
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stability of the zwitterionic liquid butyl-methyl-imidazol-2-ylidene borane†
Steffen
Tröger-Müller
 ,
Markus
Antonietti
and
Clemens
Liedel
*
,
Markus
Antonietti
and
Clemens
Liedel
*
Department of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces Research Campus Golm, 14476 Potsdam, Germany. E-mail: Clemens.Liedel@mpikg.mpg.de
First published on 27th March 2018
Abstract
Modification of the C2 position of the standard 1-butyl-3-methyl imidazolium cation by a borohydride group leads to a zwitterionic liquid (ZIL). The resulting imidazol-2-ylidene borane ZIL is liquid at room temperature. Dynamic viscosity as well as thermal and electrochemical stability are investigated. Thermal decomposition follows a similar pathway as in comparable imidazolium ionic liquids. The surprisingly low viscosity and good reductive stability make it a promising candidate for electrochemical applications.
Introduction
Imidazolium ionic liquids (ImILs) are a well understood and versatile class of ionic liquids. As most ILs they are usually of low-flammability, have negligible vapor pressure, and can be easily tuned to their prospective applications by proper design and choice of anion and cation.1 As an additional benefit, they may be obtained without the use of alkylation reactions2 and from biomass,3 which improves their ecological footprint and allows for truly green chemistry. They are applied in various fields ranging from industrial chemistry1 and catalysis4 over biomass treatment5,6 to electrochemistry and energy storage R&D.7 In all those applications, viscosity and stability play a prominent role. Both may be optimized by proper choice of cation and anion and are often based on the special reactivity of the imidazolium ring.Thermal stability is usually limited by cleavage of the N–C bonds of the alkyl chains pendant on the imidazolium ring (thermally induced or by nucleophilic attack of the counterion with increased reactivity at elevated temperatures).8 Attempts to increase the thermal stability of imidazolium ionic liquids consequently include the introduction of non-nucleophilic counterions.
The carbon atom at the 2-position (C2, between both nitrogen atoms) is the most electron deficient atom of the aromatic heterocycle and the main reason for concerns about its chemical stability.9,10 Its electron deficiency makes it prone to nucleophilic attack which may lead to degradation by ring opening.11 It also weakens the bond to its pendant proton, causing it to be rather acidic (with typical pKa values around 20–22).12 Abstraction of the proton eventually leads to the formation of N-heterocyclic carbenes (NHC) which are highly reactive species themselves11 even though their stability is higher when compared to other carbenes.13
Limited electrochemical stability bears on the electron deficiency at the C2 carbon where reduction preferentially occurs. This is also the most probable location of the unpaired electron in the resulting neutral radical,10 which may then undergo further reaction, e.g., with itself to form dimers, disproportionate, or devolve into a carbene.9
In order to increase the (electro-)chemical stability of ImILs, protection of the C2 position with alkyl groups, most notably simple methyl groups, is a viable approach. Resulting C2-methylated ImILs are more stable against Grignard reagents14 or against chemical15 and electrochemical reduction.10 Unfortunately, protection with methyl groups has been shown to increase the viscosity,16,17 which is a big trade-off in the context of possible applications and general manageability of the resulting compounds. The unusually large increase of viscosity may be attributed to a suppression of the mobility of the counterion. Its ability to move around the imidazolium ring via favorable interaction with the C2 position is impeded upon C2 substitution.17 To decrease viscosity and lower the glass transition temperature, bulky, less interacting counterions like bis(trifluoromethanesulfonyl)imide (TFSI) are commonly introduced. In contrast, Gardner et al. have recently published a facile route to substitute the hydrogen at the C2 position with a BH3-group.18,19 The resulting imidazol-2-ylidene borane compounds, so far as they are liquid, show comparatively low viscosities.20
NHC-boranes combine good stability with reducing ability and have even been shown to participate in metal complexes.21 This makes them interesting candidates for electrochemical applications. Excited by the simple route to access those unique compounds, we set out to evaluate the stability of one such imidazol-2-ylidene borane zwitterionic liquid (ZIL). We investigate thermal and electrochemical decomposition and compare it to similar imidazolium ionic liquids with TFSI counterion or methylated C2 position.
Results and discussion
We synthesized 1-butyl-3-methylimidazol-2-ylidene borane (ZIL) from 1-butyl-3-methylimidazolium iodide (IL-H-I) by reaction with sodium borohydride at elevated temperature. Structurally similar imidazolium ionic liquids with different anions and unprotected (IL-H-I and 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (IL-H-TFSI)) or protected C2 position (1-butyl-2,3-dimethylimidazolium iodide (IL-Me-I) and 1-butyl-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide (IL-Me-TFSI)) were synthesized according to the literature for comparison.19,20 In the abbreviations of ILs, the middle part denotes the group at C2 while the last part indicates the counterion. All compounds were dried in vacuum. Chart 1 displays the different ionic liquids. | ||
| Chart 1 Overview of the relevant ionic systems compared in this work. Colors are used in figures throughout this work to refer to the corresponding compounds. | ||
Even though C2 modification of ImILs is known to increase the glass transition temperature TG,14,22 the ZIL stays liquid even after prolonged drying in vacuum. Differential scanning calorimetry (DSC) measurements in Fig. 1 demonstrate that TG is as low as −64.9 °C, similar to TG of IL-H-I (−68.3 °C). The anticipated and observed increase of TG upon C2 methylation (IL-Me-I, which is solid at room temperature, vs.IL-H-I or IL-Me-TFSIvs.IL-H-TFSI) hence does not occur when the C2 position in imidazolium is protected by a BH3 group instead of a methyl group. As expected, ionic liquids with TFSI anion have a lower TG (and in the case of IL-H-TFSI exhibit cold crystallization).23 Substitution of the C2 position with a methyl group in IL-Me-TFSI seems to suppress the cold crystallization.
 | ||
| Fig. 1 Differential scanning calorimetry of the ZIL as well as IL-H-I, IL-H-TFSI, and IL-Me-TFSI (IL-Me-I is a solid). The figure shows the temperature region between −120 °C and 100 °C, revealing glass transitions in all displayed graphs (ZIL: −64.9 °C; IL-H-I: −68.3 °C; IL-H-TFSI: −85.5 °C; IL-Me-TFSI: −75.4 °C) as well as cold crystallization (−41.5 °C, −20.1 °C) and consecutive melting (1.3 °C) in the case of IL-H-TFSI (individual graphs and further details see ESI†). | ||
Since the notion of a liquid zwitterionic compound is somewhat counterintuitive, we will discuss this in more detail. Lewis acid/Lewis base pairs based on N-heterocyclic carbenes and boron compounds have been described in the literature, including discussions on stability, X-ray structures, and spectroscopy.21 These so called NHC-boranes find applications as co-initiators in radical polymerization, reagents, or catalysts. Interestingly, the structures are quite stable, and the borane compounds do not usually hydroborate themselves.21 Concerning the charge, in the resonance structure of 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene borane, the positive charge is not only located at the nitrogen but also partly at the C2 position, and the orbital model resembles an N-heterocyclic carbene with a C–B bond formed from the electron rich C2 σ orbital. As for NHC-boranes of lesser steric demand, Huang et al. have used electrostatic potential analysis to show that the Hirshfield charge on 1,3-dimethylimidazol-2-ylidene borane (which is very similar to the ZIL described here) is (i) indeed separated and (ii) the positive charge (0.456 e) is delocalized over the imidazolium ring, while the negative charge (−0.456 e) is carried by the BH3-group.20 Furthermore, X-ray crystallographic analysis has confirmed the structure of said NHC-borane and revealed a C2–B bond length of 1.596 Å in the same study. Since the ZIL is liquid up to very low temperatures, crystallographic analysis is not readily available (investigations using combined X-ray diffraction studies with zone melting as described by Choudhury et al.24 would in our case require infeasible setups with temperatures in the range of −100 °C inside the spectrometer), but the presence of a C2–B bond can be deduced from 13C nuclear magnetic resonance (NMR) spectra. Fig. 2 shows the proton decoupled 13C NMR spectra of the ZIL and IL-H-I. The spectra show the same peaks with minor differences in chemical shift with one exception. The signal of the C2 carbon is shifted downfield in the ZIL, the intensity is drastically reduced, and it is split into a quartet with approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 intensities, just as expected for C–B bonds in proton decoupled 13C NMR spectra.25 Drawing upon these results, the ZIL qualifies as a room temperature zwitterionic liquid. IL-H-I, IL-H-TFSI, and IL-Me-TFSI are room temperature ionic liquids. We note that the synthesized ZIL is a reducing agent,18 and the presence of reducing agents interferes with Karl–Fischer titration.26 Hence we could not quantitatively determine the water content, which may also contribute to liquidity of ILs.27 Instead, water content was estimated by thermogravimetric analysis (TGA) to be within the range of expectation for ionic liquids. While upon heating the extensively vacuum dried species to 120 °C in the TGA the ZIL loses 1.93% of its weight, within this temperature range several decomposition products appear (see below and ESI†), so the weight loss only partly may denote to water, and the water content is significantly lower than that.
:1:1:1 intensities, just as expected for C–B bonds in proton decoupled 13C NMR spectra.25 Drawing upon these results, the ZIL qualifies as a room temperature zwitterionic liquid. IL-H-I, IL-H-TFSI, and IL-Me-TFSI are room temperature ionic liquids. We note that the synthesized ZIL is a reducing agent,18 and the presence of reducing agents interferes with Karl–Fischer titration.26 Hence we could not quantitatively determine the water content, which may also contribute to liquidity of ILs.27 Instead, water content was estimated by thermogravimetric analysis (TGA) to be within the range of expectation for ionic liquids. While upon heating the extensively vacuum dried species to 120 °C in the TGA the ZIL loses 1.93% of its weight, within this temperature range several decomposition products appear (see below and ESI†), so the weight loss only partly may denote to water, and the water content is significantly lower than that.
 | ||
| Fig. 2 13C nuclear magnetic resonance spectra of the ZIL and IL-H-I. The inset shows the signal of the C2 carbon of the ZIL. | ||
The performance of ionic liquids depends on their viscosity in all fields that rely on mass transport properties, where low viscosity is usually favorable. On a general level a multitude of factors, such as temperature, nature of the anion, cationic structure, or additives, may influence the viscosity of ionic liquids.6,28 As noted above, substitutions at the C2 position may effect a specially strong increase in viscosity. To investigate the influence of the BH3 group, we evaluated the dynamic viscosity at room temperature using shear stress–shear rate curves of the dried samples (see ESI†). Table 1 lists the obtained viscosities in comparison to the viscosity of water.
As expected, comparison of IL-H-I and IL-H-TFSI to their C2 methylated counterparts IL-Me-I and IL-Me-TFSI reveals a drastic increase in viscosity (viscosity increases by 125% in the TFSI based ILs, and IL-Me-I is even a solid). This agrees with the model explored by Izgorodina et al.17 whereby C2 substitution decreases the mobility of the counterion in 1-methyl-3-propylimidazolium iodide.
Hydroboration of the C2 position does not follow this trend. The viscosity of the ZIL is drastically lower than that of IL-H-I and IL-Me-I. It follows a similar trend as exchange of the iodine in IL-H-I and IL-Me-I by TFSI in IL-H-TFSI and IL-Me-TFSI, respectively, which also leads to drastically reduced viscosity. While in the latter case decreased interaction between anion and cation and hence higher mobility is probably responsible for decreased viscosity, this explanation would intuitively result in the opposite behavior, i.e., increasing viscosity, when comparing the ZIL to IL-H-I or IL-Me-I.
Hence, a different physical mechanism must be responsible for the decreased viscosity in the ZIL: in comparison to IL-H-I and IL-Me-I, where the anion is preferentially located above or below the plane of the imidazolium ring,17 the anion is fixed perpendicular to the ring in the ZIL. The 2-dimensionality of the structure creates a fundamentally different situation compared to traditional ionic liquids where anion–cation interactions play a bigger role. Instead of an ionic species, the ZIL may also be described as polar molecule with a lower molecular weight (152.15 g mol−1) than the ionics IL-H-I (266.03 g mol−1), IL-Me-I (280.04 g mol−1), IL-H-TFSI (419.04 g mol−1), and IL-Me-TFSI (433.06 g mol−1). Low molecular weight often also implies high mobility30 and may contribute to the comparably low viscosity observed for the ZIL. We note that low viscosity of the ZIL may not be attributed to high water content since it is much lower than in IL-H-I (with the exact amount unfortunately unknown as described above) while viscosity is also significantly lower. We also note that the viscosity of all samples was recorded using the dried samples and decreases upon exposure to air and uptake of water from the atmosphere (see ESI†). Summarizing this part, hydroboration of the C2 position of an imidazolium IL leads to low viscosity without the introduction of bulky anions.
In order to evaluate the stability of the ZIL in comparison to the standard ionic liquids IL-H-I and IL-H-TFSI as well as their C2 methylated counterparts IL-Me-I and IL-Me-TFSI, thermal and electrochemical decomposition were evaluated by thermogravimetric analysis (TGA) and linear sweep voltammetry (LSV), respectively. Fig. 3 displays the results of TGA measurements. While methylation of the C2 position has only a little effect on the thermal stability, hydroboration of the C2 position clearly leads to decreased thermal stability (onset of decomposition at 202 °C, compared to 284 °C for IL-H-I and 425 °C for IL-H-TFSI). High thermal stability of IL-H-TFSI may be attributed to the almost negligible nucleophilicity of the bulky TFSI anion compared to the far more nucleophilic iodide in IL-H-I.
 | ||
| Fig. 3 Thermogravimetric analysis of the ZIL and the ILs. Data was acquired at a scan rate of 10 K min−1. | ||
In order to understand the reason for the decreased stability, we investigated decomposition products of the ZIL during heating in more detail. We especially focused on three possible decomposition pathways as outlined in Fig. 4. In short, we anticipated (1) dissociation of the carbon–boron bond; (2) the decomposition by ring opening after attack at the C2 position; (3) the cleavage of carbon–nitrogen bonds. With regard to the pathways 2 and 3 we assumed that the borohydride group of the ZIL shows similar behavior as borohydride salts and acts as donor of formally nucleophilic hydrogen.
 | ||
| Fig. 4 The main decomposition pathways of the ZIL investigated by TGA–MS and masses of main fragments: (1) dissociation of the carbon–boron bond. (2) Decomposition by ring opening. (3) Cleavage of the carbon–nitrogen bond. Grey boxes show assumed decomposition mechanisms that could not be verified. | ||
We evaluated the proposed decomposition pathways of the ZIL by TGA coupled with mass spectroscopy (TGA–MS). The mass profile of the TGA–MS curve and ion current graphs that show representative shapes are displayed in Fig. 5. Further ion currents indicating other decomposition products are shown in the ESI.† We note that TGA–MS was performed at significantly decreased heating rate of 2.5 K min−1 (compared to 10 K min−1 in Fig. 3), resulting in different apparent thermal stability.
 | ||
| Fig. 5 Decomposition of the ZIL: some representative examples of TGA–MS analysis recorded at a heating rate of 2.5 K min−1. From the data we obtain two general regions of decomposition. More ion currents are summarized in the ESI.† Left axis of ordinates corresponds to mass loss (black line), right axis of ordinates to the other curves. | ||
Fig. 5 shows two plateaus/peaks in the ion current curves which correspond to two steps of mass loss from TGA–MS measurement, which may indicate two distinct decomposition steps. In the first step, decomposition products from pathway 3-a (cleavage of the carbon–nitrogen bond at the butyl side chain) prominently appear, together with H2 as the main decomposition product. In the second step, decomposition products from pathways 3-a (cleavage of the carbon–nitrogen bond at the methyl side chain) and 1 (cleavage of the carbon–boron bond) appear. TGA–MS data could not confirm decomposition by ring opening (pathway 2). In contrast, occurrence of imidazole (m/z 68) already in the first decomposition step indicates that cleavage of the C–B bond (pathway 1) also happens already at this temperature. The two step detection of mass loss may thus also be explained by different volatility. Recombination of decomposition products also happens, since dimethyl-2-H-imidazole was also detected, as was the original cation 1-butyl-3-methylimidazolium.
Pathway 3 is also the main thermal decomposition mechanism for normal (not zwitterionic) imidazolium ionic liquids.8 In the ZIL, the BH3 group acts as reducing agent and initiates nucleophilic attack,18 leaving saturated butane and methane as well as dealkylated imidazol-2-ylidene boranes as decomposition products. The lower overall thermal stability implies that the nucleophilic attack by BH3 actually is favored compared to conventional counterions.
Two things should be noted on TGA–MS data. (i) From the m/z values it is not possible to distinguish the imidazoles (see Fig. 4, decomposition products 1-c) from their isomeric carbenes. We assume the detected species to be the imidazoles because they are thermodynamically more stable than the carbenes. (ii) The dissociation of the carbon–boron bond does not seem to result in the formation of the 1,3-dialkylated carbene (1-a), or the carbene reacts faster than it can be detected. Moreover, removal of the BH3 group seems to result in formation of the ionic liquid cation (1-b).
Besides thermal stability, we also assessed the electrochemical stability of the ZIL by linear sweep voltammetry on 316 stainless steel working electrodes vs. lithium counter electrodes and compared it to IL-H-I, IL-H-TFSI, and IL-Me-TFSI. The results are shown in Fig. 6. In this experiment, we define stability for a current flow only below 0.15 mA (dashed lines in Fig. 6). With this limit, we found the ZIL to be quite stable with a reductive stability of −0.14 V vs. Li+/Li and an oxidative stability of 3.72 V vs. Li+/Li. The resulting electrochemical window is 3.86 V.
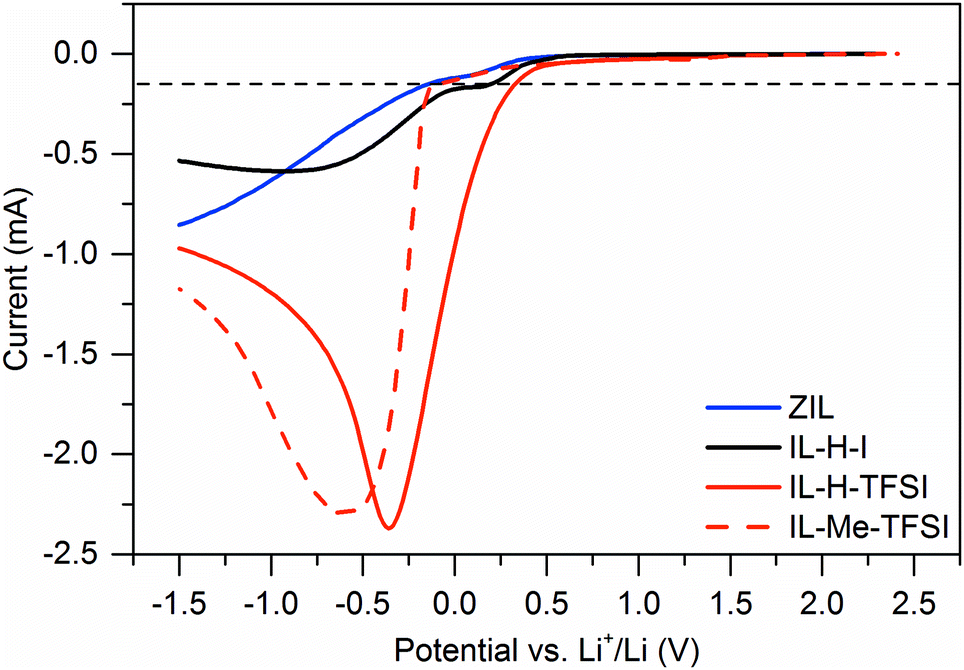 | ||
| Fig. 6 Linear sweep voltammograms of the ZIL, IL-H-I, IL-H-TFSI, and IL-Me-TFSI. Curves were recorded at 5 mV s−1 on 316 stainless steel electrodes. | ||
Concerning reductive stability, it is known to be enhanced upon protection of the C2 position in ImILs by methyl groups10 (here: −0.08 V vs. Li+/Li in the case of IL-Me-TFSI compared to 0.33 V vs. Li+/Li for IL-H-TFSI), as shown also in Fig. 6. Protection by borohydride groups results in roughly the same (even slightly better) enhancement in reductive stability. Stability in both cases (ZIL and IL-Me-TFSI) is significantly better than in the unprotected comparable ILs IL-H-I (0.20 V vs. Li+/Li) and IL-H-TFSI, indicating that C2 protection with a borohydride group has a similar effect on stability as using ILs with methylated C2 positions. The shape of the LSV curve also reveals that the electrochemical decomposition behavior of the ZIL is similar to decomposition of the iodide containing IL-H-I and differs from the TFSI containing IL-H-TFSI and IL-Me-TFSI, where TFSI decomposition may appear.
Oxidative stability of the ZIL is higher than the stability limit of IL-H-I. In the latter, decomposition is attributed to the onset of iodide oxidation (here: 3.19 V vs. Li+/Li). Still, as expected the ZIL is a reducing agent, and the stabilities of both TFSI based ionic liquids are higher (5.66 V and 5.48 V vs. Li+/Li for IL-Me-TFSI and IL-H-TFSI, respectively). Finally, it is noteworthy that water content of the ZIL or the ILs does not seem to negatively influence the electrochemical behavior (current flow below 0.15 mA may result from water reduction).
Experimental
Materials
1-Methylimidazol (≥99%, purified by redistillation) and dichloromethane (99.9% by GC) were obtained from Sigma-Aldrich, 1-iodobutane (99%) and chloroform-d (99.8%D) were obtained from Aldrich, toluene (99.7% by GC) was obtained from Honeywell, sodium borohydride (99%) was obtained from Acros, and lithium bis(trifluoromethanesulfonyl)imide (99%) was obtained from Io-Li-Tec.Methods
Synthesis of ionic liquids
Conclusions
With significant enhancement of the electrochemical reductive stability similar to C2-methylation and decrease of the viscosity similar to anion exchange by a bulky anion, zwitterionic liquids with borohydride groups may be an interesting alternative to C2-methylated and TFSI-containing imidazolium ILs. While physical properties are similar, they have several distinct advantages. For example the molecular weight is much lower (152 g mol−1 for 1-butyl-3-methylimidazol-2-ylidene borane, vs. 433 g mol−1 for 1-butyl-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide), making high loading with salts in an electrolyte more likely. The zwitterionic liquid is also far more environmentally benign (not fluorinated), much cheaper, and easy to synthesize. Thermal and oxidative stabilities are lower, owing to the nucleophilicity of the BH3 group, but sufficient at or around room temperature or moderately increased temperature. Hence, in applications which do not require heating to high temperatures or exposure to strong oxidizers, the presented ZIL may be a cheaper and more sustainable alternative to some of the best performing imidazolium ILs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Wuqi Guo for preliminary experiments. We further thank Olaf Niemeyer for fruitful support with NMR measurements, Alberto Sanz de León for rheometer support, Antje Völkel for TGA and TGA–MS, Jessica Brandt and Ursula Lubahn for DSC, as well as Eva Settels and Sylvia Fürstenberg for high resolution MS measurements. Financial support by the Max Planck Society is gratefully acknowledged. Open Access funding provided by the Max Planck Society.Notes and references
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- D. Depuydt, A. van den Bossche, W. Dehaen and K. Binnemans, RSC Adv., 2016, 6, 8848 RSC; S. Tröger-Müller, J. Brandt, M. Antonietti and C. Liedel, Chem. – Eur. J., 2017, 23, 11810–11817 CrossRef CAS PubMed.
- S. Kirchhecker, M. Antonietti and D. Esposito, Green Chem., 2014, 16, 3705 RSC; D. Esposito, S. Kirchhecker and M. Antonietti, Chem. – Eur. J., 2013, 19, 15097 CrossRef CAS PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS PubMed; P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef CAS PubMed; A. Brandt, J. Gräsvik, J. P. Hallett and T. Welton, Green Chem., 2013, 15, 550 RSC.
- K. C. Badgujar and B. M. Bhanage, Bioresour. Technol., 2015, 178, 2 CrossRef CAS PubMed.
- S. Song, M. Kotobuki, F. Zheng, Q. Li, C. Xu, Y. Wang, W. D. Z. Li, N. Hu and L. Lu, J. Electrochem. Soc., 2017, 164, A741 CrossRef CAS; K. A. Francis, C.-W. Liew, S. Ramesh and K. Ramesh, Ionics, 2016, 22, 919 CrossRef; F. Lu, X. Gao, A. Wu, N. Sun, L. Shi and L. Zheng, J. Phys. Chem. C, 2017, 121, 17756 Search PubMed; J. F. Vélez, L. V. Álvarez, C. del Río, B. Herradón, E. Mann and E. Morales, Electrochim. Acta, 2017, 241, 517 CrossRef.
- H. Ohtani, S. Ishimura and M. Kumai, Anal. Sci., 2008, 24, 1335 CrossRef CAS PubMed.
- N. de Vos, C. Maton and C. V. Stevens, ChemElectroChem, 2014, 1, 1258 CrossRef CAS.
- G. H. Lane, Electrochim. Acta, 2012, 83, 513 CrossRef CAS.
- B. Wang, L. Qin, T. Mu, Z. Xue and G. Gao, Chem. Rev., 2017, 117, 7113 CrossRef CAS PubMed.
- Y. Chu, H. Deng and J.-P. Cheng, J. Org. Chem., 2007, 72, 7790 CrossRef CAS PubMed.
- B. Gorodetsky, T. Ramnial, N. R. Branda and J. A. C. Clyburne, Chem. Commun., 2004, 1972 RSC; A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- T. Kakibe, N. Yoshimoto, M. Egashira and M. Morita, Electrochem. Commun., 2010, 12, 1630 CrossRef CAS.
- A. Bakac, V. Butkovic, J. H. Espenson, J. Lovric and M. Orhanovic, Inorg. Chem., 1989, 28, 4323 CrossRef CAS.
- V. Strehmel, A. Laschewsky, H. Wetzel and E. Görnitz, Macromolecules, 2006, 39, 923 CrossRef CAS.
- E. I. Izgorodina, R. Maganti, V. Armel, P. M. Dean, J. M. Pringle, K. R. Seddon and D. R. MacFarlane, J. Phys. Chem. B, 2011, 115, 14688 CrossRef CAS PubMed.
- S. Gardner, T. Kawamoto and D. P. Curran, Org. Synth., 2015, 92, 342 CrossRef CAS.
- S. Gardner, T. Kawamoto and D. P. Curran, J. Org. Chem., 2015, 80, 9794 CrossRef CAS PubMed.
- S. Huang, X. Qi, T. Liu, K. Wang, W. Zhang, J. Li and Q. Zhang, Chem. – Eur. J., 2016, 22, 10187 CrossRef CAS PubMed.
- D. P. Curran, A. Solovyev, M. Makhlouf Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294 CrossRef CAS PubMed.
- A. S. M. C. Rodrigues, C. F. R. A. C. Lima, J. A. P. Coutinho and L. M. N. B. F. Santos, Phys. Chem. Chem. Phys., 2017, 19, 5326 RSC.
- E. Gómez, N. Calvar, Á. Domínguez and E. A. Macedo, Ind. Eng. Chem. Res., 2013, 52, 2103 CrossRef.
- A. R. Choudhury, N. Winterton, A. Steiner, A. I. Cooper and K. A. Johnson, J. Am. Chem. Soc., 2005, 127, 16792 CrossRef CAS PubMed.
- B. Wrackmeyer, Prog. Nucl. Magn. Reson. Spectrosc., 1979, 12, 227 CrossRef CAS.
- E. Scholz, Karl Fischer Titration, Springer, Berlin, Heidelberg, 1984 Search PubMed.
- S. Fendt, S. Padmanabhan, H. W. Blanch and J. M. Prausnitz, J. Chem. Eng. Data, 2011, 56, 31 CrossRef CAS; J. Jacquemin, P. Husson, A. A. H. Padua and V. Majer, Green Chem., 2006, 8, 172 RSC.
- S. Tang, G. A. Baker and H. Zhao, Chem. Soc. Rev., 2012, 41, 4030 RSC.
- J. C. Crittenden, R. R. Trussell, D. W. Hand, K. J. Howe and G. Tchobanoglous, MWH's Water Treatment, John Wiley & Sons, Inc, Hoboken, 2012 Search PubMed.
- R. S. Lenk, ed., Polymer Rheology, Springer, Netherlands, Dordrecht, 1978 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, DSC, shear stress–shear rate curves, TGA–MS curves. See DOI: 10.1039/c8cp00311d |
| ‡ Since ILs consist of ion pairs, M+ and M− refer to the molecular ion peaks of cation and anion, respectively. If not denoted otherwise, no additional ionization occurs. |
| § This procedure was used so that residual byproduct, which collected in a sticky lower phase, would adhere to the glass wall of the Erlenmeyer flask instead of collecting in the product flask. |
| This journal is © the Owner Societies 2018 |