
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Products and mechanism of the OH-initiated photo-oxidation of perfluoro ethyl vinyl ether, C2F5OCF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) CF2
CF2
A. J. C.
Bunkan
a,
G.
Srinivasulu
a,
D.
Amedro
a,
L.
Vereecken
 b,
T. J.
Wallington
c and
J. N.
Crowley
*a
b,
T. J.
Wallington
c and
J. N.
Crowley
*a
aDivision of Atmospheric Chemistry, Max-Planck-Institut für Chemie, Mainz 55128, Germany. E-mail: john.crowley@mpic.de
bInstitute of Energy and Climate Research, IEK-8: Troposphere, Forschungszentrum Jülich GmbH, Jülich 52425, Germany
cResearch & Advanced Engineering, Ford Motor Company, Dearborn, Michigan 48121, USA
First published on 11th April 2018
Abstract
The OH-initiated photo-oxidation of perfluoro ethyl vinyl ether (C2F5OCF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2, PEVE) in air (298 K, 50 and 750 Torr total pressure) was studied in a photochemical reactor using in situ detection of PEVE and its products by Fourier transform IR absorption spectroscopy. The relative rate technique was used to derive the rate coefficient, k1, for the reaction of PEVE with OH as k1 = (2.8 ± 0.3) × 10−12 cm3 molecule−1 s−1. The photo-oxidation of PEVE in the presence of NOx at 1 bar results in formation of C2F5OCFO, FC(O)C(O)F and CF2O in molar yields of 0.50 ± 0.07, 0.46 ± 0.07 and 1.50 ± 0.22, respectively. FC(O)C(O)F and CF2O are formed partially in secondary, most likely heterogeneous processes. At a reduced pressure of 50 Torr, the product distribution is shifted towards formation of FC(O)C(O)F, indicating the important role of collisional quenching of initially formed association complexes, and enabling details of the reaction mechanism to be elucidated. An atmospheric photo-oxidation mechanism for PEVE is presented and the environmental implications of PEVE release and degradation are discussed.
CF2, PEVE) in air (298 K, 50 and 750 Torr total pressure) was studied in a photochemical reactor using in situ detection of PEVE and its products by Fourier transform IR absorption spectroscopy. The relative rate technique was used to derive the rate coefficient, k1, for the reaction of PEVE with OH as k1 = (2.8 ± 0.3) × 10−12 cm3 molecule−1 s−1. The photo-oxidation of PEVE in the presence of NOx at 1 bar results in formation of C2F5OCFO, FC(O)C(O)F and CF2O in molar yields of 0.50 ± 0.07, 0.46 ± 0.07 and 1.50 ± 0.22, respectively. FC(O)C(O)F and CF2O are formed partially in secondary, most likely heterogeneous processes. At a reduced pressure of 50 Torr, the product distribution is shifted towards formation of FC(O)C(O)F, indicating the important role of collisional quenching of initially formed association complexes, and enabling details of the reaction mechanism to be elucidated. An atmospheric photo-oxidation mechanism for PEVE is presented and the environmental implications of PEVE release and degradation are discussed.
1 Introduction
The thermal and chemical resistance of fluoropolymers has led to their use in many industrial processes and to their production in large quantities1,2 and thus to their release to the environment. We recently presented3 the first detailed kinetic study on the reaction of perfluoro ethyl vinyl ether (C2F5OCFCF2, henceforth referred to as PEVE) with OH radicals (R1), determining the rate coefficient, k1, using absolute and relative-rate techniques. k1 was found to be independent of pressure but it has a significant negative temperature dependence, indicating that the first step is addition of OH to the CC double bond. A room temperature value of k1 close to 3 × 10−12 cm3 molecule−1 s−1 implies an atmospheric lifetime of a few days, indicating that PEVE is not an important greenhouse gas. The main environmental concern about fluorinated vinyl ethers is whether their photo-oxidation leads to formation of persistent, fluorinated pollutants in the environment.
The products formed in the atmospheric photo-oxidation of organic trace gases depends on the fate of peroxy-radicals formed in the reaction between the initially generated organic radical fragments and O2. The chemistry is typically described as either “high NOx” where the peroxy radicals react with NO to form oxy radicals, which then propagate the radical chain, or “low NOx” where the dominating fate of the peroxy radical is reaction with HO2 or other peroxy radicals. Most short lived anthropogenic pollutants are oxidized under high NOx conditions, but as the two limiting cases have different mechanisms, differences in their product distribution provide valuable information about key reaction steps.
Extending our kinetics study, this work is a detailed examination of the products formed in the OH-initiated atmospheric degradation of PEVE in air, with the aim of deriving a detailed reaction mechanism applicable to atmospheric conditions (both high and low NOx). We also present a new measurement of the rate coefficient, k1, for the title reaction.
| OH + PEVE → products | (R1) |
2 Experimental methods
The experimental set-up used to study the OH-initiated photo-oxidation of PEVE has been described in detail previously4 and only a brief summary of its central components is given here. The reaction volume is a 44 l cylindrical quartz chamber equipped with a White-type multiple reflection mirror system resulting in a 43.7 m optical path length. Infrared spectra of the cell contents were recorded using a Bruker Vector FTIR instrument with a liquid N2-cooled MCT detector. The spectrometer was operated at a resolution of 0.5 cm−1 using boxcar apodization.The quartz reactor was at room temperature (298 ± 2 K) and for most experiments 1 bar total pressure. Six external, radially mounted, UV photolysis lamps provided a homogeneous light flux within the reactor for radical generation.
For the determination of k1 and for the product study in high-NOx conditions, OH radicals were generated by photolysis of methyl nitrite (2–10 × 1014 molecule cm−3)5 in the presence of up to 4 × 1014 molecule cm−3 NO using six Philips TL08 lamps (λmax ∼ 350 nm):
| CH3ONO + hν → CH3O + NO | (R2) |
| CH3O + O2 → CH2O + HO2 | (R3) |
| HO2 + NO → OH + NO2 | (R4) |
Under these conditions, the dominant fate of the organic peroxy radicals formed will be reaction with NO.
As the methyl nitrite based OH generation scheme is unsuitable for NOx free photo-oxidation, we also generated OH radicals by photolysis of ozone (1–2 × 1014 molecule cm−3) at 254 nm in a large excess of hydrogen (up to 2 × 1017 molecule cm−3). The large excess of H2 ensures that O(1D) does not react with PEVE. As desribed previously,3 variations in [H2] in this range did not lead to changes in the observed kinetics in a related system, confirming that unwanted O(1D) reactions were not important.
| O3 + hν → O(1D) + O2 | (R5) |
| O(1D) + H2 → OH + H | (R6) |
| OH + H2 → H2O + H | (R7) |
| H + O2 + M → HO2 + M | (R8) |
| H + O3 → OH + O2 | (R9) |
| HO2 + O3 → OH + 2O2 | (R10) |
This OH radical generation scheme generates both OH and HO2 radicals, and under these conditions, the organic peroxy radicals formed in the PEVE photo-oxidation will either react with HO2 radicals or undergo self-reaction.
Chemicals
Synthetic air (Westfalen), hydrogen (Westfalen 99.999%), ethylene (Matheson, Res. Pur.) and PEVE (Merck, 99%) were used as supplied without further purification. Ozone was generated by flowing air over a low-pressure Hg “Penray” lamp (thereby dissociating O2 to ground state O-atoms, which recombine with O2 to form O3) before entering the reaction cell.Methyl nitrite was prepared by the drop-wise addition of 50% sulphuric acid (Sigma-Aldrich) to a saturated solution of sodium nitrite in 50% methanol (Merck) in a three-necked round flask with a stirring magnet at 0 °C. To collect the resulting methyl nitrite, a slow flow of nitrogen was passed over the reaction mixture and into a trap at −78 °C. The resulting yellow liquid was then purified by vacuum distillation. Finally, a gas-phase sample of CH3ONO, diluted in nitrogen was prepared and stored in a blackened glass bulb at 298 K. The IR spectrum of a dilute gas-phase sample of methyl nitrite revealed minor impurities of methanol only (<0.3%).
3 Results
3.1 Infrared spectrum of PEVE
The IR spectrum of PEVE is characterized by a progression of strong absorption bands in the range 1050–1400 cm−1 (shown in Fig. 1A), a weak CC stretching mode at 1842 cm−1 and weaker absorption features at 749 to 840 cm−1. The spectrum closely resembles that of perfluoro methyl vinyl ether (CF3OCFCF2, henceforth referred to as PMVE) previously reported and assigned.6
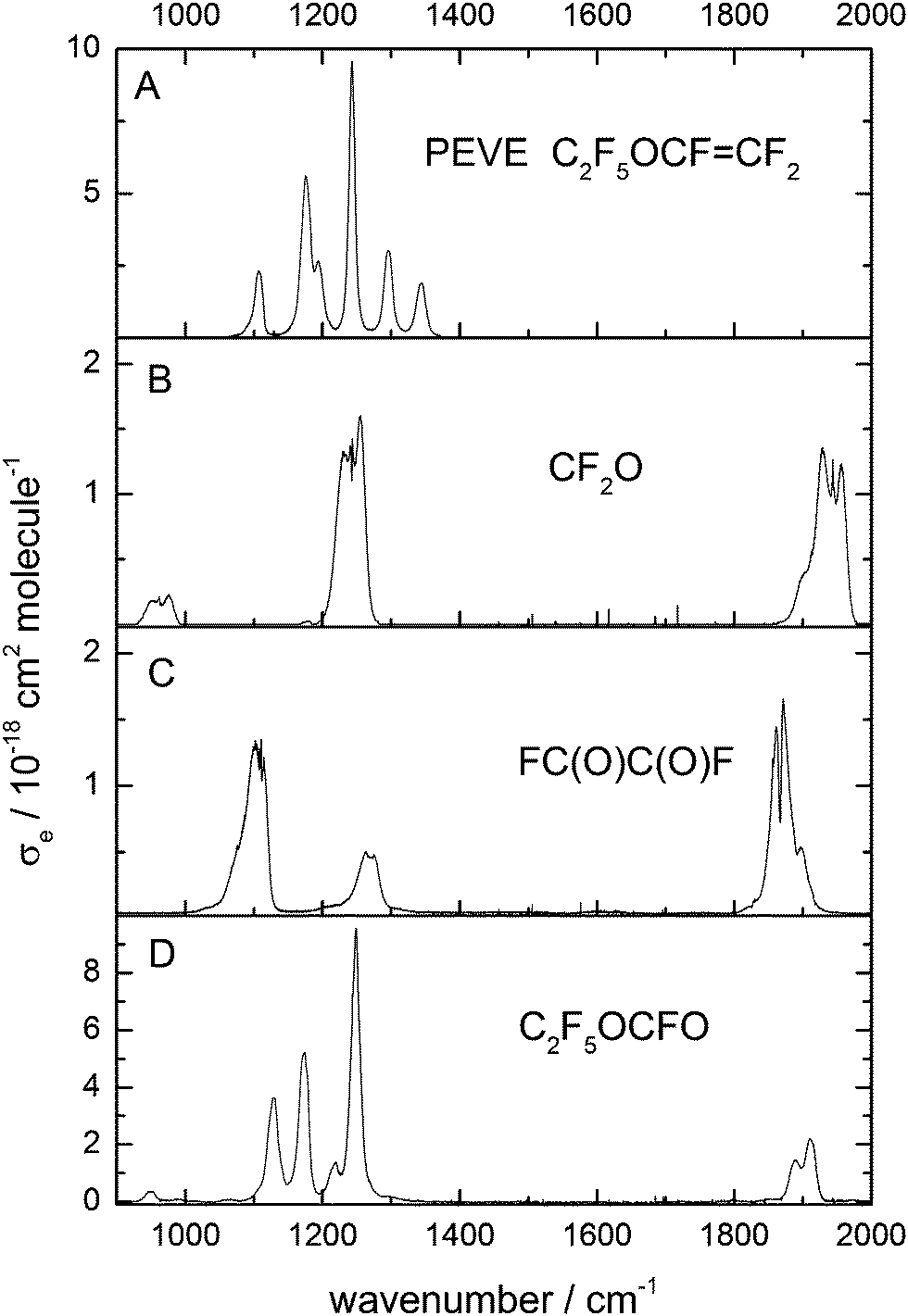 | ||
| Fig. 1 Absorption cross section (base e) of PEVE, CF2O, FC(O)C(O)F and C2F5OCFO in the C–F and C–O stretching region. The PEVE and C2F5OCFO spectra were obtained in this study, those of CF2O and CF(O)CFO have been reported previously.7 | ||
The reference spectrum of PEVE was obtained by recording spectra at different pressures of accurately diluted mixtures of PEVE (0.3 to 3.0 × 1013 molecule cm−3) and converting to absorption cross-sections after baseline correction and checking for Beer–Lambert behaviour.
3.2 Infrared spectra of photo-oxidation products
Using previously reported reference spectra (Fig. 1)7 CF2O and FC(O)C(O)F were readily identified as products of the OH-initiated oxidation of PEVE. The product spectrum obtained following PEVE oxidation showed strong absorption features at 1100–1300 cm−1 in addition to a progression of carbonyl absorption features at 1800–2000 cm−1.After subtracting features due to CF2O and FC(O)C(O)F (Fig. 1 panels B and C) the resulting spectrum is characterised by three strong absorptions at 1128, 1173 and 1249 cm−1, one at 1899 cm−1 and some weaker absorptions at 720, 757, 853, 950 and 1218 cm−1 (Fig. 1D). The spectrum has some similar spectral features to PEVE, but without the two vinylic C–F stretching modes at 1345 and 1296 cm−1. The other features in the range 1000–1300 cm−1 are somewhat blue-shifted compared to the PEVE spectrum and the weak CC-stretching absorption band at 1842 cm−1 is replaced by a much stronger absorption at 1899 cm−1. The initial assignment of this spectrum to C2F5OCFO was based on similarities to the spectra of perfluoromethyl fluoroformate.8 This assignment was independently confirmed by experiments in which the oxidation of PEVE was initiated by reaction with Cl-atoms rather than OH. In this case, only CF2O and C2F5OCFO are observed as products (see Fig. 2). This is in agreement with the results of Mashino et al.7 who observed CF3OCFO as a photo-oxidation product of the Cl initiated oxidation of PMVE with a yield of 100%. The reasons for the simplified product distribution when using Cl rather than OH to initiate the oxidation of PEVE is discussed later when we examine details of the reaction mechanism.
| PEVE + Cl(O2) → C2F5OCFO + CF2O | (R11) |
 | ||
| Fig. 2 Infrared spectrum acquired before (A) and after 360 s photolysis (B) of a mixture of 3.3 × 1013 molecule cm−3 PEVE and 2.6 × 1014 molecule cm−3 Cl2. Panel (C) shows the spectrum of C2F5OCFO obtained by subtracting water and CF2O. | ||
As each PEVE lost by reaction with Cl forms one molecule of C2F5OCFO, we were able to put the C2F5OCFO spectrum on an absolute basis. Once spectral features corresponding to water and CF2O had been subtracted, the calibrated C2F5OCFO absorption spectrum was obtained, as displayed in Fig. 1D.
3.3 Relative-rate measurements of k1
In our previous paper3 on the kinetics of the reaction between OH and PEVE we reported a rate coefficient under NOx free conditions in 50 and 100 Torr N2 and in 1 bar air. In this work we have measured the rate coefficient of PEVE with OH radicals relative to that of ethylene with NOx present.In the absence of loss processes other than reaction with OH, the depletion factors, ln(initial concentration/concentration after time t), for reactant and reference are described by:
 | (i) |
[PEVE]0, [ethylene]0, [PEVE]t and [ethylene]t are the concentrations of PEVE and ethylene at times t0 and t respectively, and k1 and k12 are the rate coefficients of reactions (1) and (12), respectively.
| PEVE + OH → Products | (R1) |
| C2H4 + OH → Products | (R12) |
The initial concentration ranges used for PEVE and ethylene were (1.3–2.9) × 1013 molecule cm−3 and (0.8–2.9) × 1014 molecule cm−3, respectively. A potential interference in relative rate measurements of fluorocarbons is the reaction of CF3O radicals with organics like ethylene.9
| CF3O + C2H4 → Products | (R13) |
To minimise the impact of (R13), high concentrations ((1–4) × 1014 molecule cm−3) of NO were added to the mixture. NO reacts rapidly with CF3O (k14(298 K) = 5.4 × 10−11 cm3 molecule−1 s−1) forming CF2O and FNO.10,11
| CF3O + NO → CF2O + FNO | (R14) |
The relative concentrations of NO and C2H4 used, and the rate coefficients for their reactions with CF3O, ensure that CF3O reacts predominantly (to 80%) with NO and variation of [NO] by a factor of four did not result in any significant changes in the measured rate coefficients. We thus conclude that our derivation of k1 is not significantly influenced by undesired reactions of CF3O radicals.
A typical experiment lasted around one hour during which the photolysis lamps were switched on for 5–10 periods of 10–300 s. The intermittent acquisition of FTIR spectra (128 scans at 0.5 cm−1 resolution) took ≈5 min. The time dependent depletion factors for PEVE and C2H4 were obtained by least squares fitting to reference spectra in the range 1080–1260 cm−1 for PEVE and 3030–3180 cm−1 for ethylene. Absorption by reaction products were accounted for by including reference spectra of CF2O, FC(O)C(O)F and C2F5OCFO as well as CH3ONO and its degradation products in the fit procedure (DOASIS12). An example spectrum with fit residuals is shown in Fig. 3.
 | ||
| Fig. 3 FTIR spectrum from a relative rate experiment in the spectral ranges used in the analysis with residuals from the fitting of reference spectra. | ||
A total of six relative rate experiments were performed under different experimental conditions, which are summarised in Table 1 and Fig. 4. A linear, least squares fit to all data (solid line in Fig. 4) gives k1/k12 = 0.361 ± 0.006 (2σ). From this k1 can be determined using an evaluated literature value13 for k11 (7.8 × 10−12 cm3 molecule−1 s−1 at 298 K and 1 bar pressure) to give: k1 = (2.8 ± 0.3) × 10−12 cm3 molecule−1 s−1 where the uncertainty includes assessment of the accuracy of the reference rate coefficient.13 The rate coefficient is in good agreement with the value (k1 = (3.0 ± 0.3) × 10−12 cm3 molecule−1 s−1) from our previous kinetic study on the title reaction3 and for other perfluoro vinyl ethers for which the OH-rate coefficients fall in the range (2.2–3.4) × 10−12 cm3 molecule−1 s−1.7,14,15 The rate coefficients and trends therein for different vinyl ethers are discussed in our previous publication3 and the interested reader is referred to that publication for details.
| Expt no. | P (Torr) | [PEVE]a | [C2H4]a | [CH3ONO]a | [NO]a | [OH]b | k rel |
|---|---|---|---|---|---|---|---|
| a The initial concentrations of PEVE (units of 1013 molecule cm−3), C3H8, NO and CH3ONO (units 1014 molecule cm−3) were determined from pressure measurements. b The OH concentration (units 108 molecule cm−3) was determined from the initial decay of PEVE. c 2σ statistical error. | |||||||
| 1 | 750 | 2.9 | 0.8 | 8.6 | 2.0 | 3.7 | 0.367 ± 0.010 |
| 2 | 750 | 2.8 | 1.5 | 8.2 | 2.1 | 7.4 | 0.368 ± 0.010 |
| 3 | 750 | 1.3 | 1.5 | 9.7 | 2.1 | 5.8 | 0.359 ± 0.020 |
| 4 | 750 | 2.9 | 1.5 | 9.8 | 1.0 | 11.9 | 0.354 ± 0.006 |
| 5 | 750 | 2.7 | 1.5 | 8.9 | 3.9 | 2.8 | 0.369 ± 0.016 |
| 6 | 750 | 2.8 | 2.9 | 8.6 | 2.0 | 5.6 | 0.358 ± 0.008 |
 | ||
| Fig. 4 Decay of PEVE versus C2H4 in the presence of OH radicals from six individual experiments in 750 Torr air diluent. The conditions for each experiment are listed in Table 1. | ||
3.4 Reaction products and yields (ΦNOx) in air (with NOx)
Products of the OH-initiated photo-oxidation of PEVE were detected and quantified in a separate series of experiments. In these experiments, NO is generated in the photolysis of CH3ONO and in some experiments was also added to the initial reaction mixture.As mentioned above, C2F5OCFO FC(O)C(O)F and CF2O were observed as products. The experiments were conducted with several photolysis periods interrupted by spectrum acquisition so that the time dependence of PEVE loss and product formation could be analysed. Time dependent concentrations of PEVE were obtained by fitting post photolysis spectra to reference spectra between 1080 and 1360 cm−1. Not only PEVE but also the fluorocarbon products (CF2O, FC(O)C(O)F and C2F5OCFO) absorb in this range, as do CH3ONO, CH3ONO2 and HNO3 and they were thus included in the spectral deconvolution procedure. Compared to PEVE, the fluorinated products absorb only weakly in this range (Fig. 1) and the concentrations of CF2O, FC(O)C(O)F and C2F5OCFO were therefore obtained by analysing the spectrum between 1855 and 2000 cm−1. In this range, the spectra of CF2O, FC(O)C(O)F and C2F5OCFO have very distinct carbonyl stretching modes (Fig. 1).
Concentration time profiles for PEVE, CF2O, FC(O)C(O)F and C2F5OCFO from a single experiment are shown in Fig. 5. A plot of Δ[product] versus Δ[PEVE] is displayed for a single experiment in Fig. 6. Fig. 6 illustrates that the plots of Δ[C2F5OCFO] and Δ[FC(O)CFO] versus Δ[PEVE] are straight lines, from which the product yields can be directly obtained. The experimental conditions and product yields (ΦNOx) from the total of seven experiments carried out in the presence of NOx are given in Table 2. In contrast to C2F5OCFO and FC(O)CFO, the plot of Δ[CF2O] versus Δ[PEVE] is non-linear and has a significant induction period before a roughly constant slope is achieved (in this case after ≈1 × 1013 PEVE has been reacted). This is a clear indication that CF2O is formed in secondary reactions. The yield of a product which is formed directly (or very rapidly) will be independent of time, whereas one which is formed in slower, secondary processes will show more complex time dependence.
 | ||
| Fig. 5 Concentration–time profiles of PEVE loss and simultaneous generation of fluorinated organic products from an experiment at 1 bar air in the presence of NOx. The error bars reflect 10% uncertinty in the cross-sections used. | ||
 | ||
| Fig. 6 Plot of formation of products versus depletion of PEVE from an experiment on the OH-initiated oxidation of PEVE in the presence of NOx. The slopes of the linear fits are the yields (±2σ) of C2F5OCFO and FC(O)CFO from this particular experiment. The polynomial fit to the CF2O data is added to emphasise the non-linearity at low PEVE conversions. | ||
| P/Torr | [PEVE]0a | [CH3ONO]0a | [NO]0a | [OH]0b | Φ NOx(CF2O)c | Φ NOx(FC(O)C(O)F)d | Φ NOx(C2F5OCFO)d |
|---|---|---|---|---|---|---|---|
| a The initial concentrations of PEVE (units of 1013 molecule cm−3), NO and CH3ONO (units 1014 molecule cm−3) were determined from pressure measurements. b The OH concentration (units 109 molecule cm−3) was determined from the initial decay of PEVE. c Φ NOx (CF2O) is the value obtained at long reaction times when the yield is constant (see text for details). d Φ NOx (FC(O)C(O)F) and ΦNOx (C2F5OCFO) were obtained from plots of Δproduct versus ΔPEVE as in Fig. 6. Uncertainties are 2σ. | |||||||
| 750 | 3.7 | 2.2 | 0.0 | 3.9 | 1.21 | 0.38 ± 0.06 | 0.49 ± 0.06 |
| 750 | 1.2 | 4.5 | 0.0 | 3.5 | 1.52 | 0.38 ± 0.04 | 0.44 ± 0.02 |
| 750 | 1.5 | 4.5 | 2.9 | 0.3 | 1.67 | 0.48 ± 0.06 | 0.46 ± 0.04 |
| 750 | 2.8 | 3.3 | 0.0 | 3.2 | 1.38 | 0.45 ± 0.16 | 0.48 ± 0.16 |
| 50 | 3.6 | 2.2 | 0.0 | 3.9 | 1.78 | 0.69 ± 0.04 | 0.11 ± 0.02 |
| 750 | 2.0 | 3.2 | 0.0 | 0.7 | 1.45 | 0.45 ± 0.18 | 0.49 ± 0.18 |
| 750 | 1.4 | 3.1 | 1.7 | 0.5 | 1.67 | 0.47 ± 0.06 | 0.56 ± 0.02 |
Time dependent yields of each of CF2O, FC(O)C(O)F and C2F5OCFO are displayed in Fig. 7. Whereas the yields of C2F5OCFO and FC(O)C(O)F are roughly constant, that of CF2O acquires a maximum value after several hundred seconds. The CF2O yields reported in Table 2 are averages obtained from the plateau after ≈240 s. Combining an estimated 10% uncertainty in absorption cross-sections with experimental scatter results in the following, average molar yields of products obtained in the presence of NOx at 1 bar pressure of air: ΦNOx(FC(O)C(O)F) = 0.46 ± 0.07, ΦNOx(C2F5OCFO) = 0.50 ± 0.07 and ΦNOx(CF2O) = 1.50 ± 0.22. The experimental CF2O yields are more variable than those of FC(O)C(O)F and C2F5OCFO, which is related to its production both in gas-phase and heterogeneous reactions (see below).
 | ||
| Fig. 7 Product yields from experiment with PEVE and CH3ONO. The results of only two experiments are displayed for C2F5OCFO and FC(O)CFO to preserve clarity of presentation. The solid, horizontal lines represent the average yield from all experiments. The CF2O yields represented by open symbols are from experiments in which NO was added. | ||
For FC(O)C(O)F and C2F5OCFO there is no significant difference in the yield between the different experiments, with or without added NO. In contrast, as illustrated in Fig. 7, the CF2O yields obtained at 1 bar pressure of air were largest when NO was added. The yields of all products were sensitive to the total pressure with values of ΦNOx(FC(O)C(O)F) = 0.69 ± 0.15, ΦNOx(C2F5OCFO) = 0.11 ± 0.03 and ΦNOx(CF2O) = 1.78 ± 0.25 obtained at 50 Torr air.
Possible reasons and mechanistic implications of the [NO] and pressure dependence are discussed below along with the overall degradation mechanism.
3.5 Reaction products and yields (Φ) in air (without NOx)
The formation of products in the OH-initiated oxidation of PEVE in the absence of NOx was studied in a separate series of experiments where OH radicals were generated by the photolysis of ozone in a large excess of hydrogen (see Section 2). The initial concentration range used for PEVE was (1.1–3.7) × 1013 molecule cm−3. The concentrations of ozone and hydrogen were (0.7–2.2) × 1014 and (0.8–2.2) × 1017 molecule cm−3, respectively. The fluorinated products observed, C2F5OCFO, CF2O and FC(O)C(O)F, were the same as those found in the CH3ONO/NO experiments. The molar yield of C2F5OCFO was similar, with Φ(C2F5OCFO) = (0.46 ± 0.07) whereas that of CF2O was slightly lower with Φ(CF2O) = 0.9–1.3. A further difference was the significant depletion of [FC(O)C(O)F] during these experiments at longer times, which appeared as a stable product in the presence of NOx.The results and experimental conditions of these experiments are summarised in Table 3, the mechanistic implications of these observations are discussed below.
| P/Torr | [PEVE]0a | [O3]0a | [H2]0a | [OH]0b | Φ(CF2O)c | Φ(FC(O)C(O)F)d | Φ(C2F5OCFO)c |
|---|---|---|---|---|---|---|---|
| a The initial concentrations of PEVE (units of 1013 molecule cm−3), O3 (units 1014 molecule cm−3) and H2 (units 1017 molecule cm−3) were determined from pressure measurements. b The OH concentration (units 109 molecule cm−3) was determined from the initial decay of PEVE. c Φ(FC(O)C(O)F) and Φ(C2F5OCFO) were obtained from plots of Δproduct versus ΔPEVE, uncertainties are 2σ. d Φ(FC(O)C(O)F) uses only data at short times and is a lower limit (see text for details). | |||||||
| 600 | 2.4 | 1.3 | 2.0 | 4.3 | 1.37 ± 0.04 | >0.36 | 0.49 ± 0.02 |
| 760 | 1.9 | 2.2 | 1.9 | 5.1 | 1.31 ± 0.02 | >0.34 | 0.49 ± 0.01 |
| 750 | 1.1 | 0.7 | 1.7 | 5.0 | 0.91 ± 0.05 | >0.30 | 0.44 ± 0.02 |
The time profiles and product yields observed in the experiments with NOx present, indicate that CF2O, FC(O)C(O)F and CF3CF2OCOF are not depleted to any significant extent by reaction with OH radicals. This is consistent with the fact that perfluorinated, saturated organic trace gases do not react rapidly with OH. In the NOx free experiments with 253.65 nm photolysis of O3/H2/air as OH precursor, a lower yield of FC(O)C(O)F was observed and it decayed significantly during the measurement. As FC(O)C(O)F does absorb weakly at 254 nm,16 some of this loss may be caused by photolysis. To examine whether other processes also play a role we stopped the photolysis after generation of a significant amount of the products and left the mixture in the dark with 2–6 × 1014 molecule cm−3 ozone for one hour with spectra recorded at regular intervals. Even in the dark, the concentration of FC(O)C(O)F decreased (with a decay constant of 2.5 × 10−4 s−1) while no significant loss of CF2O or C2F5OCFO was observed. Potential causes of the dark loss of FC(O)C(O)F could be direct reaction with ozone in the gas phase, or on surfaces, the latter mediated by surface catalysed dissociation of ozone.17 The scatter in the yields in the experiments without NOx most likely reflect variation in the removal rate and we therefore chose to report the product yield of FC(O)C(O)F as a lower limit with Φ(FC(O)C(O)F) > 0.36. The average yields of C2F5OCFO and CF2O are 0.47 ± 0.07 and 1.20 ± 0.20, respectively.
3.6 Mechanism for PEVE photo-oxidation in the presence of NO
From the observations described above, we can develop a reaction scheme for the OH-initiated oxidation of PEVE in air in the presence of NOx, which is summarised in Fig. 8. | ||
| Fig. 8 Proposed mechanism for the OH-initiated photo-oxidation of PEVE in the presence of NOx. Observed products are marked with black circles. The formation of CF2O and FC(O)C(O)F from their respective alcohols are assumed to be heterogeneous reaction and are marked with “Het”. | ||
The reaction of PEVE with OH radicals is initiated by addition of the OH radical to the double bond, which can in principle happen at both carbon atoms.
| PEVE + OH → C2F5OCFCF2OH | (R1a) |
| → C2C5OCF(OH)CF2 | (R1b) |
Computational studies on reaction of OH with PMVE18 and perfluoro propyl vinyl ether (C3F7OCFCF2, PPVE),15 indicate that addition to the terminal C-atom (R1a) dominates. The large experimental yield of FC(O)C(O)F is not commensurate with a significant contribution of (R1b) and we conclude that the OH-addition is predominantly at the terminal carbon atom (R1a). The adduct formed in (R1a) may either dissociate via C–O bond fission (R15) or react with oxygen at the neighbouring site giving a hydroxy–peroxy radical (R16).
| C2F5OCFCF2OH →C2F5 + HOCF2C(O)F | (R15) |
| C2F5OCFCF2OH + O2 → C2F5OCF(OO)CF2OH | (R16) |
| HOCF2C(O)F → FC(O)C(O)F + HF | (R17) |
The conversion of FC(O)CF2OH to FC(O)C(O)F in (R17) is unlikely to be an elementary reaction, as discussed below.
The fate of the hydroxyl–peroxy radical formed in R16 depends on the experimental conditions. In the experiments with NOx present, the radical will react with NO, forming an oxy-radical (R18) which may then dissociate to form either HOCF2C(O)F and the C2F5O radical (R19a) or the observed product C2F5OC(O)F along with the CF2OH radical (R19b).
| C2F5OCF(OO)CF2OH + NO → C2F5OCF(O)CF2OH + NO2 | (R18) |
| C2F5OCF(O)CF2OH → C2F5O + CF(O)CF2OH | (R19a) |
| → C2F5OC(O)F + CF2OH | (R19b) |
| CF2OH + O2 → CF2O + HO2 | (R20) |
As HOCF2C(O)F may dissociate to FC(O)C(O)F and HF, both reactions fit the observations, but the calculations by Vereecken et al.18 on PMVE and PPVE indicate (R19b) to be the dominant pathway. The fate of the CF2OH co-product of (R19b), is reaction with O2 to form CF2O.
As indicated above, the product yields were found to be dependent on the total pressure of air, with a substantial decrease in ΦNOx(C2F5OCFO) when going from 750 Torr to 50 Torr, accompanied by increases in ΦNOx(FC(O)C(O)F) and ΦNOx(CF2O). These trends are consistent with the results of Mashino et al.7 who (for PMVE) found that the CF3OCFO yield was reduced from 0.53 ± 0.04 at 700 Torr, to 0.08 ± 0.02 at 10 Torr. They attributed this pressure dependence to competition between HF elimination and collisional stabilization of the chemically activated PMVE–OH adduct. However, Vereecken et al.18 calculated that the barrier of this reaction is too high for it to be of any significance. Instead, they suggested that (for PMVE) the OH-adduct would preferentially dissociate to CF3 + CF(O)CF2OH, with a barrier around 125 kJ mol−1 below the entrance energy and proposed the equivalent mechanism for PPVE.15 Our observations of pressure dependent product yields can thus be understood in terms of competition between dissociation of an activated OH–PEVE complex ((R15), most important at low pressures) and thermalisation/reaction of the complex with O2 ((R16), most important at high pressures).
As addition of chlorine to double bonds is significantly less exothermic than addition of OH, the PEVE–Cl adduct is formed at lower energies. One may therefore expect a different product distribution if the conclusions drawn above are valid. This is indeed the case as only C2F5OCFO is observed, indicating that the PEVE–Cl adduct is formed below the barrier for dissociation to C2F5 + CF(O)CF2Cl and confirming that the product distribution is largely determined through the competition between dissociation and collisional stabilization of the initial PEVE-OH adduct.
The C2F5 radical formed (R15) in the dissociation of the C2F5OCFCF2OH radical will react with oxygen to give the perfluoroethyl peroxy radical (C2F5O2) which reacts with NO to form C2F5O ((R21) and (R22)). This radical is thermally unstable and will dissociate on a microsecond timescale to CF3 and carbonyl fluoride (R23).19,20 The CF3 radical reacts further with O2 and NO similar to the methyl radical giving another carbonyl fluoride molecule as final product ((R24), (R25) and (R14)).
| C2F5 + O2 + M → C2F5O2 + M | (R21) |
| C2F5O2 + NO → C2F5O + NO2 | (R22) |
| C2F5O → CF3 + CF2O | (R23) |
| CF3 + O2 → CF3OO | (R24) |
| CF3OO + NO → CF3O + NO2 | (R25) |
According to the reaction scheme ((R14)–(R25)) describing the subsequent fate of the initially formed OH-PEVE adduct, there are two main reaction routes with CF2O yields of one and two respectively, the total yield of CF2O should approximately be equal to the FC(O)C(O)F yield plus one, which is consistent with the experimental results.
Fig. 6 and 7 indicate that the formation of CF2O is delayed, whereas FC(O)C(O)F and C2F5OCFO are formed promptly. This is consistent with CF2O being formed from slow dissociation of CF3OH (R26).
| CF3OH → CF2O + HF | (R26) |
Perfluorinated primary alcohols are known to dissociate to the corresponding perfluoroacyl halide and HF.21,22 The mechanism for formation of FC(O)C(O)F in the photo-oxidation of a fluorinated vinyl ether has been discussed by Mashino et al.7 who suggested direct HF loss from CFOCF2OH in the gas phase. Later, Vereecken et al.18 calculated the barrier of this reaction to be 190 kJ mol−1 indicating that the reaction is too slow to be relevant at ambient temperatures and that FC(O)C(O)F is likely formed in a different process. The dissociation of CF3OH to CF2O + HF23 has a barrier of similar magnitude to that of CFOCF2OH, and it is reported to take place rapidly in chamber experiments with a rate depending on the history of the reaction chamber, indicating that a heterogeneous reaction is taking place.24 It has also been suggested that the dissociation of CF3OH is catalysed in the gas phase by water vapour,23 HF,25 CF3OH, or radicals.26–28
The difference in the observed CF2O yields upon changing the NO concentration can thus be rationalized as follows: When there is enough NO available for scavenging CF3O, CF2O is formed directly in the gas phase via(R14). At lower NO concentrations, a fraction of the CF3O radicals react to CF3OH which will then convert to CF2O and HF (R26). The time dependence of CF2O formation is thus controlled by the rate of diffusion of CF3OH to and from surfaces and explains the induction period of minutes. Note that neither FNO nor CF3OH were observed, and HF only in low concentrations. This reflects their high reactivity of these molecules on the quartz and metal surfaces inside the reactor.
3.7 Mechanism for PEVE photo-oxidation in the absence of NO
In the experiments with ozone and H2, there is no NO to scavenge either the CF3O radicals or the peroxy radicals. The peroxy radicals may react with HO2 to form either the hydroperoxide (R27) or with ozone to generate the oxy radical and oxygen (R28).| C2F5OCF(OO)CF2OH + HO2 → C2F5OCF(OOH)CF2OH + O2 | (R27) |
| C2F5OCF(OO)CF2OH + O3 → C2F5OCF(O)CF2OH + 2O2 | (R28) |
No evidence for formation of the hydroperoxide was found in the IR spectra and we assume that if it is formed it will either dissociate to the oxy radical and the OH radical (R29), be photolysed or be lost on surfaces.
| C2F5OCF(OOH)CF2OH → C2F5OCF(O)CF2OH + OH | (R29) |
The CF3O radicals react with hydrogen to give CF3OH and a hydrogen atom which rapidly react with O2 to form an HO2 radical
| CF3O + H2 → CF3OH + H | (R30) |
| H + O2 + M → HO2 + M | (R8) |
CF3OH will decompose to CF2O and HF as described above, but as this most likely happens on surfaces, some of the resulting CF2O may then hydrolyse to CO and HF, explaining the lower yield of CF2O in the ozone experiments.
3.8 Theoretical calculations on the fate of CF3OH and HOCF2C(O)F
As discussed above, the formation of CF2O from CF3OH and FC(O)C(O)F from HOCF2C(O)F, has been shown to be driven by heterogeneous reactions. While a detailed analysis of such reactions is outside the scope of the present paper, it is worthwhile to examine the intrinsic difference in reactivity between CF3OH and HOCF2C(O)F. We therefore examined the reactants and transition states (TS) for a set of representative reactions at the M06-2X/aug-cc-pVTZ level of theory.29,30 The relative energies were refined by single point CCSD(T)-F12a/aug-cc-pVDZ calculations.31,32 All calculations were performed using the Gaussian-09 and Molpro 2015.1 program suites.33,34For both CF3OH and HOCF2C(O)F, reaction proceeds by conversion of the –CF2OH moiety to a carbonyl group, with HF as a co-product. Literature data indicates that direct HF elimination has a large energy barrier, >160 kJ mol−1, but that the reaction can be catalysed by many co-reactants.23,25–28 The experimental data presented in this work (see Fig. 6 and 7) suggest that CF3OH eliminates HF significantly slower than HOCF2C(O)F. To study this dependence on the molecular structure, we have calculated the rate-limiting TS for HF formation both for the un-catalysed reaction, as for catalysed processes, where we examined CF3OH, CH3OH, H2O, HF, HC(O)OH, HO2, and OH as potential catalysts (see Table 4). In agreement with literature data, we find that the barrier for the un-catalysed reaction is too high to allow a significant contribution at room temperature, but that the various catalysts reduce the barrier heights, by up to 160 kJ mol−1. For nearly all catalysts, the barrier height for the HOCF2C(O)F substrate is lower than for the CF3OH compound, up to 13 kJ mol−1. The lower TS energies afford a faster rate coefficient by over an order of magnitude, affirming the experimentally observed difference in conversion rates for HOCF2C(O)F and CF3OH. The difference in the reactivity trend is expected to remain valid for nearly all reactions including heterogeneous reactions, as the underlying cause is the difference in reaction enthalpy imposed by the –C(O)F moiety in HOCF2C(O)F, which is absent in CF3OH (see Table 4). Of the gas phase co-reactants considered, we find that HO2 is the most efficient catalyst, pairing a radical reaction site with an –OO– molecular span that reduces the TS ring strain compared to the shorter –O– or –F– moieties in the other gas phase reactants. Intriguingly, we also find that CH3OH is a more efficient catalyst than CF3OH. This is related to the electron density on the –OH moiety; this aspect is not investigated in detail here. Larger catalysts consisting of complexes (e.g. HO2 with water molecules) have been shown to be even more effective, but we do not expect significant concentrations of such gas-phase complexes under our reaction conditions. Formic acid, HC(O)OH, expected to be present on the reactor walls, affords the most drastic reduction in barrier height of the catalysts considered, essentially removing any energetic barrier beyond the intrinsic reaction endoergicity. The catalysis mechanism involves donating an H-atom from the acidic hydroxy group and receiving an H-atom with the carbonyl group while shifting the double bond, thus combining a mobile H-atom with a large –O–CO– molecular span reducing TS ring strain, and strong H-bonding in the pre- and post-reaction complexes; this mechanism and its concomitant low energy barriers are likely to be applicable to all oxoacids including HNO3 and H2SO4. An in-depth study of heterogeneous reactions, be it with chemical contaminants or exposed metal surfaces, is beyond the scope of this manuscript.
| Catalyst | CF3OH | HOCF2C(O)F |
|---|---|---|
| a Calculated at the ZPE-corrected CCSD(T)-F12/aug-cc-pVDZ//M06-2X/aug-cc-pVTZ level of theory. | ||
| (None) | 186 | 186 |
| CF3OH | 97 | 93 |
| OH | 93 | 86 |
| HF | 89 | 86 |
| H2O | 75 | 64 |
| CH3OH | 52 | 39 |
| HO2 | 45 | 38 |
| HC(O)OH | 28 | 22 |
| Product energy | CF2O + HF: 30 | CF(O)C(O)F + HF: 24 |
While we cannot conclude that the reactions in Table 4 are those responsible for our observations of CF2O and FC(O)C(O)F formation, the selected catalysts are all expected to be present in our system, and combined allow for very rapid conversion of the fluoro-hydroxy moiety to a carbonyl moiety + HF, where especially low-volatility, surface-bound catalysts such as carboxylic acids or aqueous complexes have high catalytic activity. The –CFO moiety in HOCF2C(O)F lends itself to additional interactions (e.g. H-bonding), which could again enhance the interaction with a reactive surface relative to CF3OH.
Finally, we examined the reaction enthalpy of some other processes that could lead to carbonyl compounds indirectly, e.g. by formation of an –C˙FOH radical moiety that can react with O2 to form –CFO + HO2. However, the available co-reactants NO, NO2, FNO and OH do not afford accessible pathways for CF3OH forming ˙CF2OH, with endoergicities exceeding 175 kJ mol−1 in all cases. Note that such reactions would also not readily yield HF, observed as co-product in the reactions of fluorinated esters.
3.9 Persistence of the products of PEVE photo-oxidation in the atmosphere
As for other fluorinated VOCs the atmospheric photo-oxidation products of PEVE are fluorinated carbonyl compounds. Unlike their non-fluorinated analogues, fluorinated carbonyl compounds react too slowly with OH radicals for this reaction to constitute a significant atmospheric sink and their lifetime is limited by uptake on water droplets followed by hydrolysis.35Glyoxal, the non-fluorinated analogue of FC(O)C(O)F, has a very short atmospheric lifetime with respect to photolysis.36 In contrast, the first electronic excitation in FC(O)C(O)F appears at 334 nm and is very weak.16 Photolysis does therefore not constitute a significant sink of FC(O)C(O)F in the atmosphere.
To further investigate the fate of the C2F5OCFO, CF2O and FC(O)C(O)F products with respect to loss to aqueous atmospheric systems, an experiment was performed in which, following PEVE oxidation, the contents of the photochemical reactor was passed (using a chemically inert membrane pump at a flow of 5 SLM) through a wash bottle (volume = 300 cm3) containing distilled water. The gas flow did not bubble through the volume of the water, but was directed via a glass tube (velocity 4 m s−1 in the tube) towards its surface (area 50 cm2). The residence time of the gas mixture in the head-space of the wash-bottle was ≈2.4 s and the flow/geometry induced turbulent flow conditions in the head-space should ensure efficient gas–liquid contact.
Infrared spectra were recorded at 10 min intervals directly after the initial, gas-phase degradation of PEVE, then over a 60 minute period in which the contents of the cell were circulated through the membrane pump without exposure to water and then a further sixty minutes in which the water surface was available for reaction. During the first hour (no interaction with water), no significant loss of any products was observed, indicating no significant losses in the membrane pump. In contrast, the concentration of all three products decreased significantly upon contact with the water surface. Whereas CF2O was almost completely removed after ≈240 s (decay constant 6 × 10−4 s−1), the decay of C2F5OCFO (4 × 10−5 s−1) was an order of magnitude slower. FC(O)C(O)F decayed on a similar time scale to CF2O, but as the only absorptions not overlapping with the saturated water lines are very weak, we were unable to determine a decay constant.
The lifetime with respect to hydrolysis of trace gases in contact with aqueous surfaces is determined by their Henry's law solubility (H) and hydrolysis rate coefficient (kH2O) as well as their rate of transport to the surface and diffusion in the bulk. George et al.37 studied the uptake of CF2O by aqueous surfaces and report H(kH2O)½ = 350 M atm−1 s−1/2 at 273 K. A substantially lower values (at 278 K) of 4.3 M atm−1 s−1/2 was reported by de Bruyn et al.38 Subsequently, Kanakidou et al.39 modelled the atmospheric fate of CF2O based on the data of George et al.37 and reported a lifetime with respect to in-cloud losses of the order of one week, allowing for a considerable uncertainty in the Henry law coefficients and hydrolysis rates. Given that the rate of turbulent transport to the water surface is expected to be identical for all the trace gases we can reasonably assume that the similar removal rates of FC(O)C(O)F and CF2O are due to similar values for H(kH2O)½ and that they have a similar timescales for removal to aqueous surfaces in the atmosphere. In contrast, the much lower rate of removal of C2F5OCFO compared to CF2O implies a large (factor of ≈10) reduction in the H(kH2O)½ term and thus potentially longer atmospheric lifetime.
The hydrolysis of FC(O)C(O)F is likely to lead to formation of HF and CO, whereas the potential hydrolysis products of C2F5OCFO are HF, CO and CF3C(O)OH. As both HF and CF3C(O)OH are highly soluble it is expected that they would remain undetected in the gas-phase but remain in the bulk water sample in our experiments.
Our crude experiments to investigate the relative loss rates of CF2O, C2F5OCFO and FC(O)C(O)F to aqueous surfaces can only give a broad indication of the fate of these trace gases in the atmosphere and dedicated experiments to investigate the atmospheric fate of C2F5OCFO and FC(O)C(O)F with respect to gas-phase and heterogeneous processes are necessary to fully characterise the environmental impact of PEVE.
4 Conclusions
The main products formed in the OH-initiated photo-oxidation of PEVE are C2F5OCFO, FC(O)C(O)F and CF2O with respective yields (at 1 bar and in the presence of NOx) of 0.50 ± 0.07, 0.46 ± 0.07 and 1.50 ± 0.22. Variation of product yields with pressure and concentration of NO enabled a detailed mechanism to be derived, in which product branching is controlled by competition between dissociation and reaction (with O2) of the initially formed reaction complex. In the environment, CF2O formed from PEVE degradation will hydrolyse, forming HF and CO. The fate of C2F5OCFO and FC(O)C(O)F in the atmosphere remains unknown, though heterogeneous processes are likely to be significant. In addition to the mechanistic studies, we also derived a rate coefficient (2.8 ± 0.3 × 10−12 cm3 molecule−1 s−1) which is in good agreement with the values from our previous kinetic study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Merck KGaA for partial financial support of this project and for provision of samples of PEVE. We thank Uwe Parchatka for the loan of the membrane pump. Open Access funding provided by the Max Planck Society.Notes and references
- A. E. Feiring, in Organofluorine Chemistry: Principles and Commercial Applications, ed. R. E. Banks, B. E. Smart and J. C. Tatlow, Plenum Press, New York and London, 1994 Search PubMed.
- T. Hiyama and H. Yamamoto, in Organofluorine Compounds: Chemistry and Applications, ed. H. Yamamoto, Springer-Verlag Berlin, Heidelberg and New York, 2000, pp. 249–263 Search PubMed.
- G. Srinivasulu, A. J. C. Bunkan, D. Amedro and J. N. Crowley, Phys. Chem. Chem. Phys., 2018, 20, 3761–3767 RSC.
- J. N. Crowley, G. Saueressig, P. Bergamaschi, H. Fischer and G. W. Harris, Chem. Phys. Lett., 1999, 303, 268–274 CrossRef CAS.
- R. Atkinson, W. P. L. Carter, A. M. Winer and J. N. Pitts, J. Air Pollut. Control Assoc., 1981, 31, 1090–1092 CrossRef CAS.
- Z. J. Li, Z. N. Tao, V. Naik, D. A. Good, J. C. Hansen, G. R. Jeong, J. S. Francisco, A. K. Jain and D. J. Wuebbles, J. Geophys. Res.: Atmos., 2000, 105, 4019–4029 CrossRef CAS.
- M. Mashino, M. Kawasaki, T. J. Wallington and M. D. Hurley, J. Phys. Chem. A, 2000, 104, 2925–2930 CrossRef CAS.
- E. L. Vareti and P. J. Aymonino, J. Mol. Struct., 1967, 1, 39–54 CrossRef.
- C. Kelly, J. Treacy, H. W. Sidebottom and O. J. Nielsen, Chem. Phys. Lett., 1993, 207, 498–503 CrossRef CAS.
- J. Chen, T. Zhu and H. Niki, J. Phys. Chem., 1992, 96, 6115–6117 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe and T. J. Wallington, Atmos. Chem. Phys., 2008, 8, 4144–4496 CrossRef.
- S. Kraus, DOASIS, DOAS intelligent system, 2006, University of Heidelberg, https://doasis.iup.uni-heidelberg.de/bugtracker/projects/doasis/index.php Search PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2006, 3625–4055 CrossRef CAS.
- K. Tokuhashi, A. Takahashi, M. Kaise, S. Kondo, A. Sekiya and E. Fujimoto, Chem. Phys. Lett., 2000, 325, 189–195 CrossRef CAS.
- D. Amedro, L. Vereecken and J. N. Crowley, Phys. Chem. Chem. Phys., 2015, 17, 18558–18566 RSC.
- W. J. Balfour, PhD thesis, McMaster University, 1967.
- B. J. Finlayson-Pitts, S. K. Hernandez and H. N. Berko, J. Phys. Chem., 1993, 97, 1172–1177 CrossRef CAS.
- L. Vereecken, J. N. Crowley and D. Amedro, Phys. Chem. Chem. Phys., 2015, 17, 28697–28704 RSC.
- A. M. B. Giessing, A. Feilberg, T. E. Møgelberg, J. Sehested, M. Bilde, T. J. Wallington and O. J. Nielsen, J. Phys. Chem., 1996, 100, 6572–6579 CrossRef CAS.
- H. Somnitz and R. Zellner, Phys. Chem. Chem. Phys., 2001, 3, 2352–2364 RSC.
- R. E. Banks and J. C. Tatlow, J. Fluorine Chem., 1986, 33, 227–346 CrossRef.
- K. Seppelt, Angew. Chem., Int. Ed. Engl., 1977, 16, 322–323 CrossRef.
- W. F. Schneider, T. J. Wallington and R. E. Huie, J. Phys. Chem., 1996, 100, 6097–6103 CrossRef CAS.
- T. J. Wallington, M. D. Hurley, W. F. Schneider, J. Sehested and O. J. Nielsen, J. Phys. Chem., 1993, 97, 7606–7611 CrossRef CAS.
- M. T. Nguyen, M. H. Matus, V. T. Ngan, R. Haiges, K. O. Christe and D. A. Dixon, J. Phys. Chem. A, 2008, 112, 1298–1312 CrossRef CAS PubMed.
- R. J. Buszek and J. S. Francisco, J. Phys. Chem. A, 2009, 113, 5333–5337 CrossRef CAS PubMed.
- B. Long, X. F. Tan, Z. W. Long, D. S. Ren and W. J. Zhang, Chin. J. Chem. Phys., 2011, 24, 16–21 CrossRef CAS.
- B. Long, X. F. Tan, D. S. Ren and W. J. Zhang, Chem. Phys. Lett., 2010, 492, 214–219 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- T. H. Dunning, Chem. Phys. Lett., 1970, 7, 423–427 CrossRef CAS.
- T. B. Adler, G. Knizia and H. J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H. J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox and J. A. Pople, Gaussian 09, Revision B.01, Gaussian Inc., Wallington CT, 2010 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Gyorffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O’Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, T. Thorsteinsson and M. Wang, MOLPRO, version 2015.1, a package of ab initio programs, molpro, [online] Available from: http://www.molpro.net., 2015 Search PubMed.
- T. J. Wallington, W. F. Schneider, D. R. Worsnop, O. J. Nielsen, J. Sehested, W. J. Debruyn and J. A. Shorter, Environ. Sci. Technol., 1994, 28, 320A–326A CrossRef CAS PubMed.
- J. Tadić, G. K. Moortgat and K. Wirtz, J. Photochem. Photobiol., A, 2006, 177, 116–124 CrossRef.
- C. George, J. Y. Saison, J. L. Ponche and P. Mirabel, J. Phys. Chem., 1994, 98, 10857–10862 CrossRef CAS.
- W. J. de Bruyn, J. A. Shorter, P. Davidovits, D. R. Worsnop, M. S. Zahniser and C. E. Kolb, Environ. Sci. Technol., 1995, 29, 1179–1185 CrossRef CAS PubMed.
- M. Kanakidou, F. J. Dentener and P. J. Crutzen, J. Geophys. Res.: Atmos., 1995, 100, 18781–18801 CrossRef.
| This journal is © the Owner Societies 2018 |