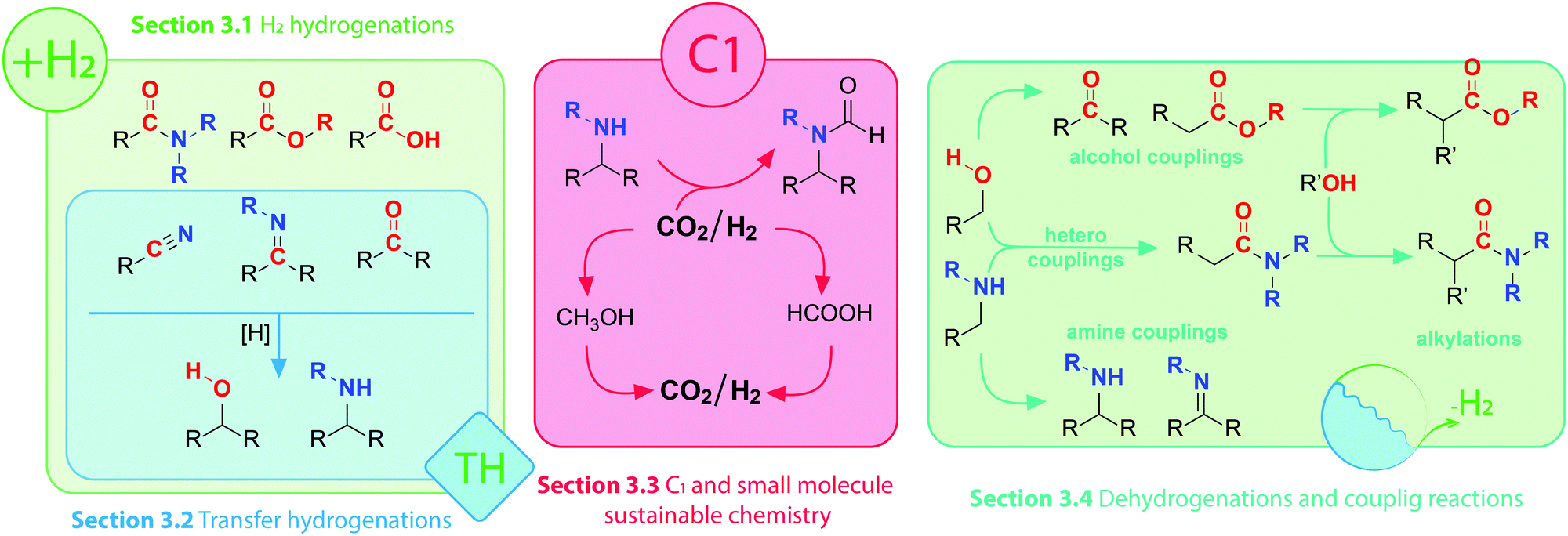 | ||
| Scheme 1 Visual guide to corresponding sub-sections of this Review and schematic layout of selected transformations of polar organic compounds. | ||
In this Review we present a critical analysis of the recent advances in catalytic (de)hydrogenation with 3d transition metal complexes enabling efficient transformations of amines, alcohols and their oxidized counterparts bearing imine, carbonyl or carboxyl functions (Scheme 1). As reactions in Scheme 1 can be split into several categories depending on their nature, we will structure our Review accordingly.
The first group of reactions are reductive transformations of carbonyl compounds, carboxylic acids and their derivatives. Important products such as alcohols and amines are produced through these transformations. Importantly, the reactivity of the carbonyl moiety in the reaction substrates varies greatly depending on the nature of the substrate and the electrophilicity of the carbon in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O unit; whereas aldehydes are relatively easy to reduce, carboxylic acid derivatives present a greater challenge for the reduction due to their significantly lower electrophilicity.1 The common lab-scale synthetic approaches for the reduction of polar oxygenates and their nitrogen-containing counterparts often rely on the use of stoichiometric highly reactive reagents. One of the early protocols for the reduction of esters and carboxylic acids involved the use of metallic sodium in ethanol, known as Bouveault–Blanc-reduction.2,3 This method was later abandoned in favour of milder and more versatile metal-hydride reagents such as LiAlH4 and NaBH4.4 Despite the high efficiency of the respective synthetic protocols, their main drawback is the stoichiometric nature and, as a result, the production of large amounts of inorganic by-products. Furthermore, the high reactivity of inorganic hydrides may present potential safety hazards limiting their large-scale applicability.5
O unit; whereas aldehydes are relatively easy to reduce, carboxylic acid derivatives present a greater challenge for the reduction due to their significantly lower electrophilicity.1 The common lab-scale synthetic approaches for the reduction of polar oxygenates and their nitrogen-containing counterparts often rely on the use of stoichiometric highly reactive reagents. One of the early protocols for the reduction of esters and carboxylic acids involved the use of metallic sodium in ethanol, known as Bouveault–Blanc-reduction.2,3 This method was later abandoned in favour of milder and more versatile metal-hydride reagents such as LiAlH4 and NaBH4.4 Despite the high efficiency of the respective synthetic protocols, their main drawback is the stoichiometric nature and, as a result, the production of large amounts of inorganic by-products. Furthermore, the high reactivity of inorganic hydrides may present potential safety hazards limiting their large-scale applicability.5
Alternatives to stoichiometric methods are catalytic processes that utilize molecular hydrogen as a reducing agent. Indeed, H2 reductant is atom efficient and cheap and so far a large number of noble metal catalysts for hydrogenation have been developed.6–8 Because they rely on the use of molecular hydrogen or a less reactive hydrogen source,9 catalytic hydrogenations pave the way for sustainable processes with a high degree of control over reaction rates and selectivity. Sections 3.1 and 3.2 of this Review will cover these reductive transformations.
The reverse process of hydrogenation is the oxidative conversion of organic molecules with the release of H2 commonly referred to as acceptorless dehydrogenation. In view of the microscopic reversibility principle, both types of processes can potentially be carried out with the same catalysts. Apart from the trivial dehydrogenative transformations, e.g. synthesis of ketones from alcohols, catalytic dehydrogenation is notable for two important applications. The first one includes dehydrogenative coupling reactions (Scheme 1)10 where reaction between several molecules yields a coupling product through a sequence of dehydrogenation and bond-forming reaction. Examples discussed in this review will among others include synthesis of esters from alcohols, or imines and amines via amine/alcohol couplings. As the products of dehydrogenative transformations can be as useful as their fully reduced counterparts we include extensive discussion on dehydrogenation catalysis in Section 3.4 of this Review.
When acceptorless dehydrogenation is performed on small molecule substrates such as methanol and formic acid,11 the released H2 is considered to be the target product of the transformation. These substrates are, therefore, acting as hydrogen carriers that produce pure H2 with carbon dioxide being the only by-product of the dehydrogenation. Such gas feed, provided it is free of catalytic poisons like CO, can be directly utilized for fuel cell applications.12,13 Such processes together with the chemical pathways to regenerate formic acid and methanol substrates from CO2 have great potential for energy applications and will be described in Section 3.3 of this Review.
With very few exceptions, Ru-based complexes are currently among the most active homogeneous (de)hydrogenation catalysts.8,14 Many of them allow stable operation at metal loadings below 100 ppm in various reductive and oxidative transformations.15–17 Nevertheless, the utilization of such catalysts has several important limitations related to the high price and low abundance of ruthenium and, more importantly, the toxicity of noble metals for the living organisms. The latter represents a particularly important issue when catalytic transformations are being developed for the production of pharmaceuticals. The removal of the toxic metal residues from the final product to an acceptable level18 can drastically increase purification costs. In recognition of these challenges, the focus of the catalytic community had been gradually shifting in the last decade to the development of new catalyst systems based on the first row transition metals, which are both abundant and less- or non-toxic in contrast to the conventional noble metal active components.19
This review will describe the rapid development of the Fe, Co and Mn-based catalysts that occurred within the last few years and gave rise to a new class of noble metal-free (de)hydrogenation protocols. As we aim at encompassing several fields where the progress has been reviewed in the past, we will guide the reader to specialized review works in the introduction to corresponding sections. This review is structured as follows: in Section 2 we will briefly introduce common synthetic and reactivity concepts for the key catalyst motives discussed throughout the review. Section 3 presents an overview of the catalytic properties and substrate scopes of Fe, Co and Mn-based homogeneous catalysts. This section is organised into subsections addressing individual reaction types that for the reader's comfort will follow the nomenclature and classification introduced in Scheme 1. Each subsection will be concluded with the summary of common reaction conditions, while the substrate scopes in full can be found in the ESI† of this Review. The overview of the catalytic data is followed by Section 4, where we discuss and critically assess the different mechanistic proposals, which have been put forward to rationalize the observed reactivity trends. The review is concluded with Section 5, where we highlight current challenges and prospects of catalytic (de)hydrogenation chemistry with non-noble metal homogeneous catalysts.
2. Ligand design, complexation and reactivity features
2.1. Ligands and metal–ligand cooperation
Ligand systems utilized for the first row transition metal catalysts often resemble or fully mimic those successfully employed for Ru-based catalysis. Among the various available ligand platforms, pincer ligands hold the upmost prominence and are featured in a vast majority of active catalysts regardless of the metal used. The application of pincer ligands in catalysis20–22 and their role in bond activation14,23 have been the subject of several excellent reviews published in the last decade. Defined as tridentate ligands,24 pincers are typically comprised of a backbone and two sidearms. Lutidine-derived pincer ligands put forward by Milstein and co-workers (Scheme 2, A) were historically among the first pincers used for catalysis by early transition metals.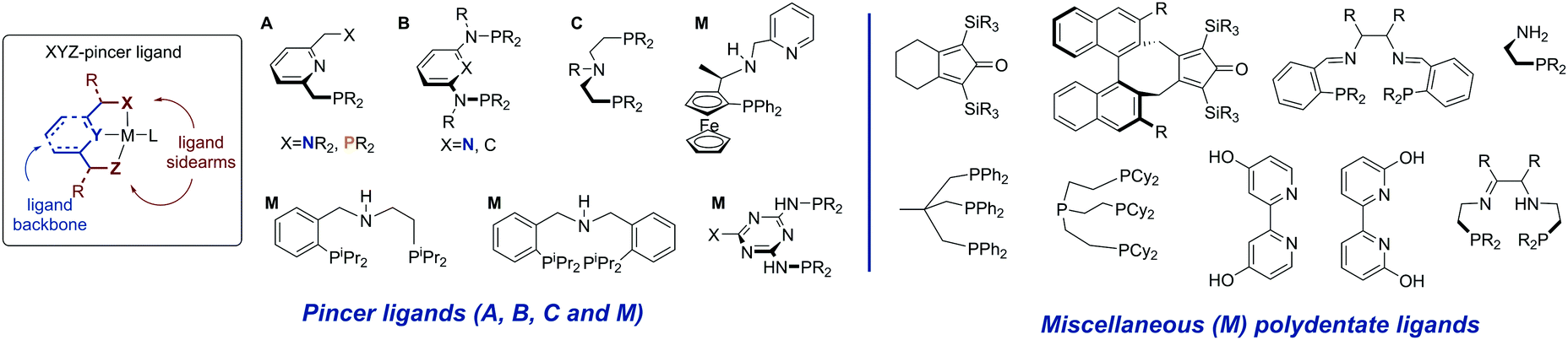 | ||
| Scheme 2 Structural overview of the pincer ligand and ligands employed for non-noble metal promoted (de)hydrogenation discussed in this Review. | ||
The replacement of the methylene arms in A for the amine linker gives rise to a well-established diaminopyridine-based ligand family B,25 while the replacement of the aromatic backbone with the aliphatic one yields the ligand family C, which in this review will be referred to as the aminopincers. The variations of these three main ligand motifs dominate the current state of the art in the base metal catalysed (de)hydrogenation chemistry. Nevertheless, other remarkable ligand systems will be given an extensive mention (M, for miscellaneous in Scheme 2).
The prevalence of pincer ligands is often explained by the combination of the high metal binding strength, expected for tridentate ligands, and their ability to form a bifunctional reactive ensemble capable of promoting chemical transformations in cooperation with the metal centre upon complexation. This property is inaccessible for the more conventional ligand platforms26 as it yields catalysts with two distinct metal- and ligand-centred functional groups that are both necessary for the catalyst to operate. Such catalysts are typically referred to as bifunctional. Setting aside the debates on the degree of involvement of the cooperative ligands in catalysis,27,28 we will discuss the basic principles behind the bifunctional catalysis phenomenon in view of the fact that the vast majority of catalyst systems discussed in this review are at least potentially bifunctional.
The acid–base bifunctionality in pincer complexes is enabled by the presence of the cooperative site in the ligand backbone or sidearm (Scheme 3). For the lutidine-based A-metal complexes, such a reactive site is provided by the pyridilmethylenic fragment, Ar-CH2-, that can be deprotonated by a strong base resulting in the dearomatization of the ligand and the formation of a coordinatively unsaturated complex. Similarly, C-metal pincers produce five-coordinate species upon deprotonation of the central NH fragment that is transformed into a metal-bound amide. For both A and C ligand platforms, the reaction with a strong base yields a reactive system containing a highly basic site on the ligand vicinal to the coordinatively unsaturated metal centre exhibiting elevated Lewis acidity. The formation of such an acid–base pair is often invoked in the mechanistic proposals for the substrate activation over bifunctional catalysts (Scheme 3).
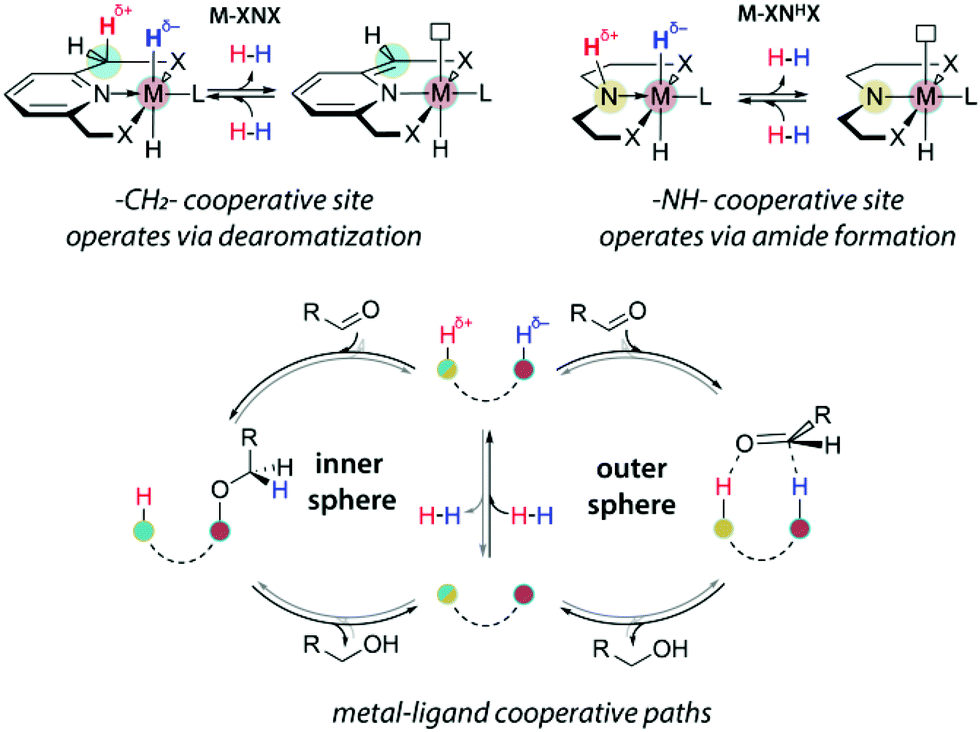 | ||
| Scheme 3 An example of MLC activation of dihydrogen and cooperative (de)hydrogenation steps. | ||
To summarize, a vast majority of ligands employed for base–metal catalysis described in this review are tridentate pincer ligands. These pincers are almost exclusively nitrogen-centred and contain strongly donating phosphine ligands as sidearms. The selection of pincer ligands in mainly motivated by their ability to engage in metal–ligand cooperative behaviour (see Scheme 3) that is known to facilitate substrate activation in both late and early transition metal complexes. The latter is also one of the major reasons for the use of the less common bidentate and tetradentate ligands (Scheme 2) that can engage in MLC behaviour as well.
2.2. From complexation to reactivity
The coordination chemistry of 3rd row transition metals can be strikingly different from that of their heavier 4d and 5d counterparts. Unlike the preparation of noble metal complexes, that is typically straightforward, complexation of Fe, Co and Mn may require different approaches for different metals and face several limitations that we will briefly describe in this section for A, B and C family of pincers. Similar to the noble metal counterparts, base metal catalysts for hydrogenation are often activated by introducing the hydride ligands, and therefore this reactivity will be briefly described. The reader is also referred to a series of recent reviews on metal hydride chemistry for further insight.29–31All the Fe complexes described in this review are Fe(II) species. Complexation of iron to lutidine-based ligands A was described in detail by the group of Milstein.32,33 The authors employed a reaction of the PNP pincer with iron bromide followed by treatment with CO (Scheme 4). Depending on the substitution pattern on the phosphine donors the resulting iron carbonyl dibromides can be transformed into the corresponding hydrides via two pathways. A mono hydride (A-Fe-1, Scheme 4) complex was prepared with the iPr substituted PNP ligand by treatment with NaHBEt3 and dihydride species (A-Fe-2, Scheme 4) were prepared from the tBu substituted PNP precursor by treatment with NaBH4.
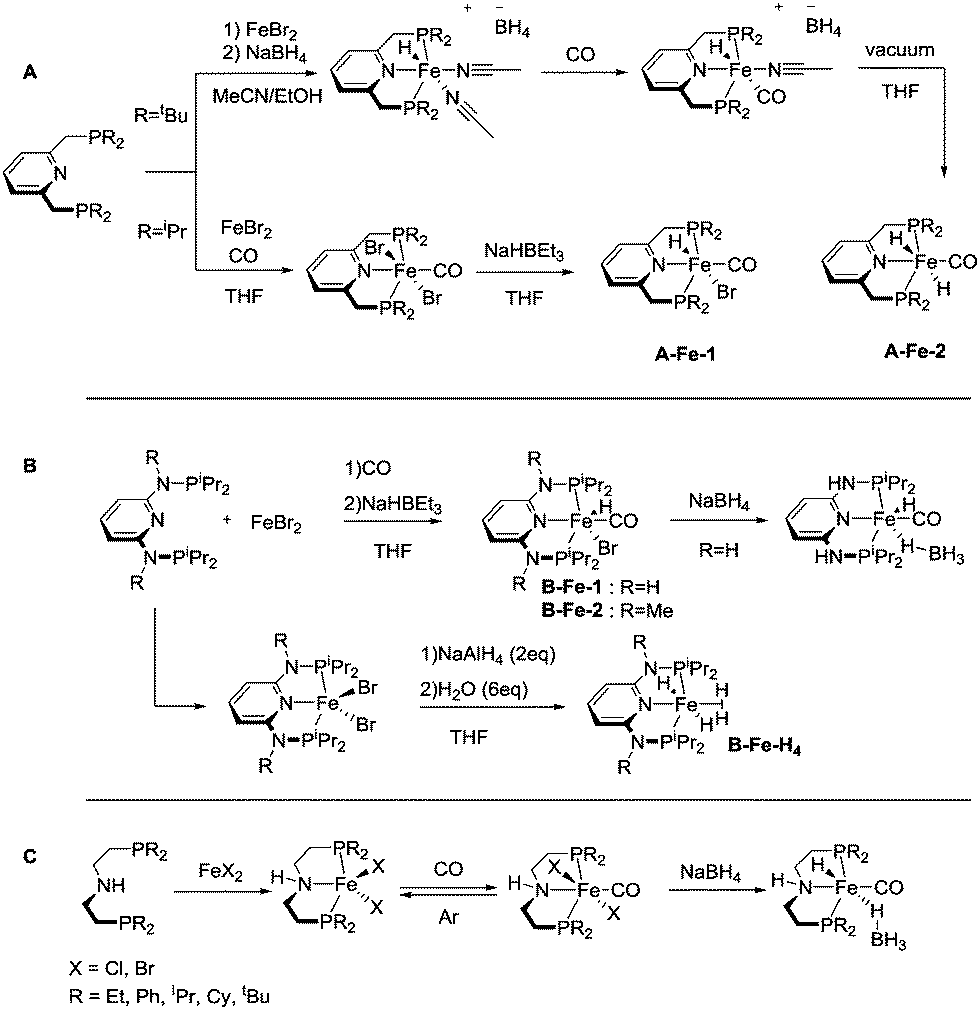 | ||
| Scheme 4 Generation of iron hydride complexes stabilised with pincer ligands A, B and C. | ||
Similarly, ligands B featuring amine linkers also complex readily with FeBr2 in the presence of CO. A one pot complexation with FeBr2 followed by NaHBEt3 treatment yields bromohydridocarbonyl species B-Fe-1. Further reaction of B-Fe-1 with sodium borohydride results in elimination of the remaining bromide ligand to produce a BH4-bound complex.34
In the absence of CO, the complexation with FeBr2 can be used to generate the five-coordinate Fe–PNP dibromide complex,35 which can be further converted to a stable polyhydride complex B-Fe-H4.36 The complexation of aminopincer ligands C with iron is typically carried out following reaction pathways analogous to those employed for ligands A and B.37
Most of the Co catalysts falling in the scope of this Review are prepared in a straightforward reaction via the direct coordination of CoCl2 with an appropriate ligand. The reaction typically yields dichloro cobalt(II) species used as a precatalyst; however other oxidation states of Co (1+ and 3+) are also encountered across the field. One of the first Co catalysts disclosed by Hanson and co-workers38 is prepared from a C-type aminopincer ligand and a rather unusual (Py)2Co(CH2SiMe3)2 precursor (Scheme 5).39 The resulting amide complex could be readily protonated with Brookhart's acid40 to form the amine-centred Co(II) complex.
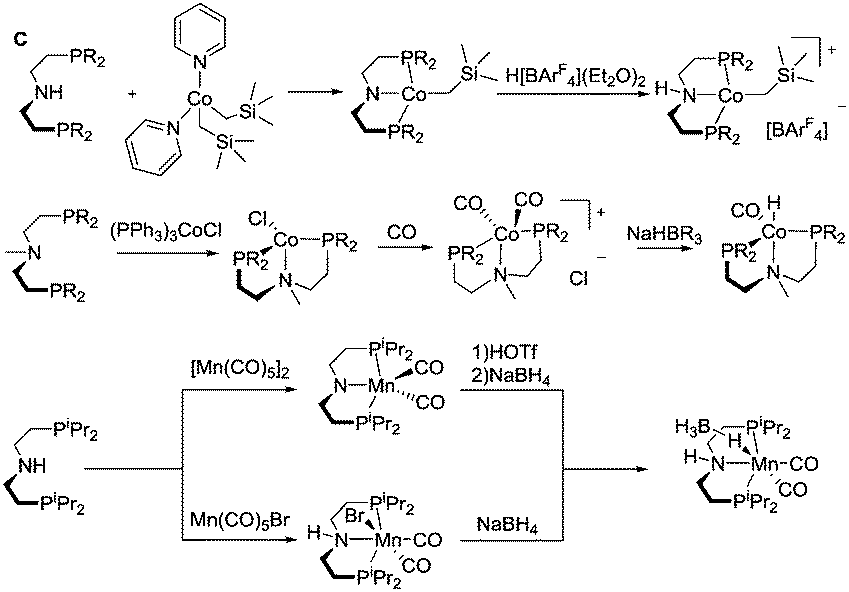 | ||
| Scheme 5 Notable examples of preparation and reactivity of Co and Mn complexes. | ||
The C aminopincers also form complexes with Co(I) centres. A typical example of such complexes was described by Bernskoetter and co-workers,41 who studied the formation of Co(I) pincers and isolation of Co(I) hydride complexes. The reaction of the methylated C ligand with the Co(PPh3)3Cl precursor (Scheme 5) led to the formation of a tetragonal C-Co chloride complex, which converts to a five-coordinate dicarbonyl C-Co species with a meridionally bound ligand in the presence of CO. The subsequent treatment with sodium trialkylborohydrides produces the Co(I) hydride complex.
Manganese, similarly to Co, is prone to forming complexes in several oxidation states. As we will discuss in the following sections, the 1+ oxidation state of Mn appears to be a strict requirement for the catalytic activity that inflicts considerable synthetic limitations. The preparation of Mn(I) complexes for hydrogenation faces a major and rather unexpected pitfall that is related to the lack of appropriate Mn(I) precursors. More importantly, Mn(II) coordination compounds that are generally prepared with ease from the corresponding halide salts MnX2 cannot be used to produce Mn(I) species, which restricts the researchers to a very limited selection of Mn(0/I) carbonyl precursors among which Mn2(CO)10 and Mn(CO)5Br are the most commonly used. As a result, the carbonyl ligands stabilising the Mn(I) centres are often retained upon the complexation with the pincer ligand. As the replacement of carbonyl ligands is usually difficult, their presence limits the potential diversity of the reactions available for the manganese complexes. Nevertheless, Mn hydrides can be prepared from the corresponding L–Mn carbonyl bromides or amide complexes as was demonstrated, for example, by Gauvin and co-workers (Scheme 5).42
3. Catalysis: activity, reaction and substrate scopes
Similar to their noble counterparts, base metal complexes catalyse a range of (de)hydrogenative transformations that were classified earlier in Scheme 1. In this section we will discuss them starting from reduction with molecular hydrogen (Section 3.1). We will further cover catalytic reduction with other hydrogen donors (transfer hydrogenation, Section 3.2) and proceed to a more convoluted chemistry.We will first discuss hydrogenation and dehydrogenation chemistry of simple C1 molecules, most notably CO2, formic acid and methanol, that is related to hydrogen storage and production (Section 3.3). The last section (Section 3.4) will deal with dehydrogenations and dehydrogenative coupling reactions that can be used in synthesis applications for the production of complex organic molecules.
For readers’ comfort, the catalytic results are introduced separately for particular ligand/catalyst classes. Within a particular class of catalysts the description is started with more facile transformations and concluded with the challenging ones. Schemes in Section 3 summarize the data described in every sub-section to provide the complete yet condensed overview of recent achievements in the field.
3.1. Reduction with molecular hydrogen
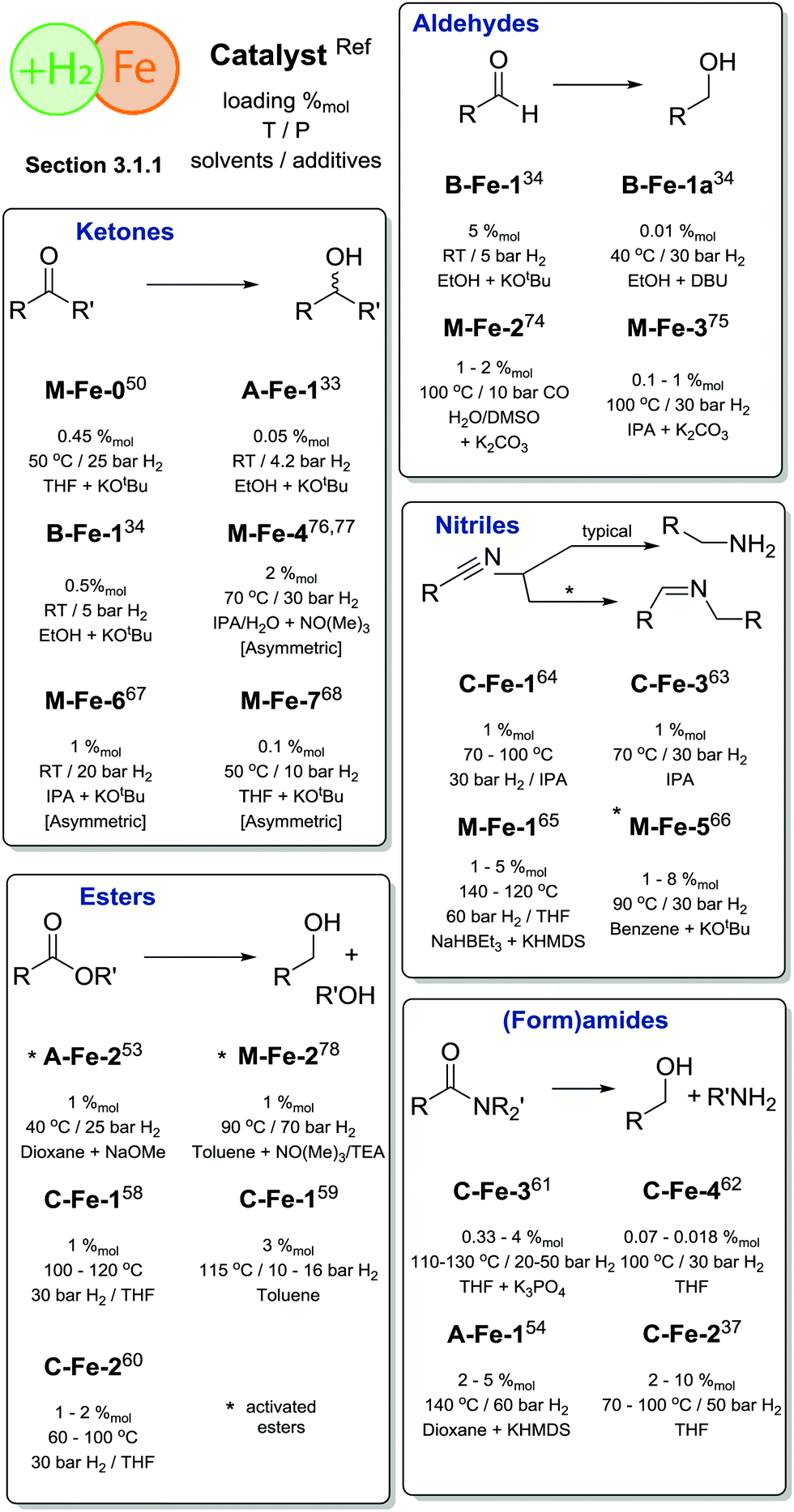
One of the first well-defined Fe catalysts for ketone hydrogenation was reported by Morris and co-workers, who described a large family of Fe complexes based on tetradentate iminophosphine ligands. A representative example of this catalyst family is the M-Fe-0 complex bearing a tetradentate PNNP ligand (Scheme 6) that allowed an efficient reduction of acetophenone to phenylmethanol under very mild conditions, namely in the presence of less than 0.45%mol Fe at 50 °C and 25 bar H2.50 Interestingly, previously described Fe complexes with a cyclohexyl linker connecting the N-donors in the PNNP ligand51 were much less active for the reduction with molecular H2, but showed a substantial activity in the transfer hydrogenation reaction, which will be the subject of the next section.
 | ||
| Scheme 6 Structures of Fe catalysts introduced in Section 3.1.1. Types of transformations described for each particular catalyst are indicated with the sub-section label. | ||
A major advance in catalytic ketone hydrogenation with Fe complexes was made by the group of Milstein, who developed an A-Fe-1 iron PNP pincer catalyst. For this catalyst, a performance comparable to that of the M-Fe-0 could already be reached when operating at a catalyst loading of only 0.05%mol at room temperature and a very low H2 pressure of 4.1 atm.33 The substrate scope of A-Fe-1 included substituted acetophenones, conjugated diketones and substrates containing CC double bonds. While the majority of the substrates were converted in good yields, the presence of amino- and nitrile-functional groups diminished strongly the activity. The A-Fe-1 catalyst showed only a moderate chemoselectivity in the hydrogenation of unsaturated ketones by retaining at best 20% of the CC double bonds present in the substrate at full conversion. Finally, the authors reported significantly lower activity of A-Fe-1 in the hydrogenation of benzaldehyde, providing 36% yields with an elevated catalyst loading of 0.125%.
Interestingly, the aldehyde hydrogenation with the A-Fe-1 catalyst was later improved. The modest activity of A-Fe-1 in the hydrogenation of benzaldehyde could be promoted in the presence of trimethylamine or acetophenone. It was proposed that the transient formation of carboxylic acid (presumably via the KOtBu-mediated Cannizzaro reaction) could deactivate the catalyst, while NEt3 and acetophenone were preventing the acid formation.52
The dihydrido-Fe complex A-Fe-2 (Scheme 6) from the same catalyst family also shows a pronounced activity in the hydrogenation of activated esters under mild conditions (40 °C, 5–25 bar H2 pressure). A range of trifluoroacetates were converted in good yields at ca. 0.5–1%molA-Fe-2 catalyst loading in the presence of NaOMe base.53 Subsequent work by the same group has demonstrated that activated amides can also be converted using related A-Fe-1 catalysts in the presence of the KHMDS base promoter.54
Bifunctional iron complexes with ligand motif B55 have also been successfully applied in the hydrogenation catalysis. Catalysts B-Fe-1 reported by Kirchner and co-workers34 in 2014 showed very good activity in the reduction of ketones and aldehydes at loadings of 0.5%mol at 5 bar H2 pressure. Remarkably, near quantitative yields of alcohols were achieved in ethanol solvent at room temperature. Building upon these results the authors have conducted a scrupulous mechanistic study that allowed them to improve the performance of B-Fe-1a by a large margin and establish a chemoselective reduction of aldehydes in the presence of ketones, esters, epoxides, alkynes and nitro aromatic compounds.56 In particular, guided by the insights provided by the mechanistic studies, the authors optimized the hydrogenation of 4-fluorobenzaldehyde at 30–60 bar H2 to reach TOFs up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and ultimately obtain outstanding TONs of up to 80000. A variety of other aromatic and aliphatic aldehydes could also be fully converted at 50–100 ppm catalyst loading at 30 bar H2 pressure and 40 °C temperature in the presence of the DBU base promoter. These results render the B-Fe catalysts among the most active systems reported to date for the selective hydrogenation of aldehydes and rival the performance of noble metal catalysts.57
000 h−1 and ultimately obtain outstanding TONs of up to 80000. A variety of other aromatic and aliphatic aldehydes could also be fully converted at 50–100 ppm catalyst loading at 30 bar H2 pressure and 40 °C temperature in the presence of the DBU base promoter. These results render the B-Fe catalysts among the most active systems reported to date for the selective hydrogenation of aldehydes and rival the performance of noble metal catalysts.57
The utility of iron-catalysed hydrogenation was later extended to unactivated esters with the development of the aminopincer Fe catalysts based on the ligand family C (Scheme 6). The groups of Beller58 and Guan59 independently reported the use of catalyst C-Fe-1 for the conversion of various esters to the corresponding alcohols at 1–3%mol catalyst loading under 10–50 bar H2 pressure and a temperature of 100–135 °C. It was observed that C-Fe-1 can be successfully employed under base free conditions and its activity is somewhat hampered when alkoxide bases are introduced as additives. It was also shown that the addition of lithium chloride, methylsulfonic acid or CO completely deactivates the catalyst.58 Interestingly, the less bulky complex C-Fe-2 was later found to show improved performance in ester hydrogenation by Beller and co-workers,60 who showed that the increased steric bulk was negatively affecting the catalytic activity.
Recent work by Langer and co-workers further expanded the utility of Fe–aminopincer catalysts to the selective hydrogenation of amides, which constitute particularly challenging substrates for homogeneous hydrogenation. After a careful evaluation of the ligand substituent effects, the authors identified catalyst C-Fe-2 (Scheme 6) bearing less bulky ethyl substituents as opposed to other catalysts in the study as a potent amide hydrogenation catalyst operating at 2–10%mol loadings, 50 bar H2 pressure and 70–100 °C under base-free conditions.37 Subsequently, Sanford and co-workers61 showed that the activity of an analogous catalyst C-Fe-3 can be significantly improved by using the K3PO4 additive. The combination of C-Fe-3 and K3PO4 allowed achieving a full conversion of a series of formamides to the corresponding amines and alcohols at only 0.33%mol catalyst loading that corresponds to ca. 300 catalytic turnovers (TON) at the full substrate conversion and up to 1000 turnovers at lower catalyst loadings. At the same time Bernskoetter and co-workers disclosed a similar five-coordinate Fe hydride catalyst C-Fe-4 that was particularly efficient for the hydrogenation of formamides, reaching TONs of up to 4430 at 0.018%mol catalyst loading at 30 atm H2 pressure and 100 °C for a variety of formamides.62
The utilization of Fe-based homogeneous catalysts in challenging nitrile hydrogenations including that of bisnitriles has been described by Beller and co-workers. A high activity of catalysts C-Fe-363 or C-Fe-164 (Scheme 6) could be achieved in isopropanol solvent at 30 bar H2 and 70–130 °C in the absence of base additives. It was noted that the methylation of the NH group of the pincer ligand renders the catalyst inactive in the hydrogenation of nitriles. The C-ligated iron aminopincers remain among the most versatile hydrogenation catalyst family to date with the potential to hydrogenate the vast majority of polar substrates discussed in this review.
An elegant extension of the aminopincer ligand family has been recently described by Milstein and co-workers, who implemented the bis(2-diisopropylphosphinobenzyl)amine ligand forming more flexible 6-membered ring chelates with iron centres.65 The resulting catalyst M-Fe-1 features only one phosphine donor of the “PNP” ligand bound to the iron centre. In the presence of 1–5%mol catalyst, NaHBEt3 additive and KHMDS, a wide range of aliphatic and aromatic nitriles could be successfully converted into the corresponding amines at 140 °C and 60 bar H2 pressure. Interestingly, another representative of the aminopincer platform, catalyst M-Fe-5, was able to convert nitriles to symmetric imines. Under the optimised conditions of 30 bar H2 and 90 °C, M-Fe-5 provided excellent selectivity to symmetric imines with retention of the CN double bond and no overreduction of the target product.66
Asymmetric ketone hydrogenation with Fe pincer catalysts has been recently described in several reports. Zirakzadeh and co-workers reported a series of M-Fe-6 (Scheme 6) catalysts67 operating at 1%mol loading under 20 bar H2 pressure in isopropanol. Further work by Morris and co-workers68 focusing on the use of M-Fe-7 and related catalysts reported a significant improvement in catalytic performance. The authors obtained excellent conversions of substituted acetophenones at 50 °C and 10 bar H2 pressure in the presence of only 0.1%mol catalyst and 1%mol KOtBu promotor. Catalysis was highly stereoselective allowing for ee up to 96%, an improvement over M-Fe-6 that showed a maximal ee of 81%.
Apart from the complexes with pincer ligands, a potent iron hydrogenation catalyst can utilize the cyclopentadienone ligand motif that gives rise to a broad family of efficient carbonyl hydrogenation catalysts.69–73 One of the early reports by Beller and co-workers74 demonstrated the utility of the M-Fe-2 cyclopentadienyl iron tricarbonyl catalyst for the reduction of aromatic, aliphatic and unsaturated aldehydes under water–gas shift reaction conditions. The catalyst operated at 100 °C under 10 bar CO pressure in the presence of water and 1–5%mol loading. Further research by the same group resulted in the development of a direct H2 reduction of similar aldehyde substrates using M-Fe-2 and M-Fe-3 catalysts and their analogues. The careful optimization of the reaction conditions that involved the use of iPrOH/H2O solvent with a K2CO3 additive allowed lowering the catalyst loading to <1% level.75
An elegant modification of the cyclopentadienone ligand motif with an (R)-BINOL backbone was recently described by Pignataro, Piarulli, Gennari and co-workers. The authors obtained catalyst family M-Fe-4 capable of asymmetric ketone hydrogenation with ee up to 77%.76,77 The utility of iron cyclopentadienone catalysts was recently expanded to the hydrogenation of activated esters.78 Lefort and Pignataro and co-workers used M-Fe-2 at 1%mol loading to convert a series of trifluoroacetates in quantitative yield at 70 bar H2 and 90 °C. It was found that the presence of a triethylamine base was crucial for the catalytic performance as the base was required for the neutralization of the trifluoroacetic acid that was formed as an intermediate and poisoned the catalyst.
In summary, Fe hydrogenation catalysis reached the extent of development comparable to that of noble metal counterparts when substrate scopes are concerned (Scheme 7). However, the activity of the majority of Fe-based catalysts remains inferior to that of Ru catalysts. This difference is pronounced to a large extent for hydrogenation of more challenging substrates, e.g. esters, where Ru-based catalysts outperform the Fe-based ones by a significant margin. For example, typical loadings for Ru ester hydrogenation catalysts vary in the range of 0.00125–0.01%mol16,17 under otherwise similar conditions.
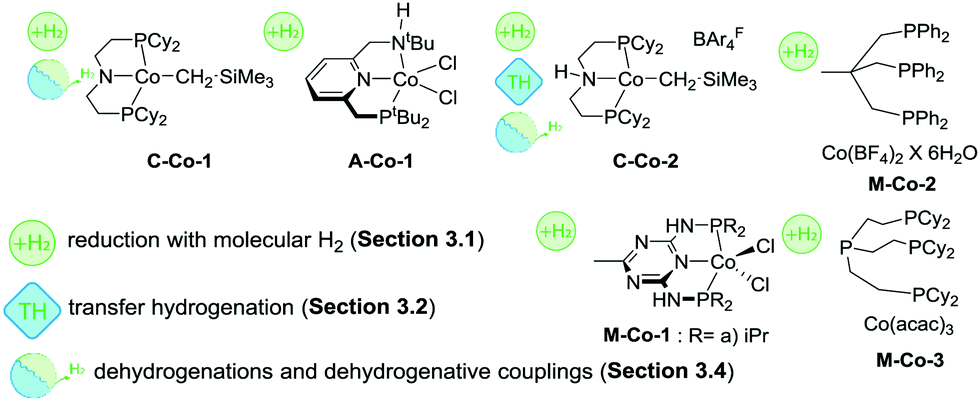
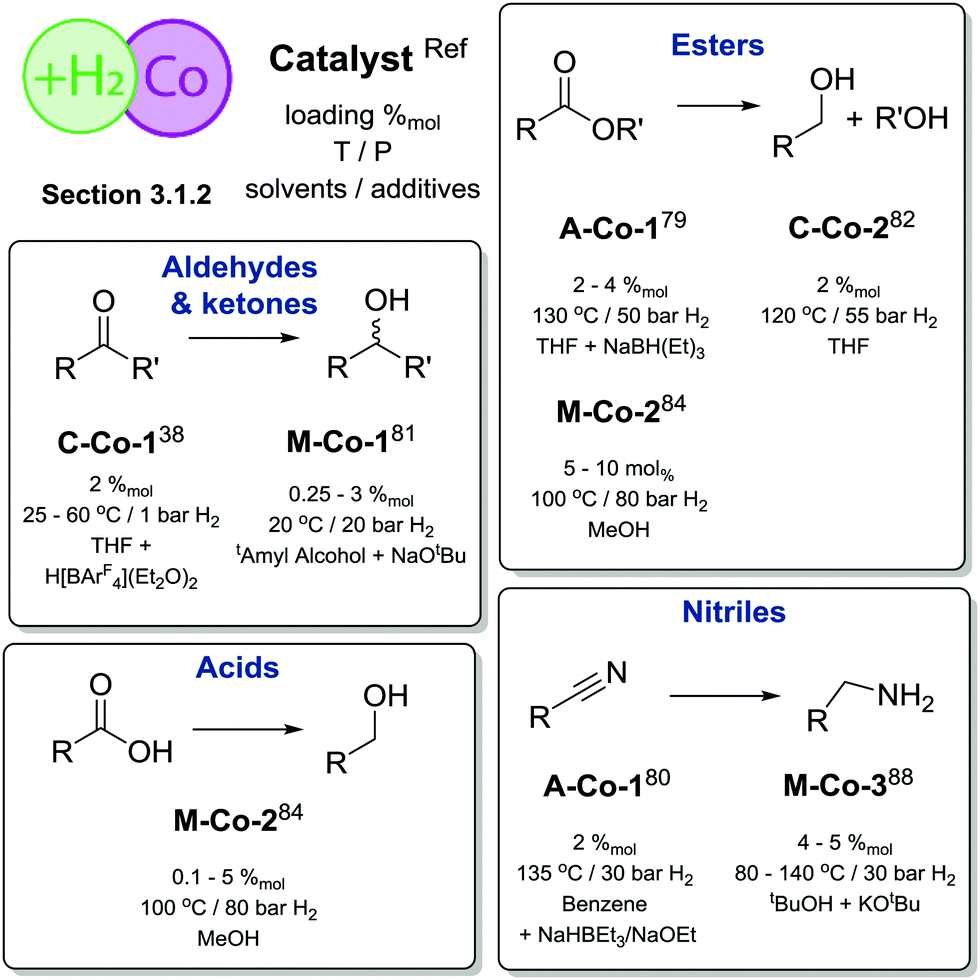
One of the first defined Co catalysts for reduction of polar CO and CN bonds with H2 was described by Hanson and co-workers as early as 2012.38 A Co(II) alkyl species C-Co-1 (Scheme 8) showed a remarkable reactivity towards hydrogenation of olefins, ketones and aldehydes. Reactions were carried out at 1–4 atm H2 pressure and at 25–60 °C and provided >90% yields for most substrates in the presence of 2%mol catalyst (Scheme 7).
 | ||
| Scheme 7 Comparative summary of Fe catalysis performance and scopes in hydrogenations described in Section 3.1.1. For the full substrate scopes see the ESI.† | ||
Reduction was found to tolerate carboxylic acid, ester, tertiary amine, halide and alcohol functional groups. Remarkably, hydrogenation of styrene was also unaltered by the addition of 10%mol water.
Later work by the group of Milstein established the activity of Co lutidine-based pincers in the hydrogenation of esters. Catalyst A-Co-1 (Scheme 8) containing a PNNH pincer ligand with a secondary amine sidearm showed a significantly higher activity compared to its PNP or tertiary amine PNN pincer counterparts.79
The catalyst operated at 2–4%mol loading in the presence of 8%mol NaHBEt3 additive required for catalyst activation and 25%mol KOtBu base promoter. To achieve moderate-to-good yields A-Co-1 required 50 bar H2 pressure and a reaction temperature of 130 °C. The same catalyst was later shown to promote nitrile hydrogenation to primary amines.80
 | ||
| Scheme 8 Co catalysts introduced in Section 3.1.2. Types of transformations described for each particular catalyst are indicated with the sub-section label. | ||
Similar to the case of ester hydrogenation, nitrile reduction with A-Co-1 required NaHBEt3 for operation as well as moderately low 4.4%mol loading of sodium ethoxide.
 | ||
| Scheme 9 Comparative summary of Co catalysis performance and scopes in hydrogenations described in Section 3.1.2. For the full substrate scopes see the ESI.† | ||
Significant improvement in the selective Co-catalysed hydrogenation of ketones and aldehydes was later reported by Kempe and co-workers.81 An easily accessible complex M-Co-1 (Scheme 8) was identified as the most active among its dichlorocobalt(II) PNP analogues with a varied substitution pattern. Catalyst M-Co-1 was active in THF and 2-methyl-2-butanol at room temperature at 20 bar H2. At 0.25–3%mol loadings, excellent yields in the hydrogenation of ketones and aldehydes were obtained for the majority of substrates. The authors also demonstrated excellent tolerance of this catalyst system to aromatic and aliphatic N-heteroatom functional groups, halides, unsaturated CC bonds in conjugation with a reduced carbonyl group as well as remote CC functions.
A more challenging ester hydrogenation was recently established with the Co catalyst by the group of Jones.82 Using the C-Co-2 catalyst that essentially is a product of Brookhart acid addition to C-Co-1 the authors managed to perform ester hydrogenation in an additive-free manner. Catalytic tests were performed at 55 bar H2 at 120 °C in THF to yield the corresponding alcohols. Very importantly, the authors observed very similar activities produced by C-Co-2 and its N-methylated counterpart, suggesting a non-bifunctional mechanism for hydrogenation of esters, while the NH-Co cooperation was anticipated for the alcohol dehydrogenation with the same catalyst.83
A large body of work on Co-catalysed hydrogenations relied on the use of tri- and tetradentate P-donor ligands colloquially known as tri- and tetraphos. Elsevier, de Bruin and co-workers84 utilized an in situ prepared Co/triphos catalyst M-Co-2 (Scheme 8) for the hydrogenation of carboxylic acids and their esters. Operating at 80 bar H2 pressure and 100 °C, the authors used 5–10%mol Co loadings to convert esters with good yields. No retention of the CC function was observed in these reactions. More importantly, carboxylic acids, that are more challenging for a homogeneous catalyst to hydrogenate compared to their esters,8 could be converted under the same conditions in the absence of any additives at a lower catalyst loading of 0.1–10%mol. A model substrate – trifluoroacetic acid – could be fully converted at only 125 ppm Co loading providing ca. 50% yield of trifluoroethanol. The performance of an in situ formed Co system was exemplary, as it was comparable to that of Ir85 and Ru86,87 based catalysts for carboxylic acid hydrogenation, which at some instances requires higher temperatures or metal loadings compared to the Co case.
Later work by Beller and co-workers88 disclosed the use of polydentate phosphine ligands in Co-catalyzed hydrogenation of nitriles to primary amines.88 It was found that the polydentate phosphine ligand identical to that in M-Co-2 gave only a low activity in the target reaction, whereas the in situ formed M-Co-3 based on a tetradentate phosphine allowed quantitative yields in benzonitrile hydrogenation at 100 °C, 5%mol Co loading and 30 bar H2. It was observed that a lower reaction temperature of 80 °C provides marginally lower yield within identical 18 h long tests but leads to a pronounced induction period of ca. 4 h associated with the formation of the active species from a Co(acac)3 precursor and a tetradentate phosphine. Under optimized conditions M-Co-3 was shown to hydrogenate aromatic and aliphatic nitriles with quantitative conversion and good isolated yields typically exceeding 80%.
Overall, Co catalysts exhibit somewhat lower activity in H2 reduction than their Fe counterparts based on identical or similar ligands (Scheme 9). However, Co can promote direct reduction of carboxylic acids inaccessible to Fe catalysts and has very few precedents in noble metal catalysis.7,8
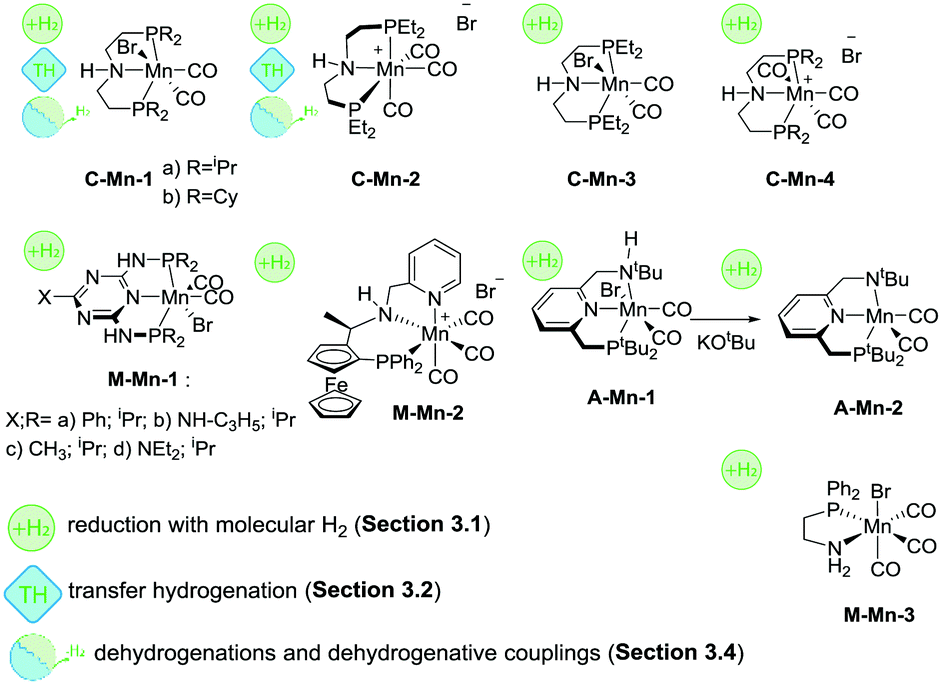
The first example of a defined Mn hydrogenation catalyst was disclosed by the group of Beller, who reported the manganese aminopincer family of catalysts C-Mn-1 (Scheme 10) that were active in the hydrogenation of aromatic and aliphatic nitriles, ketones and aldehydes.92 A slightly more active, compared to its counterparts from the same family, catalyst C-Mn-1a with isopropyl substituents on the phosphine donor groups was extensively characterized in a reactivity survey that demonstrated deprotonation and metal-hydride species formation common for Ru and Fe aminopincers with the C ligand family. The catalyst operated at 1%mol loading in the hydrogenation of ketones and aldehydes and 3%mol loading in nitrile hydrogenation with roughly 3 equivalents of NaOtBu base additive per metal loading at 100–120 °C and 30–50 bar H2 pressure. Later work by the same group expanded the scope of related Mn pincers to ester hydrogenation catalysis.93 Interestingly, catalysts C-Mn-1 were only moderately active in the hydrogenation of methyl benzoate at temperatures up to 120 °C and H2 pressures up to 80 bar at 2%mol catalyst loading. However, the use of less bulky C-Mn-2,3 catalysts allowed reaching excellent yields under significantly milder conditions.
Another example of Mn catalysts with an amine centred pincer ligand was described by Clarke and co-workers.94 At 1%mol catalyst M-Mn-2 provided excellent yields in asymmetric ketone hydrogenation and hydrogenation of esters to the corresponding alcohols. Interestingly, the reduction of both substrates was performed in alcohol solvents – ethanol for ketones and isopropanol for esters – which is typically challenging for common Ru-based catalysts. Notably, reduction of ketones was performed with high stereoselectivity allowing for ee typically over 70% and reaching 91% in particular cases.
A recent report by Beller and co-workers95 expanded the utility of C-Mn catalysts to asymmetric hydrogenation of ketones. Using C-Mn-4 at 1%mol loading a wide range of aromatic and aliphatic ketones could be converted to alcohols in excellent yields at 30–40 °C and 30 bar H2 pressure with moderate-to-good stereoselectivity up to 92/8 enantiomer ratios.
One of the most recent examples of an Mn-based ester hydrogenation catalyst, M-Mn-3 (Scheme 10) reported by our group features no pincer ligand motif, but instead utilizes a single PN bidentate ligand.96 It was noted that [Mn(CO)2(PN)2]+ cationic complexes were significantly less active than their Mn(PN) triscarbonylbromide counterpart in the hydrogenation of the methyl benzoate model substrate. Under optimized reaction conditions, loading of M-Mn-3 could be reduced to only 0.2%mol for ester hydrogenation reactions carried out at 50 bar H2 and 100 °C. M-Mn-3 allowed full retention of remote unsaturated carbon–carbon moieties in unsaturated esters. However, no chemoselectivity was observed in the reduction of α,β-unsaturated esters. An unusual feature of M-Mn-3 was the reliance on high loadings of the KOtBu base promotor necessary to achieve full ester conversion. While at 75%mol loading of KOtBu the hydrogenation of the model substrate yielded 98% of the alcohol product, the decrease of the base concentration to 10%mol resulted to a drop of the yield to 66%. It was demonstrated that the variation of the reaction temperature could not improve the catalysis, while the introduction of additional base to the stalled catalytic reaction reanimated the catalytic system and enabled further reduction of the ester until full conversion was reached.
First examples of Mn catalysts with pincer ligands having an aromatic backbone were reported shortly after the disclosure of the C-Mn catalyst family. A potent catalyst A-Mn-1 (Scheme 10) disclosed by Milstein and co-workers97 was shown to hydrogenate esters to the corresponding alcohols at 20 bar H2 pressure and 100 °C in the presence of 1%mol catalyst, which can be regarded as an improvement over the productivity of the C-Mn catalyst family. An interesting feature of the ligand design in A-Mn-1 is the presence of the secondary amine donor in the PNNH pincer, which undergoes deprotonation yielding an amido complex A-Mn-2 upon reaction with KOtBu. It was noted that the nature of the base promotor had a profound impact on the catalytic behaviour of A-Mn-1 with stronger KH base being superior to KOtBu and KHMDS bases used at 2%mol loading during the catalytic testing. Importantly, the amido complex A-Mn-2 showed catalytic activity under base-free conditions. This suggested that the base additive during catalysis with halide complex A-Mn-1 was necessary to activate the catalyst and allow for further formation of the catalytically active Mn hydride species.
A detailed investigation by Kempe and co-workers98 on Mn-catalysed ketone hydrogenation revealed the importance of the oxidation state of the Mn centre and auxiliary ligands bound to it. The authors found that while catalysts M-Mn-1 were competent ketone hydrogenation catalysts, with M-Mn-1b being the most active, the replacement of Mn(I) with the Mn(II) carbonyl free metal centre leads to inactive catalysts. Strikingly, even when Mn(II) was reduced, the resulting carbonyl-free Mn(I) species remained inactive in ketone hydrogenation under the studied conditions.
 | ||
| Scheme 10 Mn catalysts discussed in Section 3.1.3. Types of transformations described for each particular catalyst are indicated with the sub-section label. | ||
As a recent phenomenon, Mn-catalysed homogeneous hydrogenation has made great progress in a timeframe significantly shorter than that for Fe and Co catalysis. Substrate scopes and ligand systems utilized for these Mn-promoted H2 reductions are very similar to those for Fe and Co. The low catalytic activity remains the major drawback of the current state-of-the-art in manganese based hydrogenation (Scheme 11).
 | ||
| Scheme 11 Comparative summary of Mn catalysis performance and scopes in hydrogenations described in Section 3.1.3. For the full substrate scopes see the ESI.† | ||
3.2. Transfer hydrogenation
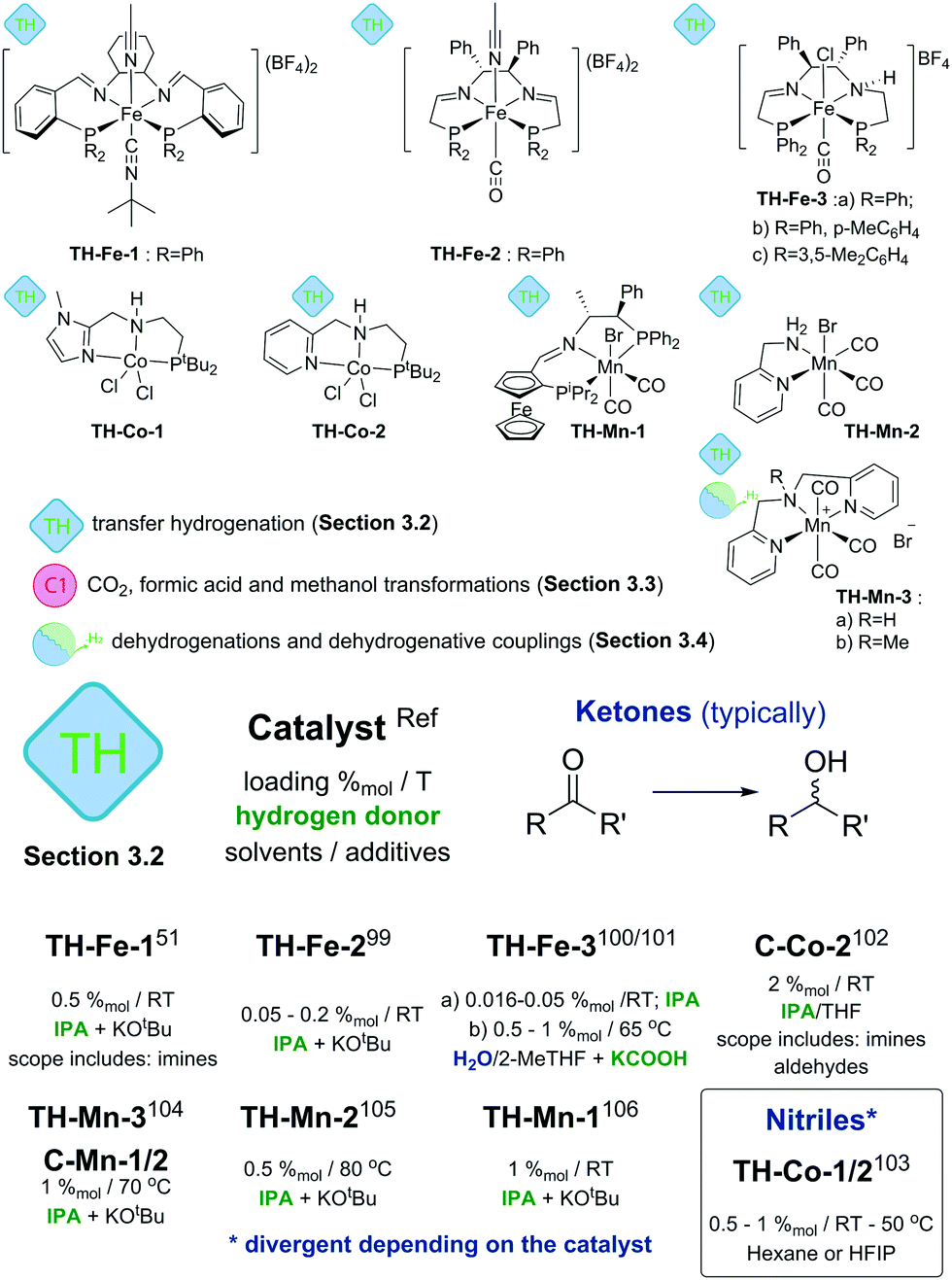
Alternatives to the use of molecular H2 for reduction are transfer hydrogenation techniques. Instead of pressurized gas they rely on the use of hydrogen donor molecules – isopropanol, formic acid, ammonia borane and others. Recent reviews9 extensively discuss transfer hydrogenation catalysis with a particular emphasis on the use of noble metals and Fe-based catalysts. Therefore, below we will focus predominantly on Mn- and Co-based systems, while the discussion on Fe catalysts will be limited to only the key seminal works.Cobalt catalysts developed by Zhou and Liu and co-workers103 show remarkable reactivity in the transfer hydrogenation of nitriles. Catalysts TH-Co-1 and -2 both utilized ammonia borane as both hydride and proton donor but showed different activity towards the reduction of nitriles. TH-Co-1 operated at 50 °C in hexane and at 1%mol loading it was capable of reducing aromatic and aliphatic nitriles to primary amines with excellent tolerance to functional groups including halides, ethers, thioethers and esters. Catalyst TH-Co-2 operating in HPIF solvent was capable of converting nitrile substrates at room temperature to symmetric secondary amines. In the presence of a primary or secondary amine, the reduction of nitriles with TH-Co-2 results in the alkylation of the former to produce unsymmetrical secondary and tertiary amines. Finally, combination of both catalysts allows stepwise synthesis of tertiary amines with three different substituents starting from several nitriles (see corresponding section in ESI†). The authors found that N-substituted analogues of this Co catalyst showed similar activities in reactions yielding primary and secondary amines, thus evidencing a non-bifunctional mechanism.
The authors found that while catalyst C-Mn-1-a (Scheme 12) showed a rather moderate activity, its less bulky analogues C-Mn-1-b and C-Mn-2 were significantly more active and provided a nearly full acetophenone conversion under similar conditions. This observation is in line with the activity difference noted for the ester hydrogenation catalysis93 suggesting the importance of the steric properties of the PNP aminopincer ligands C for the hydrogenation activity of Mn–PNP pincers. Interestingly, the authors disclosed aminopincer catalysts TH-Mn-3a,b based on an N-donor-based NNHN aminopincer that outperformed the phosphine based analogues by operating at lower catalyst loadings of 1%mol and requiring a lower amount of KOtBu additive. Strikingly, catalyst TH-Mn-3b having the methylated tertiary amine central donor was also an active transfer hydrogenation catalyst despite being not capable of NH-induced metal ligand bifunctional behaviour.
In strong contrast to conventional hydrogenation catalysis, transfer hydrogenation with Mn does not rely entirely on the use of strong donor ligands as demonstrated by TH-Mn-3 (Scheme 12) pincers. Furthermore, bidentate ligands can also enable an efficient catalysis. A simple Mn catalyst TH-Mn-2 with an aminomethyl pyridine NN chelate disclosed by Sortais and co-workers105 was shown to achieve excellent TOF values up to 3600 h−1 at 0.5%mol catalyst loadings operating in isopropanol at either 80 °C or room temperature. Room temperature operation required a prolonged reaction time of 16 hours compared to 20 minutes time necessary to achieve a full conversion at 80 °C. The authors have shown that the presence of the primary or secondary amine was critical to obtain an active catalyst as the full substitution at the amine donor led to a drastic drop in the catalytic performance.
A major improvement in enabling stereoselectivity in Mn-promoted transfer hydrogenation was described by Zirakzadeh, Kirchner and co-workers106 using an iminopincer PNP ligand modified with a ferrocenium sidearm (TH-Mn-1, Scheme 12). The authors reported asymmetric transfer hydrogenation of acetophenone with ee up to 85% using TH-Mn-1 or its monohydride analogue operating at room temperature in isopropanol solvent with 2–4 equivalents of KOtBu additive. A broad range of aromatic ketones could also be successfully converted under similar conditions with conversions of 60–96% and ee values up to 85% for the majority of the substrates.
 | ||
| Scheme 12 Transfer hydrogenation catalysts introduced in Section 3.2 and the comparative summary of their performance. Types of transformations described for each particular catalyst are indicated with the sub-section label. For the full substrate scopes see the ESI.† | ||
Transfer hydrogenation stands out among other catalytic reactions described in this review as it is the first class of reactions where base metal catalysts were shown to be highly competitive. The activity of Fe based TH-Fe-X and TH-Mn-2 catalysts (Scheme 12 and ESI†) reached several thousand turnovers per hour – values that match or exceed those of the noble-metal dominated state of the art.9 A particularly important feature of TH-Mn-2 is the absence of strong phosphine donors that are commonly used in such catalysts. The use of the N-only ligand motif renders TH-Mn-2 very practical considering low ligand and metal costs and outstanding activity.
3.3. Sustainable chemistry with C1 and small molecules substrates
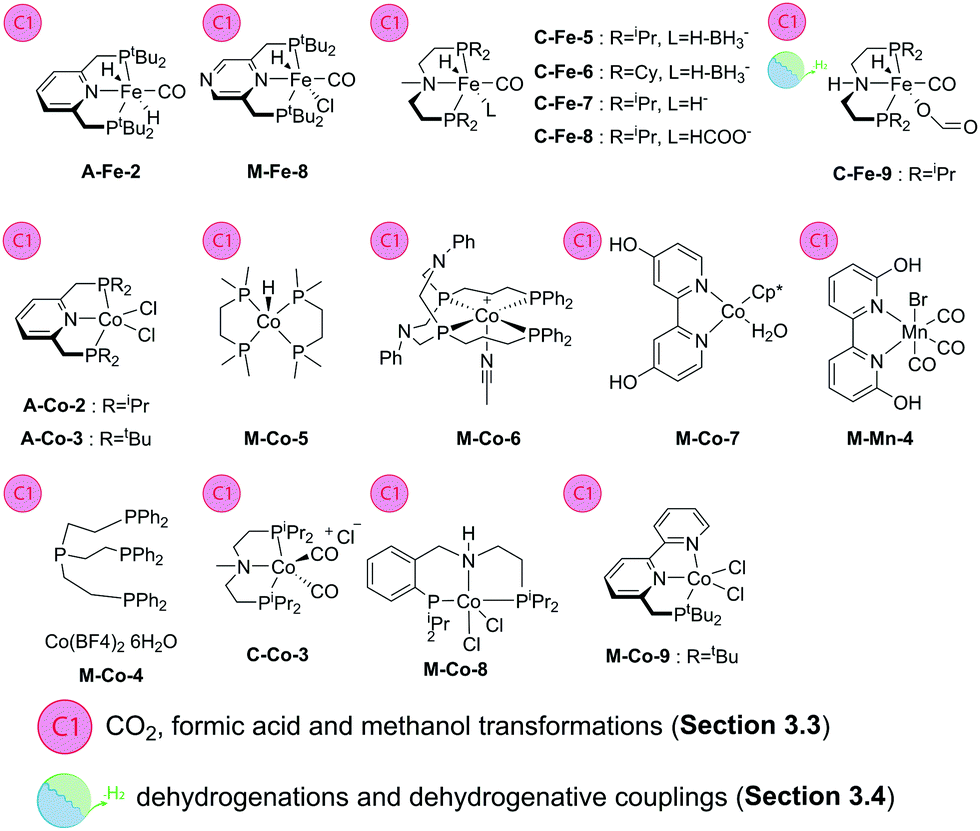
Catalytic hydrogenation processes can be classified according to their potential applications. The majority of reactions discussed in this Review are used for organic synthesis and therefore primarily aimed at fine chemical industry applications. However, there is an important group of substrates that have great potential for green energy applications. These substrates include, among others, CO2, methanol and formic acid that are known to undergo reversible (de)hydrogenations, thus acting as hydrogen carriers.10,11,107,108 In this section we will discuss recent progress of the base metal catalysis for these important transformations.Diaminopyridine-based pincer ligands B also form potent CO2 hydrogenation catalysts. Described by the group of Kirchner and Gonsalvi,110 catalysts B-Fe-1 and 2 show a high activity in producing formate salts from CO2 in THF/H2O or EtOH in the presence of organic amine bases and NaOH. The non-bifunctional complex B-Fe-2 with N-substituted sidearms of the pincer ligand allowed for TON > 10000 at 80 °C and 80 bar pressure (H2/CO2 = 1/1) outperforming its bifunctional “NH” counterpart B-Fe-1 by a significant margin. A similar reactivity trend was later observed by the same group in formate dehydrogenation reactions, where B-Fe-2 outperformed its cooperative analogue B-Fe-1 by at least a two-fold margin. The dehydrogenation of formate/amine adducts with B-Fe-2 at 40–80 °C provides up to 10000 turnovers at ca. 2635 h−1 TOF in propylene carbonate solvent.111
A large body of work was dedicated to the activity of iron aminopincer catalysts with C-type ligands in the hydrogenation of CO2 and formate dehydrogenation with a particular focus on the highly beneficial effect of Lewis acid additives on these transformations112 that, in a broader context,113 attracted significant attention of the homogeneous catalysis community. Hazari and Bernskoetter and co-workers reported on the use of catalysts similar to C-Fe-1–4 (Scheme 13) for hydrogenation of CO2 in THF in the presence of DBU at 80 °C. Interestingly, the authors found that methylation of the central secondary amine in ligands C furnished significantly more active catalysts C-Fe-5–8 capable of reaching TOF values over 23000 h−1 making in total nearly 60000 turnovers. In all the cases the addition of Lewis acids to the reaction mixture proved highly beneficial with LiOTf being the most potent promoter compared to Na and K triflates.114
The promoting effect of the Lewis acids on the Fe aminopincer catalysis has been described in earlier works on the reverse process, namely, the dehydrogenation of formic acid. Hazari and Schneider and co-workers115 reported the use of catalyst C-Fe-9 in combination with LiBF4 in dioxane at 80 °C leading to one of the best formic acid dehydrogenation catalysts reported to date. This catalytic system exhibits outstanding TOFs of >196000 h−1 and provides a stable performance for almost a million turnovers. The catalyst required no external base additive for operation, which further rendered it superior to the vast majority of the noble metal counterparts.
 | ||
| Scheme 13 Catalysts introduced in Section 3.3. Types of transformations described for each particular catalyst are indicated with the sub-section label. | ||
The representatives of the aminopincer catalyst family can promote methanol dehydrogenation which is considered a significantly more difficult process. First demonstrated by Beller and co-workers116 using C-Fe-1 (Scheme 13) and its bromide analogue, the performance of Fe–aminopincers was later improved by the group of Hazari, Bernskoetter and Holthausen,117 who developed a base free protocol for methanol dehydrogenation employing a similar C-Fe-9 in combination with Lewis acid promoter LiBF4. As a result, the authors obtained a highly productive catalyst that performed ca. 51000 turnovers in refluxing ethyl acetate solvent with no basic additives needed.
000 h−1 if the reaction takes place under 20 bar pressure. The catalyst was shown to operate in the presence of rather unusual 2,8,9-triisopropyl-2,5,8,9-tetraaza-1-phosphabicyclo[3,3,3]undecane base, commonly referred to as Verkade base, named after its inventor.120 Although limiting prospective large scale application, this clearly outlined the high potential of Co-based catalysts for CO2 conversion. Later work by Linehan and Wiedner and co-workers121 explored the hydrogenation of CO2 with the M-Co-6 catalyst containing a macrocyclic amine/phosphine ligand with two additional pendant phosphine arms structurally similar to the very active Co-based CO2 electrochemical reduction catalysts.122M-Co-6 operated in the presence of the 2-tert-butyl-1,1,3,3-tetramethylguanidine base promoter in acetonitrile at room temperature. Under a total pressure of 1.7–1.8 bar the authors obtained turnover frequencies of 87–180 h−1 depending on the H2/CO2 ratio, which varied from 15/85 to 75/25.
Another class of Co complexes, M-Co-7 (Scheme 13) inspired by their Ir-based counterparts,123 was also shown to hydrogenate CO2. Being capable of aqueous phase bicarbonate/CO2 hydrogenation, M-Co-7 operated at 60–100 °C under 40–50 bar pressure reaching moderate maximal TOF of 39 h−1.124
Aminopincer complexes of Co, C-Co, were also shown to hydrogenate CO2 to formates. A report by Bernskoetter and co-workers41 described the activity of precatalyst C-Co-3 in the presence of LiOTf Lewis acid that was crucial for the catalytic performance. Combination with the DBU base promoter and acetonitrile solvent was found to provide the best catalyst productivity that amounted to ca. 29000 turnovers at 45 °C.
If the reaction is taken a step further, the hydrogenation of CO2 can yield methanol. A recent report by Beller and co-workers125 relies on an in situ formed Co(acac)3/triphos catalyst to promote this transformation. The catalyst operating in THF/EtOH solvent requires 100–140 °C to provide up to 78 turnovers under 70/20 (bar/bar) pressure of H2/CO2. The authors identified Co(acac)3 to be the optimal precursor for catalysis and noted that the use of Co(II) tetrafluoroborate hydrate provides nearly three-fold lower activity. The crucial additive that the authors employed was HNTf2 (trifluoromethanesulfonimide) which was used in ca. 2.5-fold excess of the Co.
An important extension of the CO2 reduction chemistry is the direct utilization of CO2 as a C1 building block in organic synthesis. A very common example in noble metal catalysis – amine alkylation or formylation – can also be promoted by Co catalysts. Disclosed by Milstein and co-workers,126 a series of (L)Co(II) dichlorides were active in amine formylation with 30/30 bar H2/CO2 in the presence of NaHBEt3 and KOtBu additives at 150 °C in toluene solvent. Primary and secondary amines were converted to the corresponding formamides in the presence of the M-Co-8 (Scheme 13) catalyst at 5%mol loading. Interestingly, catalysts with lutidine-based PNP pincer ligands A-Co-2,3 and bipyridine-based M-Co-9 showed good activity as well, while PNN complex A-Co-1 featuring the secondary amine sidearm was inactive. An important observation made by the authors suggests the active species to be Co(I) species formed from the Co(II) precatalyst upon treatment with NaHBEt3. The proposed active catalyst species were isolated and shown to be active in the absence of NaHBEt3 additive.
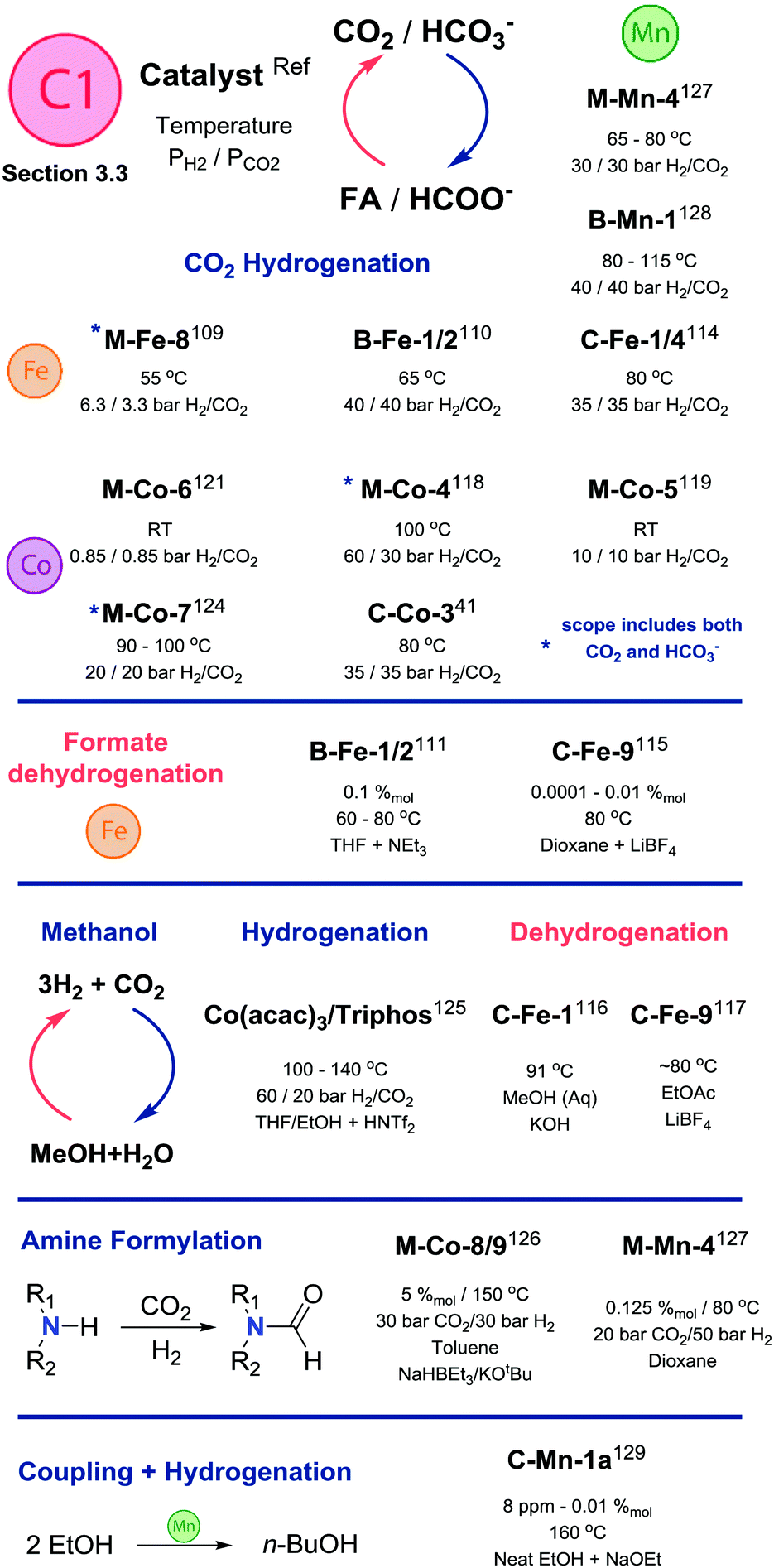 | ||
| Scheme 14 Comparative summary of catalyst performance and scopes in transformations described in Section 3.3. For the full substrate scopes see the ESI.† | ||
At the same time, the group of Kirchner and Gonsalvi described the use of the B-Mn-1 catalyst in CO2 hydrogenation to formates at 80 bar pressure and 80 °C.128 In THF/H2O solvent in the presence of lithium triflate and DBU base promoter the authors reached high TON values over 30000 using B-Mn-1 while its non-bifunctional analogue B-Mn-2 consistently provided much lower TON values beyond 1500. This behaviour of Mn pincers is particularly intriguing as their iron counterparts B-Fe-1 and 2 showed an inverse reactivity trend with NH-substituted pincers being more active.110
The latest example of a sustainable transformation promoted by Mn catalysts was reported by Liu and co-workers.129 An extensive study using several Mn catalysts identified C-Mn-1a (Scheme 13) as an excellent catalyst for ethanol upgrading that proceeds through dehydrogenative coupling and subsequent hydrogenation to 1-butanol. The authors unravelled a complex catalyst interconversion network and identified a series of intermediates in this transformation that was ultimately performed with over 114000 catalytic turnovers at an average TOF of 3078 h−1; this is a truly staggering performance for a base metal catalyst operating at 8 ppm loading at 165 °C.
Apart from the example of C-Mn-1a above, small molecule chemistry has benefited greatly from the base metal catalysis in the recent years (Scheme 14). Cobalt catalysts have been shown to promote the hydrogenation of CO2 to formates at very high TOFs and iron catalysts using C-type aminopincer ligands are currently among the most active formic acid dehydrogenation catalysts capable of base-free operation.
3.4. Dehydrogenation and dehydrogenative coupling reactions
In the final part of Section 3 we will discuss dehydrogenative transformations catalysed by base metal species. These reactions can be performed in a simple setting, e.g. producing ketones from alcohols with hydrogen liberation, or can form convoluted reaction networks where dehydrogenated substrates and liberated H2 can participate in consecutive reactions. These reactions, studied in detail for noble metal catalysts,10 are becoming increasingly important synthetic tools and currently utilize a large number of base metal catalysts.130Similar catalysts were later found to promote dehydrogenation of glycerol in the presence of alkali hydroxides to form lactic acid salts (Scheme 16).133 Complexes C-Fe-1, 4 and 9 were found to be the most effective among other analogues with different substitution patterns and provide the lactate formation TONs up to 1050 at varied loadings as low as 0.004–0.2%mol.
Amine substrates can also undergo dehydrogenative coupling in Fe- catalysed transformations. Beller and co-workers reported that C-Fe-1 can be used to catalyse the synthesis of lactones and lactams through dehydrogenative transformation of diols or aminoalcohols (Scheme 16).134 Bernskoetter and co-workers135 recently described the use of the C-Fe-4 catalyst for dehydrogenative amidation of primary alcohols including methanol with secondary amines to yield the corresponding amides with good TON values up to 790.
Iron complexes with aromatic backbones also readily catalyse dehydrogenative coupling reactions. Among the most recent examples, Milstein and co-workers disclosed catalyst M-Fe-5 promoting aldimine formation via the hydrogenative coupling of nitriles and amines.136 At 60 °C and 10–20 bar H2, catalyst loadings of 1%mol were sufficient for the selective formation of a wide range of aromatic and aliphatic aldimines. Triazine137 and diaminopyridine138 based Fe(II) pincers, e.g.B-Fe-1, developed by Kirchner and co-workers were also found to catalyse amine/alcohol couplings to yield secondary amines.
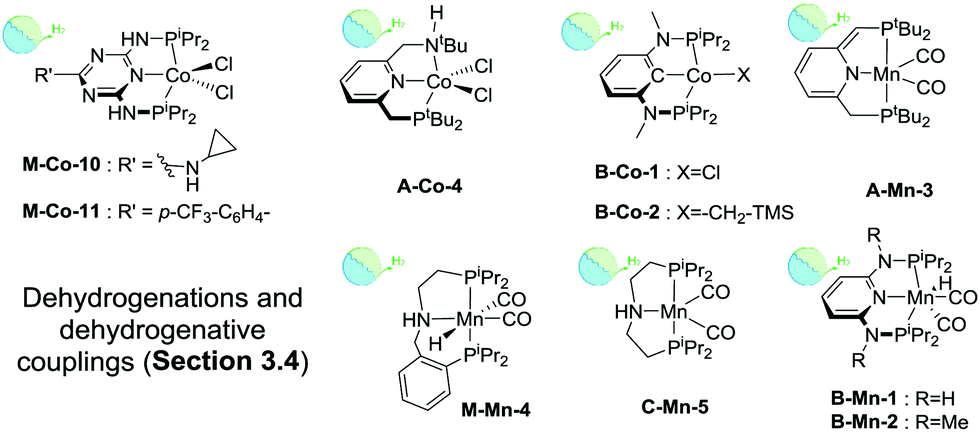 | ||
| Scheme 15 Catalysts introduced in Section 3.4. Types of transformations described for each particular catalyst are indicated with the sub-section label. | ||
Interestingly, non-pincer complexes, e.g.M-Fe-2, are potent dehydrogenative coupling catalysts as well. Feringa and Barta and co-workers reported this catalyst to efficiently promote alkylation of aliphatic or aromatic amines with aliphatic alcohols and diols.139 In the latter case the coupling leads to the cyclic amine (Scheme 16). The catalyst was shown to operate at 5%mol loading at 120–130 °C and required trimethylamine N-oxide additive for activation. Further work by Barta and co-workers extended the utility of M-Fe-2 to the alkylation of secondary and primary amines with benzyl alcohol derivatives140 and elegant synthesis of pyrroles from primary amines and unsaturated diols (Scheme 16).141
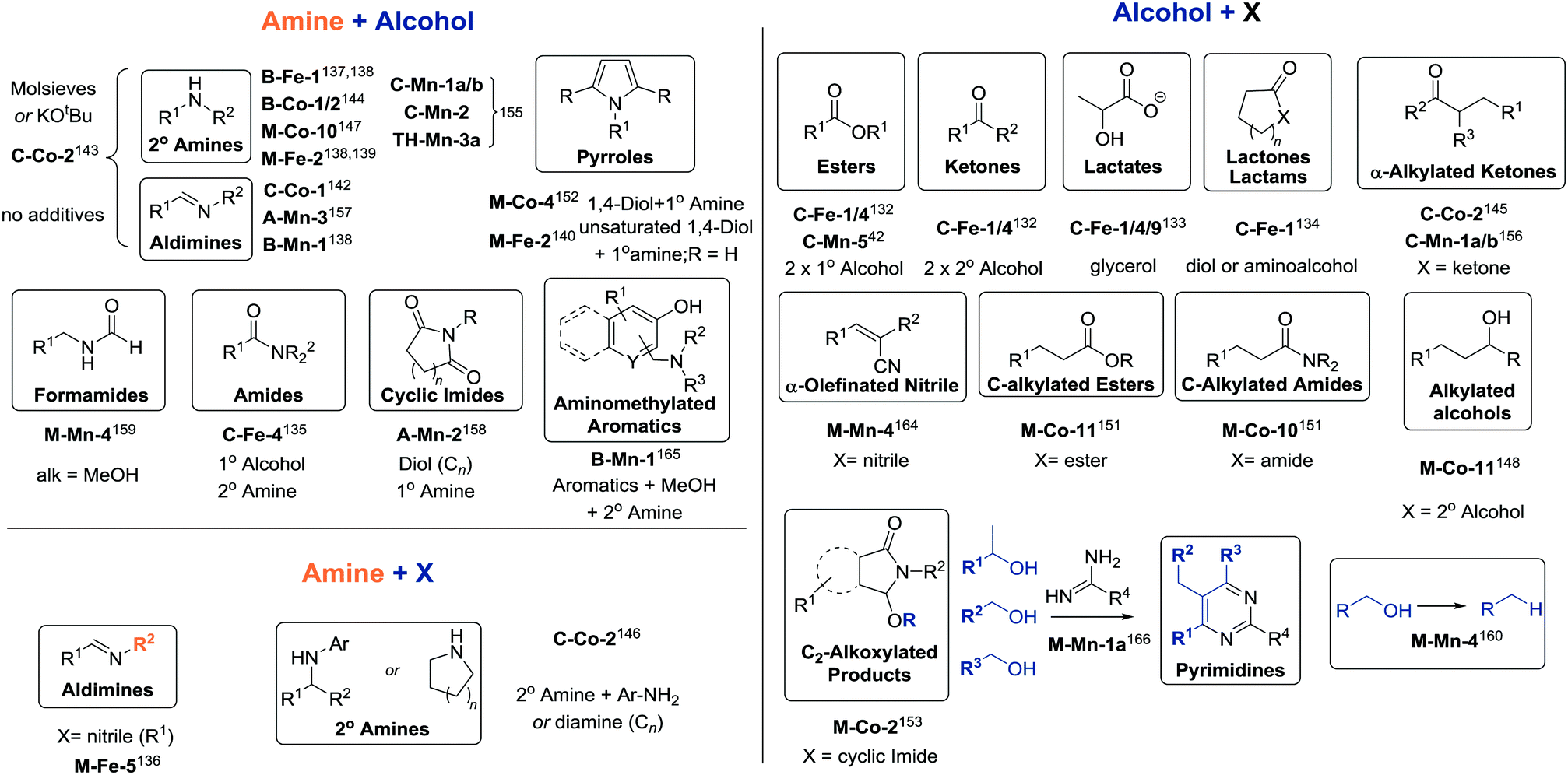 | ||
| Scheme 16 Products of various dehydrogenative coupling reactions described in Section 3.4. For the full substrate scopes see the ESI.† | ||
Zhang and co-workers143 later demonstrated that alcohol–amine coupling mediated by C-Co-2 can yield secondary amines instead of imines if the reaction was performed in the presence of molecular sieves. Catalyst loadings of 2%mol were required to obtain a variety of secondary amines in good to excellent yields with no imine byproduct obtained in most of the catalytic tests. A similar reaction could also be promoted by Co pincers with an aromatic backbone. Co(II) PCP pincers B-Co-1 and 2 (Scheme 15) reported by Kirchner and co-workers144 could catalyse efficiently aromatic amine alkylation with various aliphatic and aromatic alcohols in the presence of KOtBu or molecular sieves. Interestingly, the activity was not restricted to Co(II) species; related Co(III)PCP catalysts also showed catalytic activity, although inferior to that of Co(II)PCPs.
Alcohols can also be used for α-alkylation of ketones in Co-based catalysis (Scheme 16). Zhang and co-workers demonstrated that the C-Co-2 catalyst can be utilized in a multistep reaction involving dehydrogenation, aldol condensation and subsequent hydrogenation steps.145 Finally, the same group of authors later showed that amine homocoupling to produce secondary and cyclic amines using C-Co-2 was also possible under similar conditions.146
Before the implementation of C-ligated cobalt pincers, the “hydrogen” borrowing strategy, i.e. sequential dehydrogenation-coupling-hydrogenation, was applied to amine/alcohol coupling by Kempe and co-workers,147 who used M-Co-10 (Scheme 15) and related catalysts with varied substitution patterns for the alkylation of different aromatic amines. The authors employed M-Co-10 at 2%mol loading in toluene in the presence of the KOtBu promoter with no molecular sieves required (in contrast to C-Co-2). Very recently, a related Co catalyst M-Co-11 was shown to promote alkylation of secondary alcohols with primary ones at 2–5%mol loadings in the presence of 1.1 equivalents of KHMDS per substrate (Scheme 16).148
A more challenging C-alkylation of amides and esters known to be promoted by noble-metal catalysts149,150 was recently demonstrated by Deibl and Kempe.151 Amides were converted in the presence of 2.5%molM-Co-10 catalyst in THF at 100 °C while alkylation of esters was done with M-Co-11 in toluene at 5%mol loading. For both reactions, the addition of 1.2–1.5 equivalents of KOtBu was necessary to promote the alkylation.
Dehydrogenative coupling promoted by Co took a step further when Milstein and co-workers demonstrated that 1,4-diol and primary amine can be coupled to produce a 1,2,5-substituted pyrrole with liberation of hydrogen and water.152
Catalyst A-Co-4 (Scheme 15) used in this transformation required 5%mol loading and the same amounts of KOtBu and NaHBEt3 additives to operate at 150 °C in toluene. The scope of transformation included coupling of 2,5-hexanediol, 1,4-butanediol and its 1,4-diphenyl analogue with various aromatic and aliphatic primary amines. The authors observed no activity in the absence of sodium triethylborohydride while some variation of the base loadings and types was possible with KH and KOtBu providing the highest and nearly identical product yields.
Very recently Beller and co-workers153 reported an efficient methoxylation of cyclic imides using the Co–triphos catalyst M-Co-2 (Scheme 8). With this catalyst various substituted and/or N-alkylated succinimides and phthalimides were methoxylated at the C2 carbonyl group to produce 2-methoxy functionalized products (Scheme 16). Interestingly, intramolecular alcoxylation was also possible when the starting cyclic imide was N-substituted with a 3-hydroxypropyl unit. The same group later extended their scope of Co-catalyzed transformations to C3-alkenylation of indoles with carboxylic acids in a hydrogen atmosphere using a Co(acac)3/triphos catalyst.154
Mn-catalyzed coupling of amines and alcohols can be halted at the aldimine formation stage when no further hydrogenation takes place to yield an amine product (Scheme 16). The group of Milstein157 reported the use of a lutidine-based Mn pincer A-Mn-3 (Scheme 15) capable of promoting amine–alcohol coupling to yield aldimines and molecular H2 and water at 135 °C at 3%mol loading. Although reactions were performed in sealed vessels, no further hydrogenation of aldimines was observed and selectivity of the transformation was within 70–100%. Comparing these results to an open vessel reaction revealed no impact of a hydrogen pressure buildup on the transformation, suggesting no plausible hydrogenation pathway promoted by A-Mn-3. Interestingly, catalyst A-Mn-2 that features the NH function on the sidearm was also active in diol/primary amine coupling but instead produced cyclic imides.158
Mn pincers based on a 2,6-diaminopyridine scaffold B were also shown to be potent catalysts for coupling amines and alcohols. Aldimine formation was promoted by catalyst B-Mn-1 (Scheme 15) under conditions similar to those utilized for a lutidine-based A-Mn-3 catalyst.138 Unlike the iron complex B-Fe-1, the isoelectronic B-Mn-1 was fully selective for aldimine formation while Fe-catalysed coupling led to amines, thus, being hydrogenative.
Formylation of amines using methanol can also be promoted by manganese pincers. Milstein and co-workers159 disclosed catalyst M-Mn-4 that features a modified PNP aminopincer ligand. In methanol solvent at 110 °C, the disclosed catalyst required a 2%mol loading for near quantitative conversions of starting amines with yields of 50–86% depending on the substrate. The same catalyst was found to be highly active in the “dehydrogenative deoxygenation” of alcohols160 – a combination of alcohol dehydrogenation to produce an aldehyde and subsequent Wolff–Kishner reduction of the aldehyde with hydrazine (Scheme 16). The same catalyst M-Mn-4 was shown to be superior to related A-Mn-2 and 3 in the latest report describing α-olefination of nitriles with primary alcohols as co-substrates. In sharp contrast to noble metal promoted alcohol/nitrile couplings producing saturated α-substituted nitriles,161–163 catalysis by M-Mn-4 furnishes α,β-unsaturated products when operating at 4%mol loading in a closed-vessel setup at 135 °C in toluene solvent.164
The use of methanol as a coupling agent was also explored by Kirchner and co-workers, who described an elegant aminomethylation protocol for the conversion of aromatic compounds into their methyleneamine derivatives (Scheme 16).165 Catalyst B-Mn-1 (Scheme 15) introduced earlier was active at 4%mol loading and selectively provided amine products, while the isoelectronic analogue B-Fe-1 mainly promoted the methylation reaction.
Mn catalysts were recently shown to promote acceptorless dehydrogenative coupling of alcohols producing esters and H2. Gauvin and co-workers42 reported on the use of the C-Mn-5 (Scheme 15) catalyst operating at temperatures of 110–150 °C under base-free conditions. The extensive mechanistic study performed by the authors will be described in Section 4.
One of the latest reports on Mn-promoted dehydrogenative transformations makes use of the M-Mn-1a (Scheme 10) catalyst in a sophisticated coupling of primary alcohol, secondary alcohol and amidine to yield a variety of pyrimidines with highly complex substitution patterns (see Scheme 16).166 Operating at moderate loadings of 2%mol the catalyst could promote a three-component coupling with synthetically viable yields typically to the extent of 60–70%. At a higher loading of 5%mol even four-component coupling yielding fully substituted pyrimidine products was possible.
4. Mechanistic aspects of (de)hydrogenation
A solid base of mechanistic works on base metal promoted (de)hydrogenations has been formed in the last few years. Either integrated into experimental study or being fully theoretical investigations these works are of crucial importance for understanding the principles underlying the catalytic reactivity. Relying in part on the extensive and well-established knowledge of Ru catalysis, mechanistic works discussed in this section grew more complex with the involvement of the base metal catalysis. One of the reasons for this is that instead of one noble metal, three base metals currently promote similar transformations. The intrinsic differences between Fe, Co and Mn and their impact on the catalytic performance are perhaps the most intriguing topic in the following mechanistic investigations. In this section we will highlight recent findings regarding the reactivity and mechanistic analysis of base metal (de)hydrogenation catalysis.4.1. Aromatic pincers: chemically divergent catalysis and involvement of bifunctional action
One of the direct consequences of having different metals promoting similar reactions is their divergence in catalytic performance. The work reported by Kirchner and co-workers provides a good example of this phenomenon.138 As described above, the authors found that alkylation of amines with alcohol yields imines in the manganese catalysed process, whereas the isoelectronic iron catalyst yields amines as the sole product. The reaction pathway for the manganese-catalysed reaction was studied using DFT calculations (Scheme 17). The authors assumed a bifunctional mechanism operative in this case and the highest barrier (33 kcal mol−1) was computed for the transformation of the Mn hydride 1 to a dihydrogen complex BMn taking place via the deprotonation of the ligand NH sidearm. The hydrogen release from BMn results in a five-coordinate AMn that can either form stable alkoxide adduct CMn or promote the alcohol dehydrogenation via the formation of an alkoxide Mn–PNP complex and inner sphere β-hydride elimination to yield the initial hydride complex 1via a ca. 38 kcal mol−1 barrier. Alternative pathways with 1 also involved deprotonation of the PNP ligand at the intermediate steps and they were all found to proceed with very similar barriers.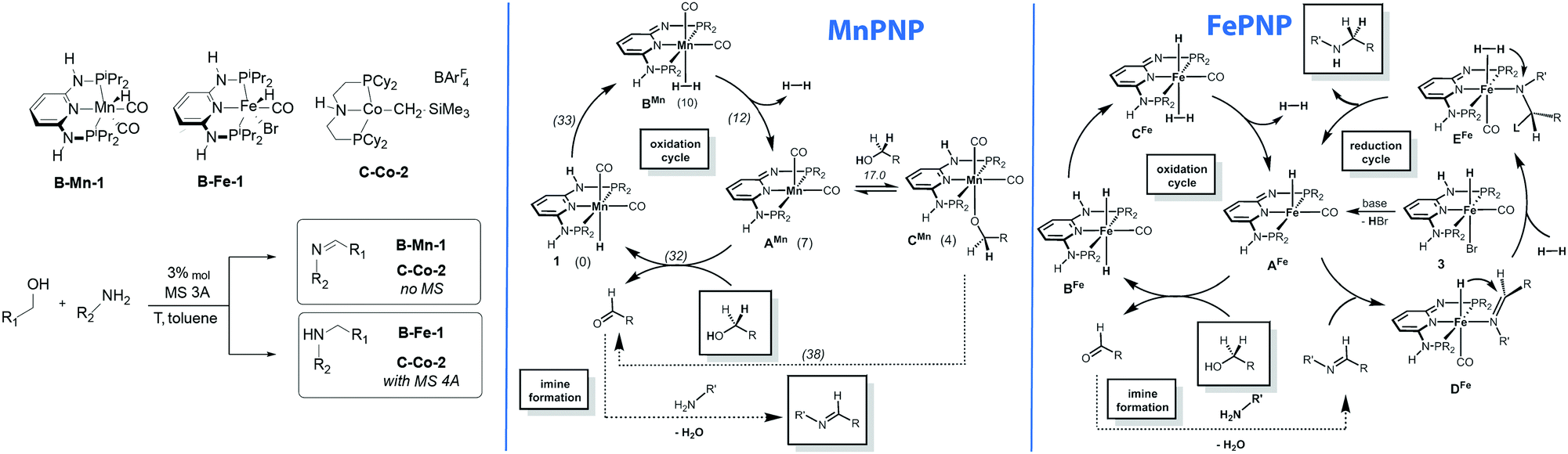 | ||
| Scheme 17 Chemically divergent amine–alcohol coupling with Fe and Mn catalysts. Catalytic cycles reproduced from ref. 138 (Mastalir et al., Chem. – Eur. J., 2016, 22, 12316) with permission from Wiley-VCH Verlag GmbH &Co. KGaA. | ||
While the manganese catalyst formally promoted the oxidation of alcohol to ketone that further formed the imine product upon water liberation, catalysis by iron complexes was proposed to promote further reduction of imines via a mechanism similar to that earlier disclosed for ketone hydrogenation.34 Most remarkably, the iron catalysed reduction is proposed to proceed via a five-coordinated AFe rather than trans-dihydride complex BFe marking a strong contrast to the report by Yang167 pointing out the intermediacy of trans-dihydrides in acetophenone hydrogenation that does not involve ligand participation. A similar behaviour was observed in related CO2 hydrogenation Ru–PNP catalysts.168
The key feature of the chemical divergence in the B-Fe/Mn catalyst family is the apparent inactivity of the Mn catalyst in imine hydrogenation also noted for another related Mn system including A-Mn-3 PNP pincer.157 The activity of C-type Mn aminopincers described in Section 3.4.3 of this Review adds another dimension to the complexity of base metal catalysis as the majority of C-Mn pincers favour formation of secondary amines, but not the imines.155
Finally, cobalt aminopincer C-Co-2 provides an example of chemical divergence controlled solely by the reaction conditions. In the presence of molecular sieves the amine–alcohol coupling produces amines.146 In the absence of molecular sieves when B-Mn catalysts lose a great fraction of their activity C-Co-2 promotes clean formation of imines.142
Cases of divergent catalytic activity can be observed for catalysts containing the same metal centres but bound to different ligands. A good example of this behaviour is ketone and aldehyde hydrogenation by Fe–PNP pincers (Scheme 18). As described above, catalysts A and B-Fe reduce both substrates if bifunctional behaviour is enabled by the ligand structure – e.g. in complexes B-Fe-1 and A-Fe-1. When the cooperative function of the PNP ligand is blocked by substitution (B-Fe-2) or complete replacement of the cooperative motive (Fe-POCOP169) the carbonyl reduction is selective to aldehydes and does not transform ketones.
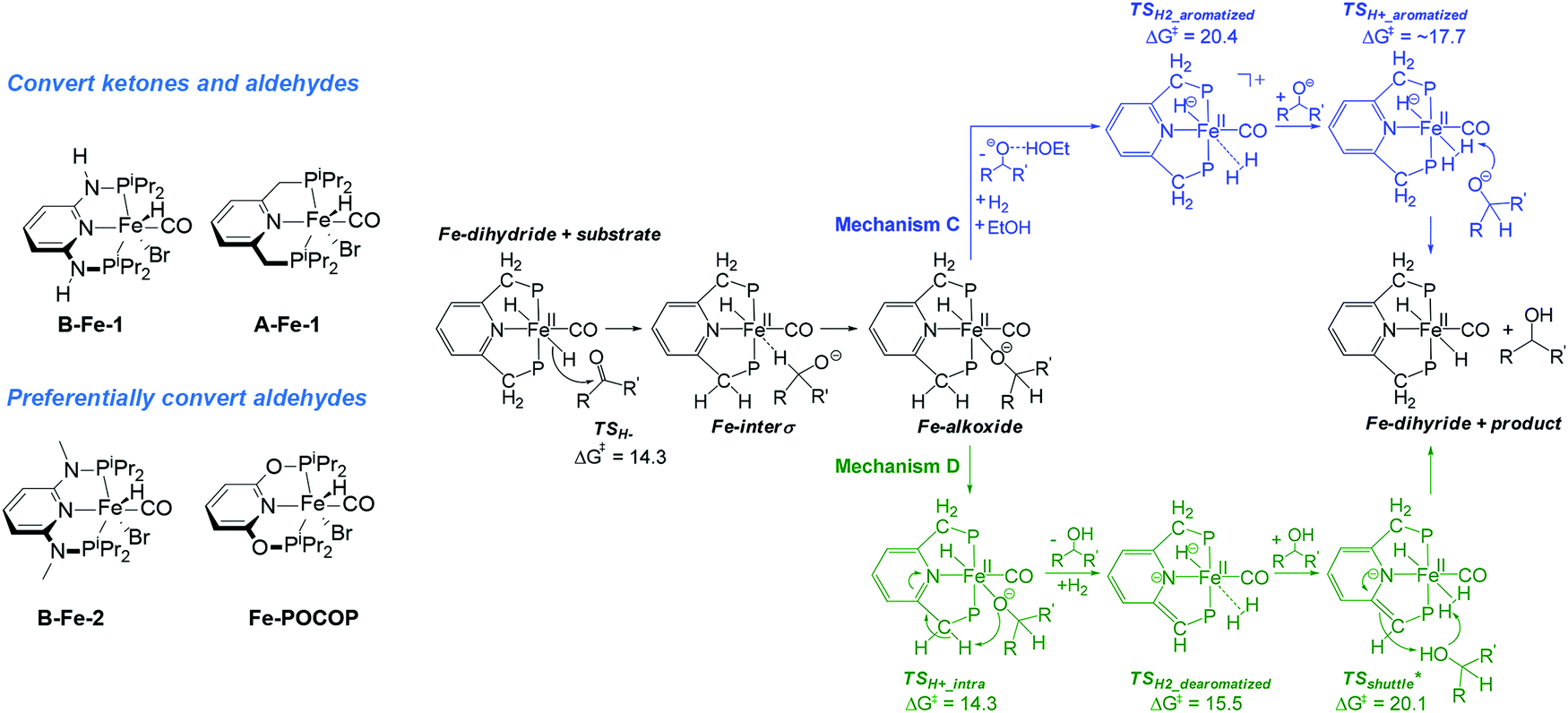 | ||
| Scheme 18 Chemically divergent aldehyde and ketone hydrogenation with Fe PNP pincers. Catalytic cycles reproduced with permission from ref. 174 (ACS Catal., 2017, 7, 5847; http://pubs.acs.org/doi/abs/10.1021/acscatal.7b00764. Further permissions related to the material excerpted should be directed to the ACS). | ||
The involvement of metal–ligand bifunctional transformations in noble metal catalysis has been extensively studied and heavily debated.27,170–173 Mechanistic studies of the Fe–PNP-catalysed chemoselective reduction of aldehydes in the presence of ketones were conducted by the authors of the original reports as well as by independent groups,33,34,167 but were not able to find a clear consensus on the nature of reduction selectivity and, importantly, the active species in catalysis.
A recent mechanistic study by Morello and Hopmann174 integrates previous findings and elegantly tackles this challenge by identifying two reaction pathways operative in aldehyde-selective and general carbonyl reduction cases. In both cases Fe cis-dihydride complex (Scheme 18) is assumed to be the active species. The bifunctional mechanism D (Green path, Scheme 18) involves a hydride transfer to the carbonyl substrate followed by a proton transfer from the ligand sidearm to the bound alkoxide ligand. This leads to the alcohol release and the formation of a five-coordinate intermediate. The latter coordinates H2 to facilitate its heterolytic dissociation assisted by an alcohol product resulting in the regeneration of the initial dihydride complex.
For the non-bifunctional pincers the reaction also starts with the Fe–alkoxide formation (Mechanism C, blue path, Scheme 18). The alkoxide–metal bond is then cleaved with the concomitant insertion of a dihydrogen molecule, which is then readily split heterolytically with the hydride taken up by the metal centre and the proton transferred to the product. As the authors rightfully noted in the paper “iron–alkoxide has earlier been dismissed in computations on FePNPCH2, because its formation would result in an increase of the hydrogenation barrier”, however, “the low energy of the iron–alkoxide intermediate and the low barrier for its formation make it very likely that this intermediate will be formed” thus making a convincing case for the intermediacy of the Fe–alkoxide species in either mechanism C or D. The authors concluded that the relative stability of Fe–alkoxide compared to that of iron-dihydride can be the main selectivity-determining factor. It was observed that non-bifunctional catalysts form alkoxides that are typically less stable than the initial dihydride, whereas bifunctional catalysts form stable alkoxide complexes. Consequently, increased hydrogenation barriers lead to reduced selectivity in line with intrinsic substrate reactivity, which is higher for aldehydes and activated ketones.
4.2. Aminopincers: substrate activation complexity and relation to Ru catalysis
Aminopincer base metal catalysts have also been subjected to thorough mechanistic studies that revealed a complexity of catalytic performance of C-Fe, Co, Mn species sometimes described as “uncanny”.175 Among representative examples are Fe aminopincers shown to promote dehydrogenation and hydrogenation of N-heterocycles131 that were later found to adopt different dehydrogenation pathways depending on the C–N bond polarity in the substrate. Bellows showed that more polar C–N bonds are dehydrogenated through a stepwise mechanism while relatively unpolarised C–N bonds are dehydrogenated in a concerted manner.175 Interestingly, the methylation of the central NH amine in C led to a complete loss of dehydrogenation activity, while the opposite took effect in C-Fe catalysed CO2 hydrogenation reported by Hazari and Bernskoetter and co-workers, who described the methylation of the aminopincer ligand to provide a significantly more active hydrogenation catalyst.114The understanding of C-aminopincer-based catalysis has progressed in recent years in part due to the presence of a large number of works addressing closely related reactions. This allowed the authors to provide extensive comparison between several state-of-the-art systems in a unified framework. An elegant example of such synergy is the extensive report by Gauvin and co-workers42 on the C-Mn promoted acceptorless dehydrogenative coupling (ADC) of alcohols. In the mechanistic section of their report, the authors construct an ADC reaction network and carefully identify similarities and differences with Mn-,92 Ru-17,176–178 and Fe-based117,132,179 catalysts for ADC or reverse hydrogenation reaction.
The authors proposed an amido complex C-Mn-5 (complex 1 in Scheme 19) to be the active species in the dehydrogenation cycle. Complex 1 was proposed to act as a hydrogen acceptor leading to the formation of the aldehyde product and a hydride complex 2 (Scheme 19). The authors observed the reversible reaction between 1 and the alcohol substrate that protonates the amide nitrogen and yields a more stable Mn-bound alkoxide complex. However, the authors found no direct reaction pathway yielding the aldehyde product from this alkoxide complex and proposed that alkoxide formation merely “masks” the active species. The catalytic cycle is further closed by the hydrogen elimination from hydride 2. The aldehyde was further proposed to be converted to a hemiacetal that is dehydrogenated to an ester in a similar cycle. Similar to the Ru-catalyzed ester hydrogenation,16 the authors experimentally observed the intermediate aldehyde formation that additionally justifies their mechanistic proposal. Although hemiacetal formation was not discussed in detail, Jones and Schneider132 in their study on the C-Fe system identified several mechanisms including direct aldehyde–alcohol coupling as well as substrate-assisted and metal-catalysed generation of hemiacetal among which the metal catalysed path was favoured by a significant margin. This observation makes an important addition to the ADC and ester hydrogenation chemistry, as the non-catalysed hemiacetal formation or decomposition, often implied in the catalytic cycles throughout the field, is not necessarily a facile process that can be omitted.180
 | ||
| Scheme 19 Mechanism of Mn-catalysed dehydrogenative coupling described by Gauvin and co-workers. Reproduced with permission from ref. 42 (Nguyen et al., ASC Catal., 2017, 7), © 2017 American Chemical Society. | ||
The DFT-computed reaction pathway for Mn-promoted dehydrogenation (Scheme 19) features two steps that are energetically demanding. First one is the alcohol dehydrogenation sequence (Scheme 19, 1-EtOH → 2-H, 17.9 kcal mol−1) leading to the aldehyde formation and generation of metal hydride aminopincer 2-H. The second event includes the dehydrogenation of 2-H to regenerate the active species and produce H2. Two pathways – unassisted and substrate-promoted – were identified for this metal–ligand cooperative transformation. The substrate-promoted dehydrogenation had a somewhat lower barrier of 19.4 kcal mol−1 that was the highest overall barrier in a computed mechanism. The presence of two closely matched energy demanding steps in Mn-catalyzed ADC marks a significant difference with Ru and Fe systems that feature a significantly lower alcohol dehydrogenation barrier compared to that for H2 release.
4.3. Metal oxidation state – cobalt and manganese
A remarkable feature of base metals that distinguishes them from late transition metals is the accessibility of multiple oxidation states that can show drastically different catalytic activity. For example, the Co/triphos precatalyst and its analogues are typically formed in situ from either Co(II) or Co(III) precursors. The group of Elsevier and de Bruin observed formation of Co(II) species that were proposed to be the resting state during carboxylic acid hydrogenation with Co/triphos.84 On the other hand, Beller and co-workers observed the accumulation of Co(I) species in a related catalytic system utilizing a Co(III) metal precursor for nitrile hydrogenation.88 Similar intermediacy of Co(I) species was also observed by Milstein and co-workers, who addressed the use of a series of Co(II) precatalysts in dehydrogenative coupling. The authors found that Co(I) was likely the actual active species in catalysis that was formed upon reaction of the Co(II) precatalyst with NaHBEt3 additive. Independent verification using the isolated Co(I) catalyst confirmed this suggestion. This trend somewhat contrasts the observation that C-Co type catalysts disclosed by Zhang and Hanson (Section 3) are believed to operate as Co(II) species. In addition, an extensive study of Kempe and co-workers on CO bond hydrogenation revealed that M-Co-1 remains in 2+ oxidation upon activation, likely preserving this oxidation state throughout the catalytic cycle.81 Finally, Co catalysts for transfer hydrogenation were recently studied by Zhao and Ke and co-workers,181 who identified the inner-sphere non-bifunctional mechanism as operational and, importantly, compared their findings to those for Fe catalysts that operate bifunctionally. The authors identified a high energy penalty for Fe catalysts to operate in the inner sphere and ascribed it to the tendency of Fe to maintain an 18 electron configuration that is in part stabilized by carbonyl ligands that are not present in the Co case.
The case of Mn catalysts may likely prove to be even more convoluted than that of cobalt as manganese tends to adopt a larger number of oxidation states. One of the notable recent works by Kempe and co-workers98 highlighted the importance of maintaining the 1+ oxidation state of the manganese centre and demonstrated that dihalide Mn(II) species were catalytically inert. More strikingly, the reduction of Mn(II) to produce the carbonyl-free Mn(I) species does not enable catalytic activity, which additionally emphasizes the importance of auxiliary ligands for the activity of Mn(I).
4.4. Notable features of base metal catalysis – the role of additives and catalyst deactivation
With the large number of works addressing the base metal catalysis it became evident that additives and promoters are often critically important to improve the activity of these catalysts. In several cases described in this Review the magnitude of this effect is many-fold greater than that observed in noble metal catalysis.One of the earliest catalysts relying heavily on the additives were C-Fe aminopincers for dehydrogenation and hydrogenation catalysis. Bernskoetter and Hazari summarized the studies on the highly beneficial effect of Lewis acid (LA) additives in their recent account.112 A truly outstanding performance of C-Fe in the abovementioned reactions was in large part defined by the LA promotion that was also demonstrated to be efficient in C-Co-promoted hydrogenations.41
Another common additive in base metal catalysis, shared with their noble metal counterparts, is the basic promoter. Commonly, alkoxide bases are employed for the activation of precatalysts and triggering the bifunctional transformations that require rather low base loadings to the extent of few equivalents with respect to the catalyst. Several Mn-based catalytic reactions, however, require much higher base concentrations. The M-Mn-3 catalyst96 shows elevated activity at 75%mol loading of KOtBu that amounts to ca. 375 equivalents per metal site, indicating the potential involvement of base in the catalytic cycle. Comparable loadings of base can be found in Co catalysis – M-Co-11 catalysing alcohol alkylation required 1.1 equivalents of KHMDS.148 Similarly high loadings of KOtBu were also required by M-Co-10,11 to promote amide and ester alkylation.151
Non-anionic bases are also often applied as additives in base metal catalysis. Nitrogen bases in particular were shown to perform several functions in enhancing the catalytic performance. For example, DBU base was successfully used as a basic promoter in CO2 hydrogenation by a series of Fe and Mn catalysts and reduction of aldehydes by B-Fe-1a.56 The latter case is a rare precedent where a nitrogen base can provide catalytic activity comparable to that achievable with anionic KOtBu base. Interestingly, while room temperature activities of B-Fe-1a in the presence of KOtBu and DBU were different, the performance of both base promoters was nearly identical at 40 °C which implies significantly different promotion mechanisms by anionic and neutral bases. Weaker bases like trimethylamine are also employed as additives. For example, NEt3 was proposed to prevent the catalyst poisoning by neutralizing the carboxylic acid intermediates formed during the Fe-catalysed aldehyde hydrogenation.52
Organic and inorganic bases together with borohydrides discussed in the previous section are the most common additives found in (de)hydrogenative catalysis. Although the exact role of these additives is often debated, the majority of works imply their involvement in catalyst activation. This activation may involve the reduction of the metal centres, as in the case of Co catalysts, or may trigger the metal–ligand bifunctional reactivity, necessary to bring the dormant precatalyst into the active cycle. Another example of the additive that is believed to be a catalyst activator is encountered in Fe catalysis, most notably, M-Fe-2. As this complex is a neutral tris-carbonyl one, decarbonylation step is necessary to liberate the vacant site and allow for the complex to function as a catalyst. A number of works demonstrated that selective and mild decarbonylation can be performed in the presence of trimethylamine N-oxide that can clearly be considered a catalyst activator.139–141
An important feature of base metal catalysts that has only been addressed recently is the catalyst deactivation. Despite the tremendous hardship associated with conducting such studies, significant progress is being made to unravel the chemistry behind catalyst activation and deactivation. For example the stability of iron transfer hydrogenation catalysts closely related to TH-Fe-2 was found to depend strongly on the substitution pattern.182 Namely, ligands with diethylphosphine donors were yielding less stable catalysts compared to diphenylphosphine-based analogues. The authors identified a major pathway for deactivation to be the partial or full reduction of the tetradentate iminophosphine ligand that was preferential for ethyl substituted complexes. Interestingly, the reduction of the iminophosphine ligand producing two metal bound NH ligands makes the complex catalytically inert despite the fact that one would expect the presence of the bifunctional NH–PR2 chelate to enable the activity to some extent.
An extensive work by Langer and co-workers37 on C-Fe-catalysed amide hydrogenation provides another deep insight into catalyst (de)activation pathways. Apart from catalytic application itself, the authors studied the reactivity of C-Fe complexes towards dehydrogenation and activation by borane elimination (Scheme 20). Importantly, it was found that the ligand structure had a profound effect on the catalyst stability with less sterically demanding ligands being superior to the bulky PNP aminopincers. It was demonstrated that the increased bulk of the PNP ligand produces less solution-stable complexes that can undergo sequential dehydrogenation to form deactivated complexes 6 and 7 (Scheme 20). Complexes with low steric demand were shown to be activated via the BH3 loss to form catalytically competent Fe dihydride species 4 that readily interconvert with isolable amido complex 5. The accessibility of 4, however, does not necessarily provide a stable complex as 4 can in fact lose hydrogen and decompose as well. In addition, formation of inactive Fe(0) PNP complexes and the free ligand was observed by Langer in the case of C-Fe and A-Fe33 hydrogenation catalysts, suggesting the reduction of the metal centre to be highly unfavourable.
 | ||
| Scheme 20 Activation and deactivation pathways of an Fe–PNP catalyst described by Langer and co-workers. Reproduced with permission from ref. 37 (Schneck et al., Organometallics, 2016, 35), © 2017 American Chemical Society. | ||
5. Present standing and future prospects
In the last three years we have witnessed a tremendous burst in research devoted to base metal promoted catalysis. Especially remarkable is the surge in the catalytic systems using manganese that were disclosed within the last year, while the comparable base of reports took five years for Co- and nearly a decade for Fe-based catalysis to build up. To a large extent we owe this progress to the universal ligand platforms that allow using seemingly different metals in a surprisingly similar framework from a synthetic and a catalytic perspective. As a result, at this point nearly all (de)hydrogenative catalytic transformations accessible to noble metals can be promoted by at least one of the base metal catalysts based on similar ligand systems. This renders the demand for replacement of the noble metals in (de)hydrogenative catalysis with base metals well met indeed.Mechanistic investigations of the base metal catalysis have played a notable role in establishing the field at the current level. In particular, we have started to understand the origins of unusual selectivity patterns that distinguish base metal catalysts from their noble counterparts. However, as we inherited much of the methodology from the noble metal based catalytic systems, our outlook on the base metal chemistry under study may be limited. In this respect, deviation from the conventional mechanistic framework may bring about new exciting knowledge on the reactivity of base metals in catalysis that could have been overlooked. As we currently have numerous catalytic systems based on several distinctly different early transition metals, we may expect new mechanistic proposals to take root in the field.
With metals utilized in (de)hydrogenative catalysis becoming increasingly cheaper, the future incentive might be aimed at improving the ligands that still rely heavily on the use of strong phosphine donors. We believe that the development of simple yet efficient ligands based on C,H,N,O elements could be the next step towards making base metal catalysis practical. The use of cheap and simple ligands could generally justify lower activities of base metal catalysts. In addition, the rational ligand design also holds promise for improving the performance of base–metal catalysts in the first place. Indeed, while the well-established pincer ligand platforms could very rapidly furnish sound proof-of-principle catalysts, they often fell short in providing the activity matching that of noble metals, for which the conventional pincer ligands have been originally developed. In this respect, the development of new metal- and reaction-tailored ligands is truly promising.
In recent years we have been given several examples of a departure from conventional ligands that was highly rewarding. This is best exemplified by the case of Fe catalysis that is now unmatched in transfer hydrogenation100 and formic acid dehydrogenation reactions.115 Having these reports as an inspiration we can expect the coming breakthrough in Co and Mn catalysis in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 725686). G. A. F. is sincerely thankful to J. Hutt for valuable discussions during the preparation of this manuscript. E. J. M. H. acknowledges the support from the Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC). Partial support from the Government of the Russian Federation (Grant 074-U01) and the Ministry of Education and Science of the Russian Federation (Project 11.1706.2017/4.6) is acknowledged.Notes and references
- J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, 2000 Search PubMed.
- L. Bouveault and G. Blanc, Compt. Rend., 1903, 136, 1676–1678 CAS.
- J. J. Li, Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications, Springer, 3rd edn, 2007, p. 77 Search PubMed.
- B. S. Bodnar and P. F. Vogt, J. Org. Chem., 2009, 74, 2598–2600 CrossRef CAS PubMed.
- P. Urben, Bretherick's Handbook of Reactive Chemical Hazards, Academic Press, 6th edn, 2006, 2, p. 8 Search PubMed.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS PubMed.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- J. Pritchard, G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
- F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS.
- Y.-N. Li, R. Ma, L.-N. He and Z.-F. Diao, Catal. Sci. Technol., 2014, 1498–1512 CAS.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. N. Schulpen, E. J. M. Hensen and E. A. Pidko, ChemCatChem, 2014, 6, 1526–1530 CrossRef CAS.
- G. A. Filonenko, M. J. B. Aguila, E. N. Schulpen, R. van Putten, J. Wiecko, C. Müller, L. Lefort, E. J. M. Hensen and E. A. Pidko, J. Am. Chem. Soc., 2015, 137, 7620–7623 CrossRef CAS PubMed.
- D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS PubMed.
- (a) Guideline on the specification limits for residues of metal catalysts or metal reagents, European Medicines Agency guideline in effect as per September 1st, 2008; http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003586.pdf; (b) Elemental Impurities in Drug Products, Food and Drug Administration draft guidance; https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm509432.pdf.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed.
- S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 12226–12250 CrossRef CAS PubMed.
- G. Bauer and X. Hu, Inorg. Chem. Front., 2016, 3, 741–765 RSC.
- M. Asay and D. Morales-Morales, Dalton Trans., 2015, 44, 17432–17447 RSC.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- G. van Koten, in Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013 Search PubMed.
- D. Benito-Garagorri and K. Kirchner, Acc. Chem. Res., 2008, 41, 201–213 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- P. A. Dub, B. L. Scott and J. C. Gordon, J. Am. Chem. Soc., 2017, 139, 1245–1260 CrossRef CAS PubMed.
- P. A. Dub and J. C. Gordon, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- S. J. C. Robinson and D. M. Heinekey, Chem. Commun., 2017, 53, 669–676 RSC.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- N. V. Belkova, L. M. Epstein, O. A. Filippov and E. S. Shubina, Chem. Rev., 2016, 116, 8545–8587 CrossRef CAS PubMed.
- R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef CAS PubMed.
- R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120–2124 CrossRef CAS PubMed.
- N. Gorgas, B. Stöger, L. F. Veiros, E. Pittenauer, G. Allmaier and K. Kirchner, Organometallics, 2014, 33, 6905–6914 CrossRef CAS PubMed.
- D. Benito-Garagorri, L. G. Alves, M. Puchberger, K. Mereiter, L. F. Veiros, M. J. Calhorda, M. D. Carvalho, L. P. Ferreira, M. Godinho and K. Kirchner, Organometallics, 2009, 28, 6902–6914 CrossRef CAS.
- N. Gorgas, L. G. Alves, B. Stöger, A. M. Martins, L. F. Veiros and K. Kirchner, J. Am. Chem. Soc., 2017, 139, 8130–8133 CrossRef CAS PubMed.
- F. Schneck, M. Assmann, M. Balmer, K. Harms and R. Langer, Organometallics, 2016, 35, 1931–1943 CrossRef CAS.
- G. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed.
- D. Zhu, F. F. B. J. Janssen and P. H. M. Budzelaar, Organometallics, 2010, 29, 1897–1908 CrossRef CAS.
- M. Brookhart, B. Grant and A. F. Volpe, Organometallics, 1992, 11, 3920–3922 CrossRef CAS.
- A. Z. Spentzos, C. L. Barnes and W. H. Bernskoetter, Inorg. Chem., 2016, 55, 8225–8233 CrossRef CAS PubMed.
- D. H. Nguyen, X. Trivelli, F. Capet, J.-F. Paul, F. Dumeignil and R. M. Gauvin, ACS Catal., 2017, 7, 2022–2032 CrossRef CAS.
- S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed.
- D. S. Mérel, M. L. T. Do, S. Gaillard, P. Dupau and J.-L. Renaud, Coord. Chem. Rev., 2015, 288, 50–68 CrossRef.
- L. C. Misal Castro, H. Li, J.-B. Sortais and C. Darcel, Green Chem., 2015, 17, 2283–2303 RSC.
- T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687–1695 CrossRef CAS PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- E. McNeill and T. Ritter, Acc. Chem. Res., 2015, 48, 2330–2343 CrossRef CAS PubMed.
- C. Sui-Seng, F. N. Haque, A. Hadzovic, A.-M. Pütz, V. Reuss, N. Meyer, A. J. Lough, M. Zimmer-De Iuliis and R. H. Morris, Inorg. Chem., 2009, 48, 735–743 CrossRef CAS PubMed.
- C. Sui-Seng, F. Freutel, A. J. Lough and R. H. Morris, Angew. Chem., Int. Ed., 2008, 47, 940–943 CrossRef CAS PubMed.
- T. Zell, Y. Ben-David and D. Milstein, Catal. Sci. Technol., 2015, 5, 822–826 CAS.
- T. Zell, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 4685–4689 CrossRef CAS PubMed.
- J. A. Garg, S. Chakraborty, Y. Ben-David and D. Milstein, Chem. Commun., 2016, 52, 5285–5288 RSC.
- B. Bichler, C. Holzhacker, B. Stöger, M. Puchberger, L. F. Veiros and K. Kirchner, Organometallics, 2013, 32, 4114–4121 CrossRef CAS PubMed.
- N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2016, 6, 2664–2672 CrossRef CAS PubMed.
- L. Bonomo, L. Kermorvan and P. Dupau, ChemCatChem, 2015, 7, 907–910 CrossRef CAS.
- S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed.
- S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed.
- S. Elangovan, B. Wendt, C. Topf, S. Bachmann, M. Scalone, A. Spannenberg, H. Jiao, W. Baumann, K. Junge and M. Beller, Adv. Synth. Catal., 2016, 358, 820–825 CrossRef CAS.
- N. M. Rezayee, D. C. Samblanet and M. S. Sanford, ACS Catal., 2016, 6, 6377–6383 CrossRef CAS.
- U. Jayarathne, Y. Zhang, N. Hazari and W. H. Bernskoetter, Organometallics, 2017, 36, 409–416 CrossRef CAS.
- S. Lange, S. Elangovan, C. Cordes, A. Spannenberg, H. Jiao, H. Junge, S. Bachmann, M. Scalone, C. Topf, K. Junge and M. Beller, Catal. Sci. Technol., 2016, 6, 4768–4772 CAS.
- C. Bornschein, S. Werkmeister, B. Wendt, H. Jiao, E. Alberico, W. Baumann, H. Junge, K. Junge and M. Beller, Nat. Commun., 2014, 5, 1–11 Search PubMed.
- S. Chakraborty, G. Leitus and D. Milstein, Chem. Commun., 2016, 52, 1812–1815 RSC.
- S. Chakraborty and D. Milstein, ACS Catal., 2017, 7, 3968–3972 CrossRef CAS.
- A. Zirakzadeh, K. Kirchner, A. Roller, B. Stöger, M. Widhalm and R. H. Morris, Organometallics, 2016, 35, 3781–3787 CrossRef CAS.
- S. A. M. Smith, P. O. Lagaditis, A. Lüpke, A. J. Lough and R. H. Morris, Chem. – Eur. J., 2017, 23, 7212–7216 CrossRef CAS PubMed.
- C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816–5817 CrossRef CAS PubMed.
- C. P. Casey and H. Guan, J. Am. Chem. Soc., 2009, 131, 2499–2507 CrossRef CAS PubMed.
- J. P. Hopewell, J. E. D. Martins, T. C. Johnson, J. Godfrey and M. Wills, Org. Biomol. Chem., 2012, 10, 134–145 CAS.
- A. Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard and J.-L. Renaud, Angew. Chem., Int. Ed., 2012, 51, 4976–4980 CrossRef CAS PubMed.
- S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120–5124 CrossRef CAS PubMed.
- A. Tlili, J. Schranck, H. Neumann and M. Beller, Chem. – Eur. J., 2012, 18, 15935–15939 CrossRef CAS PubMed.
- S. Fleischer, S. Zhou, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 5120–5124 CrossRef CAS PubMed.
- P. Gajewski, M. Renom-Carrasco, S. V. Facchini, L. Pignataro, L. Lefort, J. G. de Vries, R. Ferraccioli, A. Forni, U. Piarulli and C. Gennari, Eur. J. Inorg. Chem., 2015, 1887–1893 CrossRef CAS.
- P. Gajewski, M. Renom-Carrasco, S. V. Facchini, L. Pignataro, L. Lefort, J. G. de Vries, R. Ferraccioli, U. Piarulli and C. Gennari, Eur. J. Inorg. Chem., 2015, 5526–5536 CrossRef CAS.
- P. Gajewski, A. Gonzalez-de-Castro, M. Renom-Carrasco, U. Piarulli, C. Gennari, J. G. de Vries, L. Lefort and L. Pignataro, ChemCatChem, 2016, 8, 3431–3435 CrossRef.
- D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed.
- A. Mukherjee, D. Srimani, S. Chakraborty, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2015, 137, 8888–8891 CrossRef CAS PubMed.
- S. Rösler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef PubMed.
- J. Yuwen, S. Chakraborty, W. W. Brennessel and W. D. Jones, ACS Catal., 2017, 3735–3740 CrossRef.
- G. Zhang, K. V. Vasudevan, B. L. Scott and S. K. Hanson, J. Am. Chem. Soc., 2013, 135, 8668–8681 CrossRef CAS PubMed.
- T. J. Korstanje, J. Ivar van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed.
- T. P. Brewster, A. J. M. Miller, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2013, 135, 16022–16025 CrossRef CAS PubMed.
- F. M. A. Geilen, B. Engendahl, M. Hölscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2011, 133, 14349–14358 CrossRef CAS PubMed.
- T. vom Stein, M. Meuresch, D. Limper, M. Schmitz, M. Hölscher, J. Coetzee, D. J. Cole-Hamilton, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2014, 136, 13217–13225 CrossRef CAS PubMed.
- R. Adam, C. B. Bheeter, J. R. Cabrero-Antonino, K. Junge, R. Jackstell and M. Beller, ChemSusChem, 2017, 10, 842–846 CrossRef CAS PubMed.
- J. R. Carney, B. R. Dillon and S. P. Thomas, Eur. J. Inorg. Chem., 2016, 3912–3929 CrossRef CAS.
- B. Maji and M. K. Barman, Synthesis, 2017, 3377–3393 CrossRef CAS.
- M. Garbe, K. Junge and M. Beller, Eur. J. Inorg. Chem., 2017, 4344–4362 CrossRef CAS.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 15364–15368 CrossRef CAS PubMed.
- M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825–5828 CrossRef CAS PubMed.
- M. Garbe, K. Junge, S. Walker, Z. Wei, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS PubMed.
- R. van Putten, E. A. Uslamin, M. Garbe, C. Liu, A. Gonzalez-de-Castro, M. Lutz, K. Junge, E. J. M. Hensen, M. Beller, L. Lefort and E. A. Pidko, Angew. Chem., Int. Ed., 2017, 56, 7531–7534 CrossRef CAS PubMed.
- N. A. Espinosa-Jalapa, A. Nerush, L. J. W. Shimon, G. Leitus, L. Avram, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2017, 23, 5934–5938 CrossRef CAS PubMed.
- F. Kallmeier, T. Irrgang, T. Dietel and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 11806–11809 CrossRef CAS PubMed.
- A. Mikhailine, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2009, 131, 1394–1395 CrossRef CAS PubMed.
- W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS PubMed.
- K. Z. Demmans, O. W. K. Ko and R. H. Morris, RSC Adv., 2016, 6, 88580–88587 RSC.
- G. Zhang and S. K. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC.
- Z. Shao, S. Fu, M. Wei, S. Zhou and Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 14653–14657 CrossRef CAS PubMed.
- M. Perez, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, ChemSusChem, 2017, 10, 83–86 CrossRef CAS PubMed.
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, Org. Lett., 2017, 19, 3656–3659 CrossRef CAS PubMed.
- A. Zirakzadeh, S. R. M. M. de Aguiar, B. Stöger, M. Widhalm and K. Kirchner, ChemCatChem, 2017, 9, 1744–1748 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed.
- S. Enthaler, ChemSusChem, 2008, 1, 801–804 CrossRef CAS PubMed.
- O. Rivada-Wheelaghan, A. Dauth, G. Leitus, Y. Diskin-Posner and D. Milstein, Inorg. Chem., 2015, 54, 4526–4538 CrossRef CAS PubMed.
- F. Bertini, N. Gorgas, B. Stöger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, ACS Catal., 2016, 6, 2889–2893 CrossRef CAS.
- I. Mellone, N. Gorgas, F. Bertini, M. Peruzzini, K. Kirchner and L. Gonsalvi, Organometallics, 2016, 35, 3344–3349 CrossRef CAS.
- W. H. Bernskoetter and N. Hazari, Acc. Chem. Res., 2017, 50, 1049–1058 CrossRef CAS PubMed.
- A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS PubMed.
- Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed.
- E. A. Bielinski, M. Förster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. C. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
- C. Federsel, C. Ziebart, R. Jackstell, W. Baumann and M. Beller, Chem. – Eur. J., 2012, 18, 72–75 CrossRef CAS PubMed.
- M. S. Jeletic, M. T. Mock, A. M. Appel and J. C. Linehan, J. Am. Chem. Soc., 2013, 135, 11533–11536 CrossRef CAS PubMed.
- P. B. Kisanga and J. G. Verkade, Tetrahedron, 2001, 57, 467–475 CrossRef CAS.
- S. A. Burgess, K. Grubel, A. M. Appel, E. S. Wiedner and J. C. Linehan, Inorg. Chem., 2017, 56, 8580–8589 CrossRef CAS PubMed.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- Y. M. Badiei, W.-H. Wang, J. F. Hull, D. J. Szalda, J. T. Muckerman, Y. Himeda and E. Fujita, Inorg. Chem., 2013, 52, 12576–12586 CrossRef CAS PubMed.
- J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 1890–1893 CrossRef CAS PubMed.
- P. Daw, S. Chakraborty, G. Leitus, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2017, 7, 2500–2504 CrossRef CAS.
- A. Dubey, L. Nencini, R. R. Fayzullin, C. Nervi and J. R. Khusnutdinova, ACS Catal., 2017, 7, 3864–3868 CrossRef CAS.
- F. Bertini, M. Glatz, N. Gorgas, B. Stoger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, Chem. Sci., 2017, 8, 5024–5029 RSC.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- J.-L. Renaud and S. Gaillard, Synthesis, 2016, 3659–3683 CrossRef CAS.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed.
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS.
- L. S. Sharninghausen, B. Q. Mercado, R. H. Crabtree and N. Hazari, Chem. Commun., 2015, 51, 16201–16204 RSC.
- M. Peña-López, H. Neumann and M. Beller, ChemCatChem, 2015, 7, 865–871 CrossRef.
- E. M. Lane, K. B. Uttley, N. Hazari and W. Bernskoetter, Organometallics, 2017, 36, 2020–2025 CrossRef CAS.
- S. Chakraborty, G. Leitus and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 2074–2078 CrossRef CAS PubMed.
- M. Mastalir, B. Stöger, E. Pittenauer, M. Puchberger, G. Allmaier and K. Kirchner, Adv. Synth. Catal., 2016, 358, 3824–3831 CrossRef CAS.
- M. Mastalir, M. Glatz, N. Gorgas, B. Stöger, E. Pittenauer, G. Allmaier, L. F. Veiros and K. Kirchner, Chem. – Eur. J., 2016, 22, 12316–12320 CrossRef CAS PubMed.
- T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed.
- T. Yan, B. L. Feringa and K. Barta, ACS Catal., 2016, 6, 381–388 CrossRef CAS.
- T. Yan and K. Barta, ChemSusChem, 2016, 9, 2321–2325 CrossRef CAS PubMed.
- G. Zhang and S. K. Hanson, Org. Lett., 2013, 15, 650–653 CrossRef CAS PubMed.
- G. Zhang, Z. Yin and S. Zheng, Org. Lett., 2016, 18, 300–303 CrossRef CAS PubMed.
- M. Mastalir, G. Tomsu, E. Pittenauer, G. Allmaier and K. Kirchner, Org. Lett., 2016, 18, 3462–3465 CrossRef CAS PubMed.
- G. Zhang, J. Wu, H. Zeng, S. Zhang, Z. Yin and S. Zheng, Org. Lett., 2017, 19, 1080–1083 CrossRef CAS PubMed.
- Z. Yin, H. Zeng, J. Wu, S. Zheng and G. Zhang, ACS Catal., 2016, 6, 6546–6550 CrossRef CAS.
- S. Rösler, M. Ertl, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef PubMed.
- F. Freitag, T. Irrgang and R. Kempe, Chem. – Eur. J., 2017, 23, 12110–12113 CrossRef CAS PubMed.
- L. Guo, X. Ma, H. Fang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2015, 54, 4023–4027 CrossRef CAS PubMed.
- Y. Iuchi, Y. Obora and Y. Ishii, J. Am. Chem. Soc., 2010, 132, 2536–2537 CrossRef CAS PubMed.
- N. Deibl and R. Kempe, J. Am. Chem. Soc., 2016, 138, 10786–10789 CrossRef CAS PubMed.
- P. Daw, S. Chakraborty, J. A. Garg, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 14373–14377 CrossRef CAS PubMed.
- J. R. Cabrero-Antonino, R. Adam, V. Papa, M. Holsten, K. Junge and M. Beller, Chem. Sci., 2017, 8, 5536–5546 RSC.
- J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Chem. Sci., 2017, 8, 6439–6450 RSC.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- M. Peña-López, P. Piehl, S. Elangovan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 14967–14971 CrossRef PubMed.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben David, N. A. Espinosa Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 CrossRef CAS PubMed.
- N. A. Espinosa-Jalapa, A. Kumar, G. Leitus, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11722–11725 CrossRef CAS PubMed.
- S. Chakraborty, U. Gellrich, Y. Diskin-Posner, G. Leitus, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 4229–4233 CrossRef CAS PubMed.
- J. O. Bauer, S. Chakraborty and D. Milstein, ACS Catal., 2017, 7, 4462–4466 CrossRef CAS.
- M. L. Buil, M. A. Esteruelas, J. Herrero, S. Izquierdo, I. M. Pastor and M. Yus, ACS Catal., 2013, 3, 2072–2075 CrossRef CAS.
- B. Anxionnat, D. Gomez Pardo, G. Ricci and J. Cossy, Org. Lett., 2011, 13, 4084–4087 CrossRef CAS PubMed.
- K. Motokura, D. Nishimura, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 5662–5663 CrossRef CAS PubMed.
- S. Chakraborty, U. K. Das, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11710–11713 CrossRef CAS PubMed.
- M. Mastalir, E. Pittenauer, G. Allmaier and K. Kirchner, J. Am. Chem. Soc., 2017, 139, 8812–8815 CrossRef CAS PubMed.
- N. Deibl and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 1663–1666 CrossRef CAS PubMed.
- X. Yang, Inorg. Chem., 2011, 50, 12836–12843 CrossRef CAS PubMed.
- G. A. Filonenko, E. J. M. Hensen and E. A. Pidko, Catal. Sci. Technol., 2014, 4, 3474–3485 CAS.
- S. Mazza, R. Scopelliti and X. Hu, Organometallics, 2015, 34, 1538–1545 CrossRef CAS.
- G. A. Filonenko, M. P. Conley, C. Copéret, M. Lutz, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2013, 3, 2522–2526 CrossRef CAS.
- H. Li and M. B. Hall, ACS Catal., 2015, 5, 1895–1913 CrossRef CAS.
- F. Hasanayn and A. Baroudi, Organometallics, 2013, 32, 2493–2496 CrossRef CAS.
- X. Yang, ACS Catal., 2012, 2, 964–970 CrossRef CAS.
- G. R. Morello and K. H. Hopmann, ACS Catal., 2017, 7, 5847–5855 CrossRef CAS.
- S. M. Bellows, S. Chakraborty, J. B. Gary, W. D. Jones and T. R. Cundari, Inorg. Chem., 2017, 56, 5519–5524 CrossRef CAS PubMed.
- X. Yang, ACS Catal., 2013, 3, 2684–2688 CrossRef CAS.
- X. Yang, ACS Catal., 2014, 4, 1129–1133 CrossRef CAS.
- E. Alberico, A. J. J. Lennox, L. K. Vogt, H. Jiao, W. Baumann, H.-J. Drexler, M. Nielsen, A. Spannenberg, M. P. Checinski, H. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 14890–14904 CrossRef CAS PubMed.
- S. Qu, H. Dai, Y. Dang, C. Song, Z.-X. Wang and H. Guan, ACS Catal., 2014, 4, 4377–4388 CrossRef CAS.
- L. M. Azofra, I. Alkorta, J. Elguero and A. Toro-Labbé, J. Phys. Chem. A, 2012, 116, 8250–8259 CrossRef CAS PubMed.
- C. Hou, J. Jiang, Y. Li, Z. Zhang, C. Zhao and Z. Ke, Dalton Trans., 2015, 44, 16573–16585 RSC.
- P. O. Lagaditis, P. E. Sues, A. J. Lough and R. H. Morris, Dalton Trans., 2015, 44, 12119–12127 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Extracts of full substrate scopes and reaction conditions for Section 3. See DOI: 10.1039/c7cs00334j |
| This journal is © The Royal Society of Chemistry 2018 |