
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design and properties of functional zwitterions derived from ionic liquids
Hiroyuki
Ohno
 *a,
Masahiro
Yoshizawa-Fujita
b and
Yuki
Kohno
c
*a,
Masahiro
Yoshizawa-Fujita
b and
Yuki
Kohno
c
aDepartment of Biotechnology, Tokyo University of Agriculture and Technology, Naka-cho, Koganei 184-8588, Tokyo, Japan. E-mail: ohnoh@cc.tuat.ac.jp
bDepartment of Materials and Life Sciences, Sophia University, 7-1 Kioi-cho, Chiyoda-ku, Tokyo 102-8554, Japan
cNational Institute of Advanced Industrial Science and Technology (AIST), 4-2-1, Nigatake, Miyagino-ku, Sendai 983-8551, Japan
First published on 22nd March 2018
Abstract
A zwitterion, an ion pair where cation and anion are covalently tethered, is known to be a type of salt. These ions have not been recognised as interesting, but they are physicochemically unique and fascinating ions. In the present review, some functional zwitterions derived from ionic liquids are mentioned to emphasise the usefulness of the tethering of the component cations and anions of ionic liquids. Basic properties, advantages and disadvantages after the functional design of zwitterions, and some applications are summarised.
 Hiroyuki Ohno | Hiroyuki Ohno received his PhD degree in 1981 from Waseda University, Tokyo, Japan. After working at Waseda University and Case Western Reserve University (USA), he moved to Tokyo University of Agriculture and Technology (TUAT) as an associate professor in 1988. He was promoted to professor in 1997. Then, he served as a director of the university library, vice dean, and dean for his university. He is now the president of TUAT. FRSC since 2008. His recent research activities are concentrated on the science of ionic liquids, especially the design of functional ionic liquids. He has published more than 660 papers, reviews, and book chapters. |
 Masahiro Yoshizawa-Fujita | Masahiro Yoshizawa-Fujita was born in Hitachi, Japan in 1973. He received his PhD degree (2002) from Tokyo University of Agriculture and Technology (Japan). During his PhD studies, he received a Research Fellowship for Young Scientists at the Japan Society for the Promotion of Science (JSPS). He spent two years as a postdoctoral research fellow at Monash University (Australia). He moved to Sophia University as an Assistant Professor in 2006. He was promoted to Associate Professor in 2011. His recent research activities are concerned with the design of ionic liquids, especially for battery research and biomass processing. |
 Yuki Kohno | Yuki Kohno received his PhD degree from Tokyo University of Agriculture and Technology (TUAT), Japan in 2012. After working as a post-doctoral research fellow in TUAT, he joined University of Colorado Boulder (CU), USA in 2013. During this period, he received a fellowship from the Japan Society for the Promotion of Science (JSPS Postdoctoral Fellowships for Research Abroad). He moved to the National Institute of Advanced Industrial Science and Technology (AIST) as a researcher in 2016. His current research focus is on the design of ionic liquid-based reaction–separation systems. |
1. Introduction
Textbooks of physical chemistry tell us that salts such as sodium chloride (NaCl) are solid at room temperature. Their melting points (Tm) are quite high but this is the function of ion–ion interaction forces. The Tm of NaCl is about 800 °C and with increasing the radii of the component ions, the Tm decreases. For example, Tm of cesium chloride (CsCl) is 645 °C, which further decreases when organic ions are used instead of alkali metal cations. The Tm values of tetrapropylammonium chloride and 1-ethyl-3-methylimidazolium chloride are 241 °C and 87 °C, respectively. This tendency can be comprehended as the dawn of low-temperature melting salts known as “ionic liquids”. There are increasing numbers of reports on these unique materials and many applications have been proposed in quite diverse areas. With the progress of ionic liquids (ILs), there have been several demands to improve on the drawbacks of ILs. The science of functional zwitterions (ZIs) originated from this viewpoint and is growing wildly. The present review mentions the initial stages of functional ZIs arising from IL studies, and their development targeted toward some applications. Our aim is to summarise our fundamental studies based on ZIs derived from ILs. Unlike well-known and general zwitterionic compounds such as amino acids, the IL-derived ZIs are quite a new class of materials that have unique properties reflecting those of ILs. This paper contains studies of ZIs prepared from ion pairs of ILs, and the interesting properties of the ZIs are described in terms of ion conductive materials, liquid–liquid biphasic systems, and model interfaces of cell membranes.2. Ionic liquids and zwitterions
ILs, salts with very low melting points empirically below 100 °C, are quite interesting liquid-state materials composed only of ions. Since there are many reviews and books on the ILs,1–5 we do not want to go into the details of the physicochemical properties of ILs. Here, we briefly compare the ILs and functional ZIs. The word “zwitterion” means an ion pair where the cation and anion are covalently tethered. It is easy to compare ordinary ion pairs (especially ILs) and ZIs as shown in Fig. 1. In ordinary ILs, cations and anions are independently mobile under the influence of not only electrostatic forces but also hydrogen bonding and dispersion forces from other ions. There are many chances to exchange partners of ions in such ILs. However, ZIs keep their ion pairs in spite of many interactions from other ions, since the ion pair has been tethered covalently. This is a big difference between ZIs and ILs, especially mixed ILs. ZIs are not new and have been known from a long time ago; for example, amino acids are typical ZIs at moderate pH in an aqueous medium. As shown in Fig. 1, functional ZIs derived from ILs are expected to show properties of ILs. The initial motivation to develop ZIs was from the need to develop ILs as solvents for electrochemical devices. For example, only lithium ions should be transported in the electrolyte solution for lithium-ion batteries. In such a case, for the efficient transportation of target ions, other component ions should not be transported.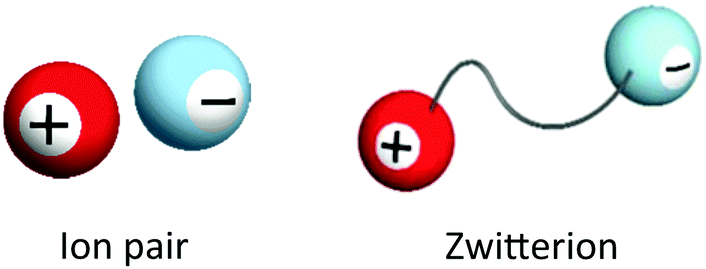 | ||
| Fig. 1 An ordinary ion pair and ZI. | ||
Obviously, all ions are mobile and migrate along with a potential gradient in electrochemical cells. To suppress the migration of component ions under the potential gradient, there are two prospective candidates for satisfying the requirements, namely, ZIs and polymers. Since the net charge of ZIs is neutral, they do not migrate, even under a potential gradient.
Charged polymers, especially both cations and anions are covalently bound to the chains, are quite large and their diffusion is extremely small since there are no free counterions. There are a few reviews on polymerised ILs,5–8 and one can find many interesting scientific areas surrounding the polymerised ILs.
In comparing the basic properties of functional ZIs with ordinary ILs, as shown in Table 1, there are some similar properties as well as completely different ones. The most distinct difference is seen in the bulk ionic conductivity. It is obvious that ZIs show small DC conductivity, but they show reasonably high AC conductivity. A detailed discussion will be given later, but it should be noted here that the use of ZIs is effective for suppressing long-range ion migration when ILs are used as electrolyte solutions for DC-driven devices.
| Properties | ILs | ZIs |
|---|---|---|
| Melting point | Low | High |
| Decomposition temperature | High to moderate | High to moderate |
| Vapour pressure | Quite low | Quite low |
| Ionic conductivity | High | Quite low |
| Density | Low to high | Low to high |
3. Zwitterions as electrolyte materials
3.1. The design of non-mobile ions under the potential gradient
Since ILs have high ion densities and high mobility of their component ions, they also have a high ionic conductivity. Furthermore, ILs can dissolve a variety of salts and have been actively investigated as new electrolyte materials.5,9 Although ILs have a number of physical properties superior to organic solvents, they are not suitable solvents for transporting the ions produced by the dissociation of added salts like lithium salts and sodium salts. Since the IL itself, which is used as a solvent, is composed of ions, their component ions also migrate along with the potential gradient. Generally, the number of component ions of IL used as a solvent is much larger than that of added ions; i.e., when it is desired to transport only specific ions, for example, lithium cations and protons as the electrolyte material of rechargeable batteries and fuel cells, respectively, a low target-ion transference number is a serious problem. To achieve a high target-ion transference number, ZIs have been proposed as new electrolyte materials to critically suppress the migration of the component ions of ILs.10,11 ZI, in which both cation and anion are tethered by a covalent bond, is considered to provide a unique environment to suppress the migration of the component ions under a potential gradient as shown in Fig. 2. In fact, the ionic conductivity of ZIs is very low, which is below 10−6 S cm−1, even at 200 °C,11 because the independent migration of cations and anions is prohibited; i.e., ZIs have no carrier ions, despite providing a high ion density, and provide a unique ionic environment to transport only the target ions.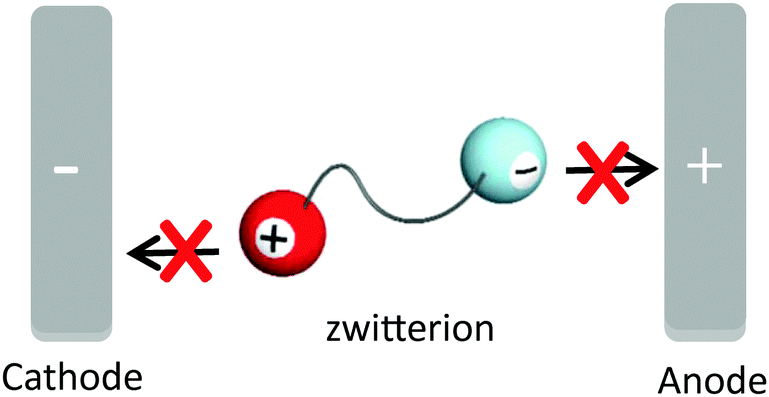 | ||
| Fig. 2 A design concept of non-mobile ions under the potential gradient. | ||
3.2. Design of target ion conductors
Most electrochemical devices require target carrier ions such as lithium cations, protons, sodium cations, or iodide anions for the construction of the corresponding electrochemical cells. In other words, a matrix that predominantly transports only these target ions is required for electrochemical devices. ZIs, where both cation and anion are tethered, are candidates for ion conductive matrices for target ion transport. The chemical structure of ZIs mentioned in this section is summarised in Fig. 3.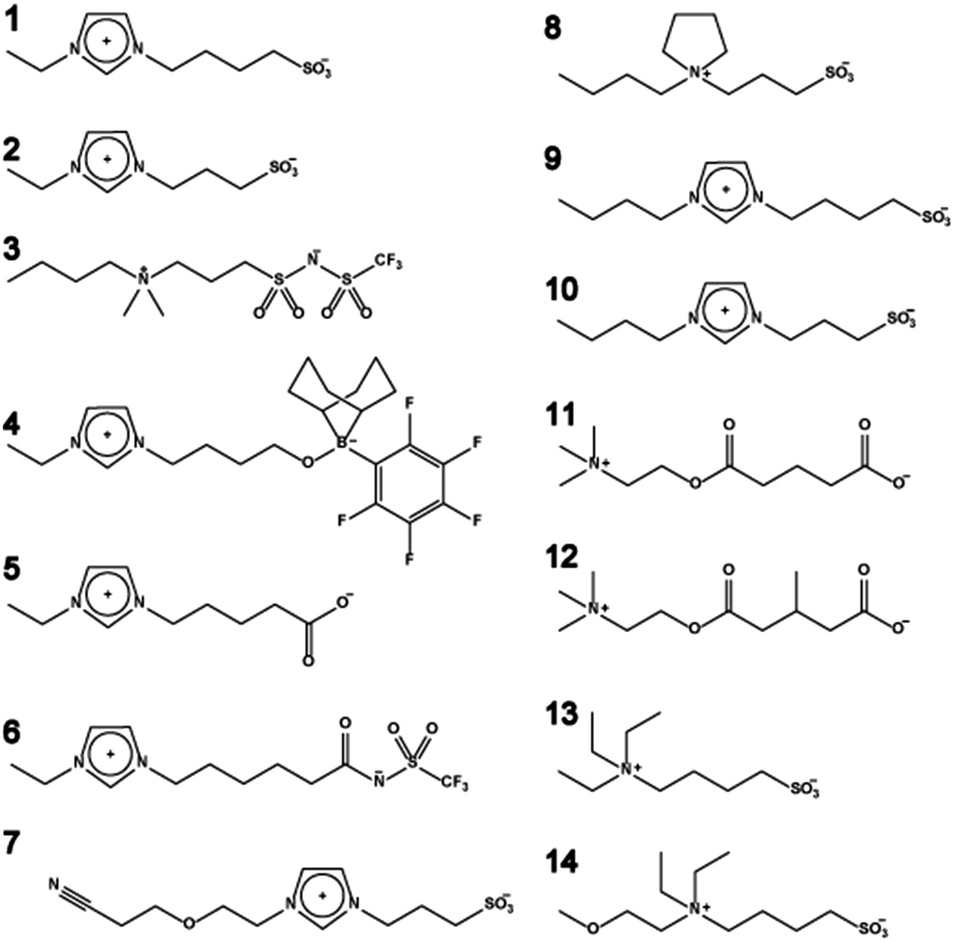 | ||
| Fig. 3 Chemical structure of typical functional ZIs. | ||
ZIs have been studied as novel electrolyte materials for lithium-ion batteries12–14 and fuel cells.15 As a result of the addition of ZI to polymer electrolytes having lithium ions, lithium-ion transport is promoted.16,17 Moreover, as a result of the addition of ZI to IL/lithium-salt complexes, the coulombic efficiency for the dissolution/precipitation of lithium was significantly improved.18 These results reveal that ZIs are excellent candidates for use as electrolyte materials. It is also an advantage of electrolyte materials that ZIs can dissolve many kinds of inorganic salts and dissociate them into ions.11,19
Fig. 4 exhibits the Arrhenius plots of ionic conductivity for neat 1 and its lithium-salt complexes. A curved relationship in the Arrhenius plots was observed for all the lithium-salt complexes as shown in Fig. 4. In general, an ion conductive process is expressed by the Vogel–Fulcher–Tamman (VFT) eqn (1), which empirically explains the temperature dependence of viscosity in amorphous materials.
σi = σ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−B/(T − T0)] exp[−B/(T − T0)] | (1) |
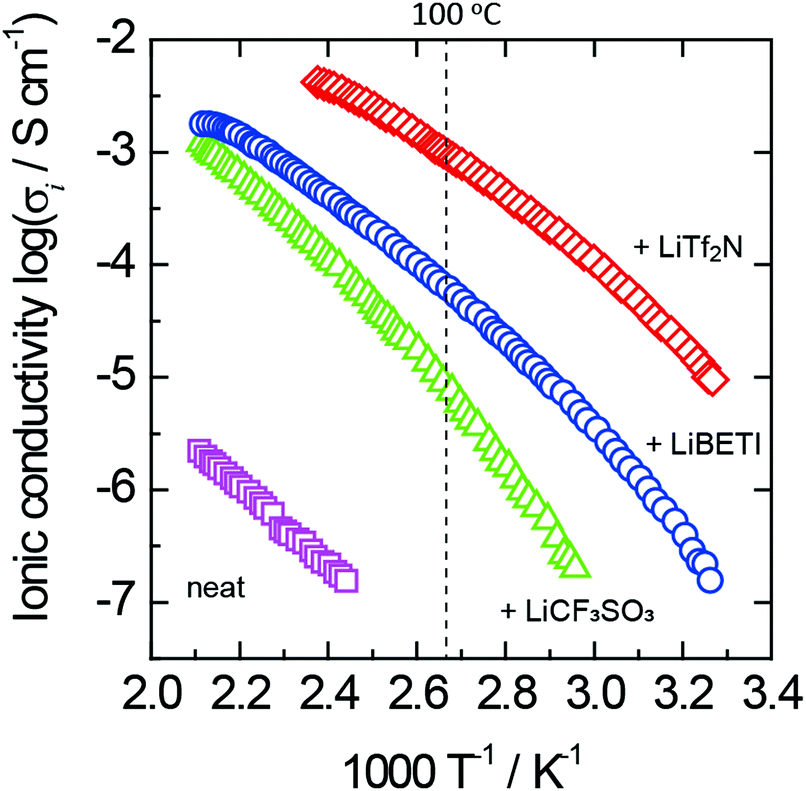 | ||
| Fig. 4 Arrhenius plots of ionic conductivity for neat 1 and its lithium-salt complexes. The lithium-salt content is 50 mol% for all the complexes. | ||
Fig. 5 exhibits the relationship between the ionic conductivity at 50 °C and Tg for various ZIs mixed with an equimolar amount of lithium salts. The ionic conductivity of the resulting ZI/lithium-salt mixtures increased with the decrease in Tg. The Tg was therefore re-confirmed to be closely related to the ionic conductivity of ZIs, similar to other ion conductive materials.21 Of course, the absolute value of the ionic conductivity is affected by other factors such as ion concentration, the degree of dissociation of the salts existing in the matrix, the size of the component ions, and so on.22 In the case of ZIs, added salt species significantly affected the physicochemical properties of the complexes. As mentioned above, it is important to select suitable ion pairs between ZI and added salts. When some of these ions formed an IL-like environment, such as the imidazolium cationic part of the ZI-1 and Tf2N anion, this ion pair is typically known to be an excellent IL with low Tg. The newly formed ion pairs with low Tg after mixing ZI and added salt is the reason for high ionic conductivity.
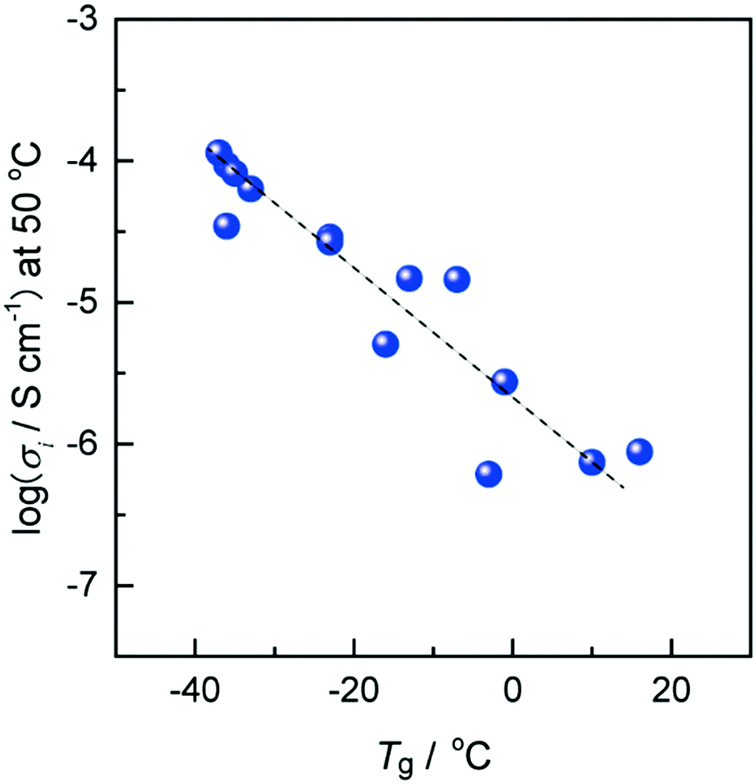 | ||
| Fig. 5 The relationship between ionic conductivity at 50 °C and Tg for ZI/lithium-salt mixtures. | ||
Since ZI/salt mixtures show characteristic ion conduction as mentioned above, these are expected to be used as functional ion conductors. In order to apply these ZI/salt mixtures as practical electrolyte materials for lithium batteries, the lithium-transference number (tLi+) is quite an important parameter. The tLi+ of the complexes of ZI/salts, i.e., the trifluoromethyl-sulfonylamide-type ZI 323 or the organoborate-type ZI 424 with LiTf2N, was determined by electrochemical techniques to be over 0.5, which was much higher than that of common organic solvent/salt mixtures. The IL moiety was formed by the combination of the onium cation and Tf2N anion, and the migration of remaining lithium cations would occur in the IL moiety. On the other hand, the tLi+ of the imidazolium cation-based ZI having the carboxylate anionic tail as the counter anion, 5, was only 0.14,14 which was quite a low value as compared with those of 3 and 4. The strong binding force between the lithium ion and sulfonate group was confirmed by the existence of highly mobile Tf2N anions and less mobile ZI/lithium-ion complexes through the study with PFG-NMR measurements.25 The tLi+ was strongly affected by the anion structure of the ZIs. From these results, when ZIs were used as ion conductive matrices, the highly dissociable anion (weak Lewis-basic anion like trifluoromethylsulfonylamide as a counter anion) was necessary to increase the cation transference number, as well as lower the Tg of ZIs themselves.
New types of polymer gel electrolytes were prepared by mixing various polymers and ZIs. Since ZIs are ionic compounds but do not migrate along with the potential gradient, they were expected to work as non-volatile polar solvents for guest salts. ZI/lithium-salt complexes were mixed with poly(vinylidene fluoride-co-hexafluoropropylene) (PVdF-HFP),26,27 poly(methyl acrylate) (PMA),28 poly(lithium acrylate) (PLiAC),28 or nitrile rubber (poly(acrylonitrile-co-1,4-butadiene) (NBR)).29 Thermal analysis revealed that these polymer gel electrolytes were thermally stable over 300 °C. Thermally stable polymer gel electrolytes composed of polyimide and ZI/IL complexes were also prepared by in situ polycondensation.30 PLiAC and NBR-based gel electrolytes containing 6/LiTf2N exhibited the ionic conductivity of about 10−5 S cm−1 at 50 °C, with tLi+ of 0.44 and 0.32, respectively. The PLiAC system exhibited higher tLi+ due to the existence of non-mobile anions fixed on the polymer chains. Tiyaboonchaiya et al. also reported that the addition of ZI improved lithium-ion transport properties in a polyanion, poly(lithium-2-acrylamide-2-methyl-1-propanesulfonate) (PAMPSLi).16
In order to investigate the effect of ZIs on the electrochemical properties of poly(ethylene glycol)dimethyl ether (PEGDME)-type electrolytes, known amounts of ZIs were added to the PEGDME electrolytes.31,32 The electrochemical stability of PEGDME/LiTf2N with ZIs was over 5 V vs. Li/Li+, which was better than that of PEGDME/LiTf2N without ZIs (below 4.5 V vs. Li/Li+), even at low ZI content. The cycle stability of PEGDME/LiTf2N with 7 was better than the electrolyte without the ZI within the cut-off voltage range of 3.0–4.6 V for the charge–discharge tests of Li/LiCoO2 cells.32 AC impedance spectra indicated that the increase in interface resistance between the cathode and electrolyte was suppressed in the presence of 7.
ILs containing ZIs have also been studied as the electrolytes for lithium-metal and lithium-ion batteries. Byrne et al. reported that ionic conductivity was improved by the addition of ZIs and inorganic nanofillers.17 The addition of ZIs to ILs led to the improvement of the diffusion coefficient of lithium ions, the formation of a thinner solid electrolyte interface (SEI), and the improvement of coulombic efficiencies for the lithium plating–striping reaction.18,33 The effect of the addition of pyrrolidinium ZI, 8, in an IL electrolyte on the charge–discharge tests of Li/LiCoO2 and graphite/Li cells has been investigated.34 The Li/LiCoO2 cells containing 8 exhibited stable coulombic efficiency and much higher discharge capacities in the cut-off voltage range of 3.0–4.6 V, compared to the cells without ZI. The increase in the interface resistance between cathode and electrolyte was suppressed the same as PEGDME electrolytes. Moreover, the intercalation–disintercalation reaction was stable in the graphite/Li cells containing 8. These results indicated that ZIs would be useful electrolytes for improving the performance of lithium-metal and lithium-ion batteries, especially at high cut-off voltages.
ZI 10 was mixed with given amounts of 1,1,1-trifluoro-N-(trifluoromethylsulfonyl)methanesulfoneamide (HTf2N), and the proton conductivity was evaluated.40,41 ZI 10 and HTf2N were solids at room temperature, but the complexes formed were viscous liquids at room temperature. When the concentration of HTf2N was in the range of 30 and 80 mol%, their complexes showed only Tg, which decreased with increasing the acid concentration, and the lowest value of −55 °C was observed at 50 mol%. The complexes showed a constant value of about −55 °C in the range above 50 mol%. The conductivity of complexes of 10/HTf2N increased with increasing the acid concentrations. When the acid concentration was 50 mol%, the complex exhibited the highest value of about 10−4 S cm−1 at 25 °C. At above 50 mol%, the conductivity did not change and showed a constant value of about 10−4 S cm−1 at 25 °C. There is an obvious correlation between Tg and the conductivity, the same as for the complexes of ZIs and lithium salts.
PFG-NMR measurements were carried out to evaluate the self-diffusion coefficients of the component ions.41 The acid concentration dependence on self-diffusion coefficients was different from that between Tg and conductivity. For HTf2N fractions above 50 mol%, the self-diffusion coefficient of all component ions increased. When the acid concentration was 50 mol%, protons, derived from HTf2N were found to be the fastest diffusion species. These results indicate that ZIs cannot induce the dissociation of excess HTf2N, and the HTf2N behaves as a molecular acid in the complexes. Superior proton transport properties were observed when ZI 10 was equimolarly mixed with HTf2N.
3.3. Liquid zwitterions
The Tm values of ZIs are summarised in Table 2.11,33,43–45 The Tm values of the ZIs synthesised to date have been mostly above 100 °C. The chemical structure of the ions constituting ZIs was substantially the same as that of ILs, but the Tm was considerably different. Since both cations and anions are covalently tethered, the degrees of freedom of the ions are greatly restricted. Crystallisation rapidly occurs due to the strong electrostatic attraction between the ZIs themselves. This would be responsible for higher Tm. There are three notable points for the molecular design in order to lower the Tm of ZIs, namely, the cation structure, anion structure, and spacer structure connecting the cation and anion.
To analyse the cation effect, imidazolium cation, ammonium cation, and pyridinium cation have been used as the cationic unit of ZIs. When comparing the Tm of these ZIs having sulfonate anions, imidazolium cations exhibited relatively low Tm among those ZIs having three different types of cationic structure. This is the same tendency for typical ILs. The thermal decomposition was accompanied by melting for many of the ZIs consisting of ammonium cations (these ZIs melted above 300 °C).43 However, the introduction of an ether bond and a long alkyl chain on the ammonium cation part led to Tm below 200 °C.44,46 For example, the Tm of 13 with decomposition was 293.5 °C, whereas the Tm of 14 with almost the same structure as 13 was lowered to 183 °C. It is worth mentioning here that the Tm of ZIs decreased by approximately 100 °C by introducing an ether bond. ZIs having ether bonds and alkyl chain groups in the side chain of imidazolium cation, as shown in Fig. 6, were studied to compare their thermal properties.47 ZI 15, having one ether bond, and 10 having the butyl group, were obtained as white powders at room temperature. ZI 16 having two ether bonds and 17 having heptyl group were obtained as colourless clear liquids at room temperature. The introduction of these groups was effective for lowering the Tm of the ZIs. The Tg, crystallisation temperature (Tc), and Tm of 10 having the butyl group were 12, 73, and 170 °C, respectively. The Tg, Tc, and Tm of 15 having one ether bond were 7, 72, and 175 °C, respectively. The Tm of 15 and 10 were similar, but 15 exhibited a solid–solid phase transition at 166 °C below Tm. Since the freedom of molecular motion increased with the introduction of an ether bond, ZI 15 showed polymorphism. This behaviour was also observed with ammonium-based ZIs having an ether bond.44 ZIs 16 and 17 became liquid at room temperature and exhibited the only Tg at −32 °C and −10 °C, respectively. By extending the side chain of the imidazolium cation, it was possible to obtain liquid ZIs. Both functional groups (i.e., ether bonds and long alkyl chains) suppressed the crystallisation of ZIs. ZI 16 having two ether bonds showed a lower Tg. Ether oxygens strongly interacted with ions, particularly cations, through ion–dipole interactions. Since the ion–dipole interaction suppressed strong electrostatic interactions between ZIs, the degree of freedom of each ion was enhanced. Such an effect was observed as a result of lowering Tg.
 | ||
| Fig. 6 Chemical structure of ZIs. | ||
A novel series of ZIs based on the E or Z isomer of urocanic-acid derivatives have been synthesised by Bordes et al.48 Surprisingly, all the synthesised ZIs, even with carboxylate as the counter anion, showed Tm below 100 °C. The Tm of the E (18) and Z isomer (19) was 41 and −20 °C, respectively. The stereochemistry of the isomer was found to greatly affect the thermal properties of ZIs. Moreover, the Tm of urocanic-acid derivatives was much lower than that of a carboxylate-based ZI 20 (Tm = 160 °C)30 in spite of the same carbon number for the spacer between imidazolium and carboxylate. The double bond between the cation and anion may contribute to lower the Tm of ZIs.
ZIs 10, 15 to 17 were mixed with equimolar amounts of LiTf2N, respectively.47 ZIs 15 and 10 were solids at room temperature, but they became viscous liquids after the addition of an equimolar amount of LiTf2N. These complexes showed Tg only, and no Tm was found from the DSC measurements. The Tg of the complexes of 15, 10, and 17 were −12, −18, and −19 °C, respectively. The Tg of the complexes decreased by 10 to 30 °C as compared to that of the corresponding ZIs. The Tg of the 16/LiTf2N complex was −27 °C, and it was found that 16 maintained a low value even after the addition of LiTf2N. As mentioned above, the interaction between imidazolium cation and Tf2N anion was considered to form an IL moiety.11 This presumption is supported by the fact that the thermal decomposition temperature of ZI/LiTf2N complexes is about 390 °C, which is consistent with the thermal decomposition temperature of the imidazolium-based ILs having the Tf2N anion.26
The temperature dependence of the conductivity of each complex for ZIs 10, 15, 16 is shown in Fig. 7. Although the 17/LiTf2N complex was obtained as a liquid, the ionic conductivity was below 10−6 S cm−1 even at 80 °C due to crystallisation occurring in a short time. The lines in the figure are the result of fitting the VFT equation. In any complex, the temperature dependence of the ionic conductivity showed an upper convex curve and obeyed the VFT equation. The ion conduction in ZI/LiTf2N complexes should therefore be dominated by the viscosity of the complexes.
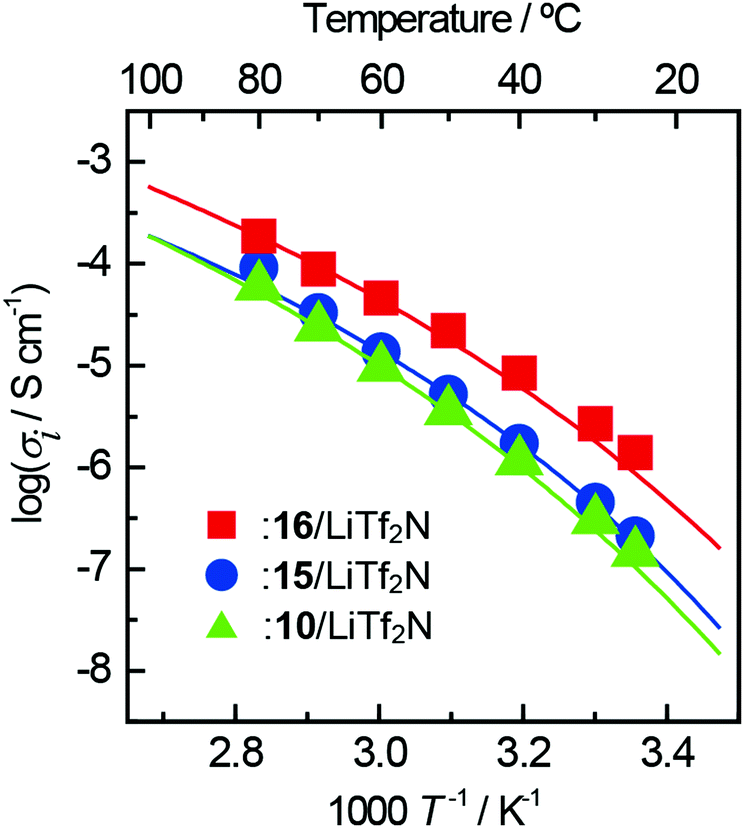 | ||
| Fig. 7 Arrhenius plots of ionic conductivity for ZI/lithium-salt complexes. The lithium-salt content is 50 mol% for all the complexes. | ||
The ionic conductivity of the 15/LiTf2N complex was 9.2 × 10−5 S cm−1 at 80 °C, which was almost the same as that of the 10/LiTf2N complex. The ionic conductivity of 16/LiTf2N complex was 3.8 × 10−4 S cm−1 at 80 °C, which was 4 times higher than that of the 15/LiTf2N complex. The 16/LiTf2N complex had the lowest Tg among the complexes prepared in this study. This could be the reason for such a high ionic conductivity. The introduction of the ether bond on the side chain was effective to not only decrease the Tm of the ZIs but also improve the ionic conductivity.
Generally, the composites having a greater amount of lithium salts showed higher ionic conductivity up to a certain amount. The lithium-salt concentration dependence of the conductivity and Tg was investigated for ZI 16.50 Two kinds of lithium salts, LiTf2N and lithium bis(fluorosulfonyl)imide (LiFSI), were used. Lithium-salt concentration was in the range between 20 and 90 mol%. When the lithium salt concentration was in the range of 20 to 50 mol%, the Tg of 16/lithium-salt complexes were in the range of −40 to −30 °C. When the lithium-salt concentration was greater than 50 mol%, the Tg of the complexes monotonously increased. However, when the lithium-salt concentration was 80 mol%, the Tg of 16/LiTf2N and 16/LiFSI was −10 and −32 °C, respectively. It is worth mentioning that the Tg decreased in such high lithium-salt conditions.
In the lithium-salt concentration range between 20 and 50 mol%, the complexes exhibited a constant conductivity of about 10−5 S cm−1 at 40 °C. In the salt-concentration range of 60 to 70 mol%, the ionic conductivity decreased due to increasing Tg. When the LiTf2N concentrations were 60 and 70 mol%, the ionic conductivity of each complex was about 10−7 and 10−8 S cm−1 at 40 °C, respectively. At the LiTf2N concentration of 80 mol%, the ionic conductivity increased drastically. The complex of 16 with 80 mol% LiFSI exhibited the highest ionic conductivity of 1.2 × 10−5 S cm−1 at 40 °C. In the lithium-salt concentration range between 20 and 80 mol%, Tg and conductivity showed a correlation according to the conventional perception. The ionic conductivity was improved by adding a small amount of ZI in LiTf2N and LiFSI.
Liquid ZIs were effective in improving the ionic conductivity. In order to apply them as electrolyte materials, tLi+ was also an important parameter as mentioned above. The tLi+ of 15, 10, and 16 with an equimolar amount of LiTf2N were 0.15, 0.10, and 0.15, respectively, at 40 °C,50 which were much lower than that of the systems described above. ZIs 15 and 16 having the ether bond exhibited the highest tLi+ among the three ZIs. The introduction of the ether bond was thought to lead to a decrease in tLi+, but the opposite results were obtained. The ion–dipole interaction between the lithium ion and ether oxygen promotes the dissociation of lithium salts, and lithium-ion conduction along with the ether oxygens may occur.14 The tLi+ of 16 with LiFSI 50 and 80 mol% were 0.20 and 0.46, respectively.50 The 16/LiFSI complexes had greater tLi+ than the 16/LiTf2N complexes due to lower coordination strength between the FSI anion and the lithium ion compared with that of Tf2N anion.51 The combination of ZIs and LiFSI represents an interesting matrix for electrochemical applications.
3.4. Polymerised zwitterions
Polymerised zwitterions, that possess both cationic and anionic groups on one repeating unit, are being considered as novel ion conductive alternatives to polyether-based solid polymer electrolytes, due to the highly dipolar character of their zwitterionic groups.43 Research on the solid polymer electrolytes have so far mainly involved the use of polyether matrices because these polyether chains can dissolve salts and transport the dissociated ions along with the intramolecular and intermolecular motion of these polyether chains.52 Polymer matrices with both high polarity and low Tg are rare because polar materials generally have high Tg. However, the ionic conductivity of certain polyether systems was approximately 10−4 S cm−1 at room temperature, and it was difficult to improve their conductivity. Although polymerised ZIs contain no carrier ions, they can provide desirable carrier ions upon the addition of salts to their matrices. In addition, some of these with low Tg have been reported.53 It is therefore possible to realise selective ion transport in the polymerised ZIs. If the resulting matrix can form a solid film over a wide temperature range, many useful ionic devices can be realised.For polyelectrolytes, the charges may be located either on the pendant side chains of different monomer units or the same monomer unit. According to the suggestion of Lowe et al.,54 we also expanded the definition of polymerised ZIs (polyZIs) to all polymers that possess both cationic and anionic groups. Polyampholytes refer to the polymers that possess the charged groups on different monomer units, while polybetaine refers to the polymers that possess the cationic and anionic groups on the same monomer unit.
 | ||
| Fig. 8 Chemical structure of polyampholytes. | ||
Lithium salts were added to both polyampholytes so as to generate carrier ions that can migrate over a long-distance. Three distinct lithium salts, LiTf2N, LiBF4, and LiCF3SO3, were used as additives, and the polyampholytes were mixed with equimolar amounts of lithium salts to the charged units on the polymer chains. It is well-known that these anions form ILs after mixing with imidazolium cations.9 The ionic conductivity of 21 mixed with LiBF4 and LiCF3SO3 were approximately 10−9 S cm−1, which was almost the insulating level. In contrast, when LiTf2N was added to 21, the mixture showed the ionic conductivity of 7.2 × 10−7 S cm−1 at 50 °C. The ionic conductivity was significantly improved by adding LiTf2N, even though the other anion species had the ability to form ILs. This difference is based on the plasticising effect of the Tf2N anion.56 An equimolar amount of LiTf2N to the charged units was added to 21 and 22 in order to investigate the effect of alkyl spacer between the polymerisable group and the sulfonate on the ionic conductivity. Compound 22 with propylene spacer showed the ionic conductivity of 1.2 × 10−6 S cm−1 at 30 °C, which was 30 times higher than that of 21. Compound 23 was prepared by the free-radical copolymerisation of two monomers with various molar ratios.57 One monomer had an imidazolium salt structure, and the other monomer had a lithium-salt structure as shown in Fig. 8. The ionic conductivity of 23 was in the range of 10−4 and 10−7 S cm−1 at 30 °C, depending on the molar ratio. Similar to the above-mentioned example, 23 with a spacer between the polymerisable groups and both cationic and anionic groups showed higher ionic conductivity than that of 22. Interestingly, the tLi+ of 23 exceeded 0.5 when the fraction of lithium-salt monomer was higher than that of the imidazolium-salt monomer. In general, the tLi+ of polyether/salt mixed systems is approximately 0.2 or less because lithium cations are trapped by the ether segments through the ion–dipole interaction.52 It can be concluded that introducing a flexible spacer between polymerisable groups and charged groups is a useful strategy to improve the ionic conductivity and the tLi+ of polyampholytes. Proton-conducting polyampholytes based on vinylphosphonic acid and 4-vinylimidazole were synthesised with various monomer-feed ratios via free-radical polymerisation by Bozkurt et al.58 Their ionic conductivity was in the range from 10−6 to 10−12 S cm−1 at temperatures between 0 and 160 °C under dry conditions. Compound 24 showed a higher ionic conductivity when the feed ratio of imidazole units was larger. It can be concluded that the imidazole ring acts as a proton-hopping site in the polymer matrix, as supported by Kreuer and co-workers59,60 and Watanabe and co-workers,61 who observed the effect of the imbalance of imidazole and acids on the proton-conductive behaviour. The morphology of 24 was characterised as amorphous by X-ray scattering. To realise fast proton transport in polyampholytes, it will be essential to design ion conductive paths that use the dimensional control of the IL domain.
 | ||
| Fig. 9 Chemical structures of polybetaines and monomers. | ||
The ionic conductivity and thermal properties of two kinds of polybetaines were compared, i.e., (25, 26) and (27, 28). The ionic conductivity of polybetaines with an imidazolium cation on the main-chain, 25 and 26, containing an equimolar amount of LiTf2N to the imidazolium unit, were in the range of 10−8 to 10−9 S cm−1. Polybetaines with sulfonamide anions on the main-chain, 27 and 28, showed relatively high ionic conductivity of approximately 10−5 S cm−1 at 50 °C. The compound 28 with a long alkyl spacer showed a slightly higher ionic conductivity. The difference in ionic conductivity can be attributed to the difference in freedom of the imidazolium cation. This effect has already been reported in the IL polymers.21,64,65 For a simple system such as 1-methyl-3-vinylimidazolium[Tf2N], the ionic conductivity decreased by approximately four orders upon polymerisation. The ionic conductivity of comb-like IL polymers with a flexible spacer between the polymerisable groups and imidazolium cations were almost the same before and after polymerisation. The ionic conductivity of 25 and 26, in which the imidazolium cation was fixed on the main chain, was very low even after adding LiTf2N to the matrix. Compounds 27 and 28 with the counter anion on the main chain had ionic conductivities three or four orders higher than 25 and 26. The distance between the polymer main-chain and the imidazolium cation is also essential for maintaining high ionic conductivity in polybetaines.
The dipole moments of zwitterionic side groups in polybetaines composed of aliphatic ammonium cations have been reported by Galin et al.43 Their dipole moments depend on the distance between the cationic and anionic groups. This suggests that these polybetaines have varied solvation power towards ionic guest species. Although the spacer distances between the cations and the anions were nearly identical, the ionic conductivities of 26 and 27 were very different. This supports that the distance between the polymerisable groups and the imidazolium cation is important in maintaining high ionic conductivity as mentioned above.
Recently, a polybetaine with an organoborate was synthesised via the dehydrocoupling polymerisation of lithium 9-borabicyclo[3,3,1]nonane hydride and 1,3-dihydroxyethyl-imidazolium bromide.66 The chemical structure of the polybetaine with organoborate 29 is unique because both cationic and anionic charges are located on the polymer main-chain (so-called linear-type polybetaines). The ionic conductivity of 29/LiTf2N mixture was 3.4 × 10−6 S cm−1 at 50 °C in spite of a relatively high Tg of −6 °C. Although the imidazolium cation was fixed on the main-chain for 29, the ionic conductivity was almost equal to that of 27 and 28 with low Tg. This may be due to the difference in the ion conduction mechanism between linear-type polybetaines and pendant-type polybetaines.
Galin et al. blended the tail-end-type polybetaines with alkali metal salts such as iodides, tetrafluoroborates, thiocyanates, perchlorates, trifluoromethanesulfonates, or tetraphenylborates, and performed the structural and electrochemical analyses for the mixtures.67 The tail-end type refers to polybetaines since zwitterionic groups are bound to the polymer backbone via the end of the hydrophobic tail. Compound 30, with sulfonate as the ZI moiety, was fully characterised as amorphous by X-ray powder diffraction patterns, due to a flexible spacer between the zwitterionic moiety and the polymer backbone. The polybetaine blends with alkali metal salts showed hexagonal and lamellar structures. The development of superstructures occurred upon solubilisation of salts, indicating that the microphase separation was promoted by the presence of dissociated ions. The stoichiometric blends of 30 with various alkali metal salts had Tg between 97 and 186 °C. The ionic conductivity of the stoichiometric blends was in the range from 0.4 to 4.3 × 10−3 S cm−1 at Tg + 30 °C. Although the stoichiometric blend with NaCF3SO3 or NaSCN showed higher values, the relationship between superstructure and the ionic conductivity is still unclear for amphiphilic zwitterions.
Compound 31, containing the dicyanoethenolate anion was individually blended with three different alkali metal salts, LiClO4, NaSCN, or NaCF3SO3.53 Both LiClO4 and NaSCN were miscible up to the stoichiometric mixture, while the solubility of NaCF3SO3 was less than the stoichiometric value. This was significantly different from 30 having the sulfonate anion tail, which gave a stoichiometric mixture even with NaCF3SO3. The dicyanoethenolate anion seemed to have less power to solvate alkali metal ions. The ionic conductivity of 31 was about 10−3 S cm−1 at Tg + 30 °C, which was almost the same as that of 30 containing various other salts.
Recently, sulfobetaine-containing copolymers were synthesised in order to control their morphology. Strehmel et al. reported the free-radical copolymerisation of 32 with n-butylmethacrylate (BMA) in ILs.68–70 The miscibility of the monomer segments in their copolymers was influenced by the chemical structure of the ILs. The maximum content of the less polar monomer segments, BMA, in the copolymers increased when ILs that contain a long alkyl chain as a less polar group were used for copolymerisation. The copolymerisation of BMA is quite effective for decreasing the Tg of polysulfobetaines.71 On the other hand, the tail-end-type polybetaines were used as nanostructural templates in water.72 The mixtures consisting of 33, methylmethacrylate, a sulfonic monomer, and water formed a basic bicontinuous microemulsion and were polymerised to obtain transparent membranes. These strategies can be quite appealing for obtaining highly ion conductive polybetaines.73
4. Zwitterions for aqueous biphasic systems
In addition to the remarkable progress in the design of ZIs as electrolyte materials, we recently focused on the physicochemical properties of ZIs after mixing with water. Since many ZIs have high melting points, many ZIs undergo solid/liquid-type phase separation even after adding water. In contrast, suitably designed ZIs show liquid/liquid-type phase separation to form aqueous biphasic systems (ABS). In this section, recent developments for the ZI-based ABS are summarised.4.1. Thermoresponsive phase behaviour of zwitterion/water mixtures
The IL-based ABS, in which ILs are phase-separated from water to form two liquid–liquid phases, have been widely investigated to apply them as alternatives to volatile organic solvent-based ABS. ILs, consisting of fluorinated anions such as Tf2N anions and hexafluorophosphate anions, are immiscible with water, and easily give the ABS.74 Water-miscible ILs also have a chance to phase-separate from water by further addition of phase-splitting promoters such as inorganic salts.75 The separated phases of these IL-based ABS are generally stable regardless of outer stimuli such as temperature. On the other hand, our studies demonstrated that suitably designed ILs with moderate hydrophobicity underwent highly temperature-sensitive lower critical solution temperature (LCST)-type phase behaviour after mixing with water.76–78 These IL/water mixtures exhibited homogeneous states in a certain temperature range but were found to show phase-separation by heating for only a few degrees. This phase change between the homogeneous and phase-separated state is reversible, and the transition temperature can be controlled by the subtle balance of hydrophobicity and hydrophilicity of the ILs.79For practical application of the above-mentioned IL-based ABS, especially in the field of bioengineering, it is of great importance to maintain their properties even in the presence of other salts, since many bio(macro)molecules are handled in buffered aqueous solutions. The reported thermoresponsive IL/water systems, however, have the potential to detract their properties as a result of unexpected ion pair formation through ion exchange after being mixed with other ions. We therefore envision that designing ZI/water mixtures showing the thermoresponsive behaviour should be attractive for the IL-based ABS since the ion pairs of ZIs are always fixed by covalent linkage even after adding different charged components.
With regard to the necessary factors for ILs to show LCST behaviour, we have revealed that ILs with moderate hydrophobicity between water-miscible ILs and water-immiscible ILs potentially acquired thermoresponsiveness. More precisely, relatively hydrophobic ILs with more than 7 hydrated water molecules per one ion pair were strongly expected to undergo LCST-type phase behaviour after mixing with water.79,80 According to the same design criteria, we have synthesised a series of phosphonium or ammonium-type ZIs containing sufficiently long alkyl chains on the cations. Sulfonate or phosphonate anions were chosen as the partner for every cation. The newly prepared ZIs were mixed with a certain amount of pure water and their phase behaviour was visually analysed as a function of temperature.
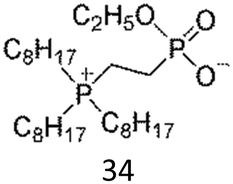
As expected, a few ZI/water mixtures showed LCST-type phase transition. Table 3 summarises the reported ZIs showing thermoresponsive behaviour with water. The first example we reported was phosphonium phosphate-type ZI, 34.81 This ZI was prepared via the quaternisation reaction of alkyl phosphine, followed by anion exchange and hydrolysis of ethyl ester. Although an anhydrous 34 was solid at room temperature (Tm is 162 °C), the liquid phase can be easily obtained by mixing it with a small amount of water. A typical LCST response was seen in 34/water mixtures and the saturated water content of the hydrated ZI phase varied widely with temperature.
| Structure | Phase behaviour |
T
c/°C (water:ZI mol) |
|---|---|---|
|
LCST | 11 (35:1) |
|
LCST | 10 (25:1) |
|
UCST and LCST (when two ZIs were mixed) | UCST; 17, LCST; 41 (35:0.67:0.33 for water: 36:37) |
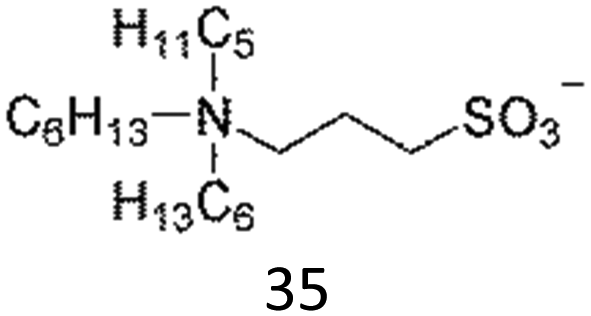
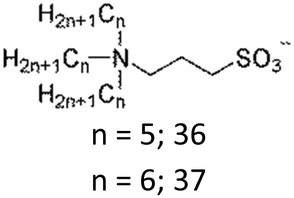
The interesting thermoresponsive behaviour was also realised in ammonium sulfonate-type ZIs (Table 3). We first clarified that 36 formed a homogeneously mixed solution with water, whereas 37 showed a solid–liquid phase separation with water, regardless of temperature. On the other hand, 35, which had intermediate hydrophobicity between 36 and 37, showed LCST-type phase transition.82 Fine control of the hydrophobicity of ZI is therefore crucial for designing such thermoresponsiveness. The structural modification of component ions is quite effective in this case. In addition, mixing ZIs with different hydrophobicity would also be useful in precisely controlling the total hydrophobicity of the ZI mixtures to meet the requirements for undergoing LCST. This method would be particularly beneficial in ZI systems since there is no fear of ion exchange reactions between the mixed ions. This is one of the advantages of ZIs. We have found that a mixture of 36 and 37 indeed exhibited LCST behaviour at a specific mixing ratio.83 Furthermore, this mixture also showed upper critical solution temperature (UCST) behaviour, in which the homogeneously-mixed phase was split into two phases upon cooling.83 The phase transition temperature (Tc) was controllable by changing the mixing ratio of the ZIs. Mixing ZIs is therefore effective for finely controlling the total hydrophobicity of ZIs.
Very recently, we have reported that addition of an equimolar amount of Brønsted acid, HTf, into hydrophobic ZI 37 converted the thermoresponsiveness and the resulting ZI/acid mixture underwent LCST-type phase behaviour with water.82 Such ZI-based thermoresponsive acid mixtures could offer simple and energy-saving catalytic systems, where the catalytic reaction takes place in the homogeneous phase and the products can then be extracted into the aqueous phase after phase separation driven by the small temperature change.
4.2. Improving the water content of hydrophobic ionic liquids
As described above, suitably designed ZIs with adequate hydrophobicity allowed phase separation with water, and the water content of the separated ZI-rich phase can be controlled. We found that ZIs acted as effective additives to improve the water content of hydrophobic ILs. General hydrophobic ILs are characterised by their very low saturated water content when mixed with water. Meanwhile, they could be more useful if various compounds were dissolved and stabilised in them. The solubility of water-soluble compounds including proteins in ILs is significantly influenced by the water content. Some hydrophilic ILs containing a small amount of water, classified as hydrated ILs, are capable of dissolving many kinds of proteins.84–86 Given the previous results on the hydrated ILs, improving the hydrated state of hydrophobic ILs would be effective for stable dissolution of proteins. Although it is possible to control the water content of hydrophobic ILs by designing component ions, it is difficult to precisely and successively tune the desired amount of saturated water amount in the ILs by this method. Simply adding hydrophilic ILs should also work for controlling the water content. However, possible ion exchange among the mixed ions could remove the unique properties of these ILs. The ZIs features (i.e., inhibition of ion exchange reaction) could also function in this case.Several imidazolium-based ZIs with sulfonate anions were mixed with a typical hydrophobic IL such as 1-butyl-3-methylimidazolium Tf2N ([C4mim][Tf2N]), and a given amount of water was then added to the mixture. While the saturated water content of neat [C4mim][Tf2N] was as low as 0.4 wt%, the amount was improved successively up to 17.8 wt% by adding hydrated 3-(1-butyl-3-imidazolio)propanesulfonate-type ZI: 7 (Fig. 3).87 The solubility and stability of cytochrome c (Cyt. c), a representative water-soluble heme protein, in the hydrophobic IL/7/water (1:1:8 by mol) were then analysed. As shown in Fig. 10, we found that Cyt. c was soluble in the hydrated mixture, while Cyt. c was totally insoluble in the neat [C4mim][Tf2N]. The addition of the 7/water mixture, therefore, facilitated the dissolution of Cyt. c in the hydrophobic IL.
 | ||
| Fig. 10 Photograph of Cyt. c (3 mg mL−1) mixed with [C4mim][Tf2N] (right), and [C4mim][Tf2N]/7/water mixture (left). | ||
The aforementioned imidazolium-type ZI systems, however, improved the water content of hydrophobic IL only in the homogeneous state. After adding an excess amount of water to obtain ABS, the increased amount of saturated water content in the IL phase was quite small. This could be because 7 was mainly dissolved in an aqueous layer due to its highly hydrophilic character. In this regard, we conceived that sufficiently hydrated ZIs being selectively partitioned in hydrophobic IL phases should allow improvement of the water content even in the separated IL phase. We then designed ZIs with finely balanced hydrophobicity that satisfied the requirements.
Fig. 11 (left) shows the structure of the as designed phosphonium-type ZI, 38. In this study, buffered aqueous potassium phosphate solution (PKB, 100 mmol L−1, pH 7.0) was used for potential bio-engineering applications. [C4mim][Tf2N]/PKB mixture with a volume ratio of 1/10 was prepared, and then 38 was added to the mixture. As shown in Fig. 11 (right), the addition of 38 effectively improved the saturated water content of the IL phase from 0.4 wt% to 62.8 wt%.88 It clearly suggests that the suitably hydrophobic 38, comprised of the phosphonium cation with long alkyl chains and sulfonate anions, was partitioned in the IL phase, promoting the increase in the maximum water content in the IL phase.
 | ||
| Fig. 11 Structure of phosphonium-type ZI, 38 (left); saturated water content of the separated IL phase as a function of mixing fractions of ZI in the IL/ZI mixture (right). | ||
To visually detect the effect of the water content, we checked the solubility of proteins in these mixtures. We chose Cyt. c as a typical protein that can easily be detected by the naked eye as red, due to the heme chromophore. The distribution behaviour of Cyt. c between the [C4mim][Tf2N]/38 mixed phase and PKB phase was then analysed to clarify the effectiveness of the addition of 38.88 Obviously, as seen in the photograph in Fig. 12, Cyt. c moved from the PKB upper phase to the IL bottom phase upon increasing the added amount of 38. The distribution ratio (D) was then calculated from the absorbance of Cyt. c in each phase. When an equimolar amount of 38 was mixed with [C4mim][Tf2N], the D value reached 94%. Since Cyt. c was not soluble in both pure and buffer-saturated [C4mim][Tf2N], it was confirmed that 38 facilitated the dissolution of Cyt. c in the [C4mim][Tf2N]. Spectroscopic analyses confirmed that both the higher-order structure of Cyt. c in the vicinity of the heme and its redox activity were maintained in the hydrated IL phase. Furthermore, the Cyt. c dissolved in the IL phase can be re-extracted into the aqueous phase by adding PKB with higher concentrations. It follows that Cyt. c can be dissolved selectively, stably and reversibly in the [C4mim][Tf2N] phase by adding 38.88 The use of suitably designed ZIs as additives to hydrophobic ILs would be a powerful strategy to control the solubility of bio(macro)molecules in both homogeneous and phase-separated states.
 | ||
| Fig. 12 Extraction of Cyt. c from the buffered aqueous upper phase to the [C4mim][Tf2N] phase by increasing the added amount of ZI 38. | ||
5. Zwitterions as a model interface for cell membranes
It is well-known that phospholipid-based cell membranes play an important role in discriminating intracellular fluid from the extracellular fluid. As initially shown in the famous fluid mosaic model of the cell membrane,89 the major component of the cell membrane is phospholipids. When looking at the structure of phospholipids carefully, we can find that they are ZI derivatives (Fig. 13). The hydrophilic part of the phospholipid is phosphorylcholine, a pair made up of the cholinium cation and phosphonate anion. This pair has already been analysed to be an excellent medium for a series of proteins when a small amount of water was added.84–86 This means that the mixture of phosphorylcholine and a small amount of water should be regarded as a liquid model of the hydrophilic part of the cell membrane. This kind of homogeneous ZI/water mixture should be used as a model fluid for membrane components, especially for membrane proteins. It is rather difficult to analyse the activity of proteins on the cell membrane; however, the hydrated phosphorylcholine is a homogeneous liquid, and it is rather easy to analyse the properties and reactivity of membrane proteins in it. This is quite a hot topic and results will be reported in the near future.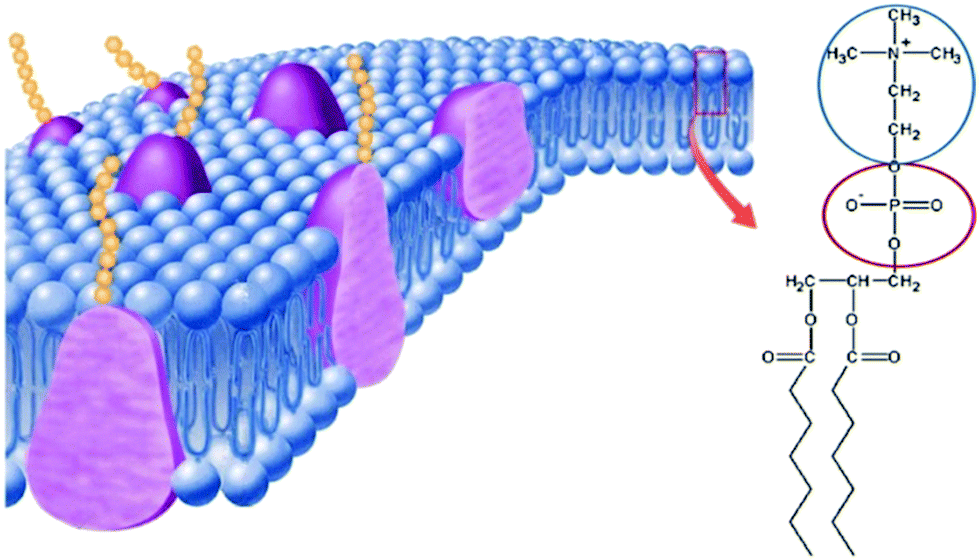 | ||
| Fig. 13 Schematic representation90 of the cell membrane, and the structure of a typical phospholipid that can be regarded as a zwitterion-derivative. | ||
It should be mentioned that the surface of the cell membrane can be comprehended to be the assembly of hydrated phosphorylcholine-type ZIs. Since there are concentrated proteins and other molecules, intracellular fluid in the cells should contain no free water. This situation is totally the same as hydrated phosphorylcholine-type ZI. This could be the reason why the pair of the cholinium cation and phosphonate anion showed excellent compatibility with many proteins.89 In both cases, there is no free water and all water molecules are strongly bound to ions. This unusual situation could contribute to the stable preservation of proteins and other biological molecules. As suggested above, this hydrated ZI is useful for analysing the dynamic and static behaviour of membrane proteins not in the membrane but in a homogeneous solution. This enables us to detect some physicochemical properties of such membrane proteins with conventional apparatus. The dissolution of membrane proteins and other membrane-components in such hydrated ZI is now in progress.
6. Future aspects
As mentioned above, ZIs derived from ILs are quite a new class of materials that have unique properties. The introduction of the properties of ILs into ZIs improved their unfavourable inherent properties such as very high Tm. Basic studies on these ZIs should increase with the attention and expectations of the functional ILs. Some expected applications of IL-derived ZIs are summarised in Fig. 14. These include electrochemical devices, bio-related materials, synthesis/catalysis, and functional surfaces. Although some of these applications have already been investigated by us and other groups, we have not mentioned the details of the studies in this review paper.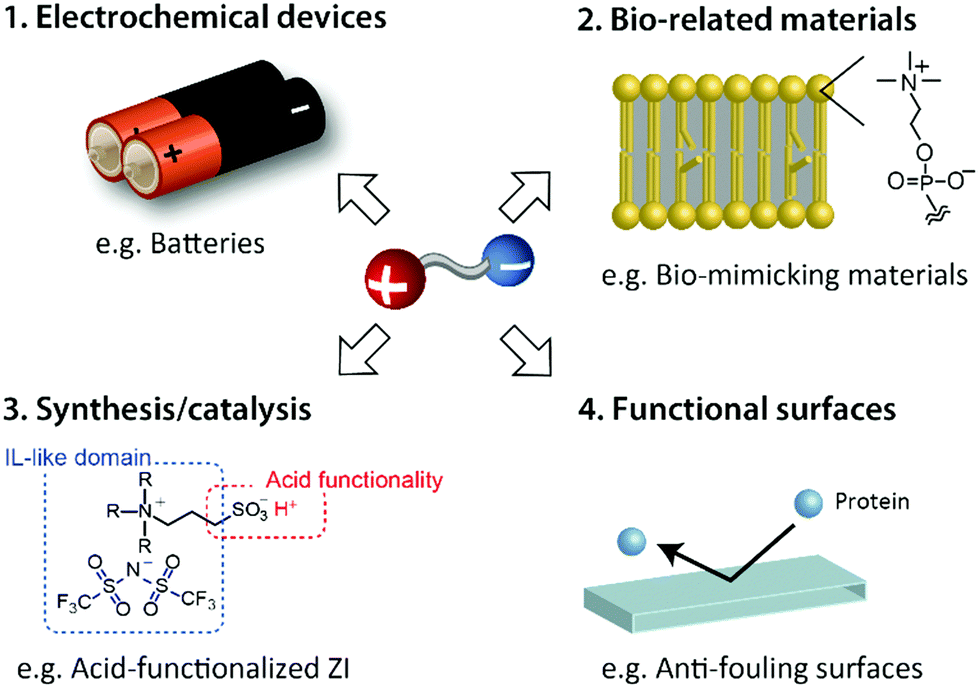 | ||
| Fig. 14 Expected applications of IL-derived ZIs. | ||
7. Conclusions
Zwitterions, derived from ionic liquids have unique properties such as relatively low glass transition temperature, small ion conductivity and unique phase behaviour after mixing with water. Furthermore, the mixture of certain zwitterions and a small amount of water can be regarded as an excellent fluid model of cell membranes. These unique properties are comprehended as arising from the nature of ILs and the tethering of both component ions. The development of such zwitterions will be accelerated with the development of ILs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Most of this mini-review is from the results of our recent study (H. O.). All these studies have been supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (KAKENHI) (17H01225, 24750112, 16K17954). Authors also acknowledge considerable efforts of staffs and students working for ILs and ZIs in our laboratory.Notes and references
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- M. Freemantle, An Introduction to Ionic Liquids, RSC publishing, 2009 Search PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- H. Ohno, Electrochemical Aspects of Ionic Liquids, 2nd edn, Wiley Interscience, New York, 2011 Search PubMed.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- N. Nishimura and H. Ohno, Polymer, 2014, 55, 3289–3297 CrossRef CAS.
- O. Green, S. Grubjesic, S. Lee and M. A. Firestone, Polym. Rev., 2009, 49, 339–360 CrossRef CAS.
- G. G. Eshetu, M. Armand, H. Ohno, B. Scrosati and S. Passerini, Energy Environ. Sci., 2016, 9, 49–61 CAS.
- M. Yoshizawa, M. Hirao, K. Ito-Akita and H. Ohno, J. Mater. Chem., 2001, 11, 1057–1062 RSC.
- M. Yoshizawa, A. Narita and H. Ohno, Aust. J. Chem., 2004, 57, 139–144 CrossRef CAS.
- D. Q. Nguyen, J. Hwang, J. S. Lee, H. Kim, H. Lee, M. Cheong, B. Lee and H. S. Kim, Electrochem. Commun., 2007, 9, 109–114 CrossRef CAS.
- D. Q. Nguyen, H. W. Bae, E. H. Jeon, J. S. Lee, M. Cheong, H. Kim, H. S. Kim and H. Lee, J. Power Sources, 2008, 183, 303–309 CrossRef CAS.
- H. Kim, D. Q. Nguyen, H. W. Bae, J. S. Lee, B. W. Cho, H. S. Kim, M. Cheong and H. Lee, Electrochem. Commun., 2008, 10, 1761–1764 CrossRef CAS.
- T. Mizumo, T. Watanabe and H. Ohno, Polym. J., 2008, 40, 1099–1104 CrossRef CAS.
- C. Tiyapiboonchaiya, J. M. Pringle, J. Sun, N. Byrne, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Nat. Mater., 2004, 3, 29–32 CrossRef CAS PubMed.
- N. Byrne, J. M. Pringle, C. Tiyapiboonchaiya, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2005, 50, 2733–2738 CrossRef CAS.
- N. Byrne, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Adv. Mater., 2005, 17, 2497–2501 CrossRef CAS.
- H. Lee, D. B. Kim, S.-H. Kim, H. S. Kim, S. J. Kim, D. K. Choi, Y. S. Kang and J. Won, Angew. Chem., Int. Ed., 2004, 43, 3053–3056 CrossRef CAS PubMed.
- H. Park, H. S. Kim and Y. M. Jung, J. Phys. Chem. B, 2011, 115, 1743–1750 CrossRef CAS PubMed.
- H. Ohno, M. Yoshizawa and W. Ogihara, Electrochim. Acta, 2004, 50, 255–261 CrossRef CAS.
- W. Xu, E. I. Cooper and C. A. Angell, J. Phys. Chem. B, 2003, 107, 6170–6178 CrossRef CAS.
- A. Narita, W. Shibayama and H. Ohno, J. Mater. Chem., 2006, 16, 1475–1482 RSC.
- A. Narita, W. Shibayama, K. Sakamoto, T. Mizumo, N. Matsumi and H. Ohno, Chem. Commun., 2006, 1926–1928 RSC.
- F. Wohde, R. Bhandary, J. M. Moldrickx, J. Sundermeyer, M. Schönhoff and B. Roling, Solid State Ionics, 2016, 284, 37–44 CrossRef CAS.
- H. Ohno, M. Yoshizawa and W. Ogihara, Electrochim. Acta, 2003, 48, 2079–2083 CrossRef CAS.
- Z. H. Li, Q. L. Xia, L. L. Liu, G. T. Xiao, D. S. Gao and X. D. Zhou, Electrochim. Acta, 2010, 56, 804–809 CrossRef CAS.
- A. Narita, W. Shibayama, M. Tamada and H. Ohno, Polym. Bull., 2006, 57, 115–120 CrossRef CAS.
- E. Marwanta, T. Mizumo and H. Ohno, Solid State Ionics, 2007, 178, 227–232 CrossRef CAS.
- M. Tamada, S. Ueda, T. Hayashi and H. Ohno, Chem. Lett., 2008, 37, 86–87 CrossRef CAS.
- M. Suematsu, M. Yoshizawa-Fujita, H. Zhu, M. Forsyth, Y. Takeoka and M. Rikukawa, Electrochim. Acta, 2015, 175, 209–213 CrossRef CAS.
- S. Yamaguchi, M. Yoshizawa-Fujita, H. Zhu, M. Forsyth, Y. Takeoka and M. Rikukawa, Electrochim. Acta, 2015, 186, 471–477 CrossRef CAS.
- N. Byrne, P. C. Howlett, D. R. MacFarlane, M. E. Smith, A. Howes, A. F. Hollenkamp, T. Bastow, P. Hale and M. Forsyth, J. Power Sources, 2008, 184, 288–296 CrossRef CAS.
- S. Yamaguchi, M. Yoshizawa-Fujita, Y. Takeoka and M. Rikukawa, J. Power Sources, 2016, 331, 308–314 CrossRef CAS.
- A. C. Cole, J. L. Jensen, I. Ntai, K. L. Tran, K. J. Weaver, D. C. Forbes and J. H. Davis, Jr., J. Am. Chem. Soc., 2002, 124, 5962–5963 CrossRef CAS PubMed.
- D. Li, F. Shi, J. Peng, S. Guo and Y. Deng, J. Org. Chem., 2004, 69, 3582–3585 CrossRef CAS PubMed.
- S. Kitaoka, K. Nobuoka and Y. Ishikawa, Chem. Commun., 2004, 1902–1903 RSC.
- K. Qiao and C. Yokoyama, Chem. Lett., 2004, 33, 472–473 CrossRef CAS.
- J. Gui, H. Ban, X. Cong, X. Zhang, Z. Hu and Z. Sun, J. Mol. Catal. A: Chem., 2005, 225, 27–31 CrossRef CAS.
- M. Yoshizawa and H. Ohno, Chem. Commun., 2004, 1828–1829 RSC.
- M. Yoshizawa-Fujita, N. Byrne, M. Forsyth, D. R. MacFarlane and H. Ohno, J. Phys. Chem. B, 2010, 114, 16373–16380 CrossRef CAS PubMed.
- Â. Rocha, T. Carvalho, P. Vidinha and N. M. T. Lourenço, ChemPlusChem, 2012, 77, 1106–1111 CrossRef.
- M. Galin, A. Chapoton and J.-C. Galin, J. Chem. Soc., Perkin Trans. 2, 1993, 545–553 RSC.
- M. Yoshizawa and H. Ohno, Chem. Lett., 2004, 33, 1594–1595 CrossRef CAS.
- M.-L. Pujol-Fortin and J.-C. Galin, Macromolecules, 1991, 24, 4523–4530 CrossRef CAS.
- A. Mathis, M. Galin, J.-C. Galin, B. Heinrich and C. G. Bazuin, Liq. Cryst., 1999, 26, 973–984 CrossRef CAS.
- M. Yoshizawa-Fujita, T. Tamura, Y. Takeoka and M. Rikukawa, Chem. Commun., 2011, 47, 2345–2347 RSC.
- R. Bordes, J.-D. Marty and N. Lauth-de Viguerie, Fr.-Ukr. J. Chem., 2016, 4, 85–94 CrossRef CAS.
- M. Hirao, H. Sugimoto and H. Ohno, J. Electrochem. Soc., 2000, 147, 4168–4172 CrossRef CAS.
- M. Suematsu, M. Yoshizawa-Fujita, T. Tamura, Y. Takeoka and M. Rikukawa, Int. J. Electrochem. Sci., 2015, 10, 248–258 Search PubMed.
- P. Johansson, L. E. Fast, A. Matic, G. B. Appetecchi and S. Passerimi, J. Power Sources, 2010, 195, 2074–2076 CrossRef CAS.
- F. M. Gray, Polymer electrolytes (RSC Materials Monographs), Royal Society of Chemistry, 1997 Search PubMed.
- M. Galin, A. Mathis and J.-C. Galin, Polym. Adv. Technol., 2001, 12, 574–582 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Chem. Rev., 2002, 102, 4177–4189 CrossRef CAS PubMed.
- M. Yoshizawa, W. Ogihara and H. Ohno, Polym. Adv. Technol., 2002, 13, 589–594 CrossRef CAS.
- S. Besner, A. Vallée, G. Bouchard and J. Prud’homme, Macromolecules, 1992, 25, 6480–6488 CrossRef CAS.
- W. Ogihara, N. Suzuki, N. Nakamura and H. Ohno, Polym. J., 2006, 38, 117–121 CrossRef CAS.
- A. Bozkurt, W. H. Meyer, J. Gutmann and G. Wegner, Solid State Ionics, 2003, 164, 169–176 CrossRef CAS.
- K. D. Kreuer, A. Fuchs, M. Ise, M. Spaeth and J. Maier, Electrochim. Acta, 1998, 43, 1281–1288 CrossRef CAS.
- M. F. H. Schuster, W. H. Meyer, M. Schuster and K. D. Kreuer, Chem. Mater., 2004, 16, 329–337 CrossRef CAS.
- A. Noda, M. A. B. H. Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2003, 107, 4024–4033 CrossRef CAS.
- P. K. Singh, V. K. Singh and M. Singh, e-Polym., 2007, 7, 335–368 Search PubMed.
- A. Laschewsky, Polymers, 2014, 6, 1544–1601 CrossRef CAS.
- M. Yoshizawa and H. Ohno, Chem. Lett., 1999, 889–890 CrossRef CAS.
- M. Yoshizawa and H. Ohno, Electrochim. Acta, 2001, 46, 1723–1728 CrossRef CAS.
- A. Narita, W. Shibayama, N. Matsumi and H. Ohno, Polym. Bull., 2006, 57, 109–114 CrossRef CAS.
- M. Galin, E. Marchal, A. Mathis and J.-C. Galin, Polym. Adv. Technol., 1997, 8, 75–86 CrossRef CAS.
- V. Strehmel, A. Laschewsky and H. Wetzel, e-Polym., 2006, 6, 131–140 Search PubMed.
- V. Strehmel, Macromol. Symp., 2007, 254, 25–33 CrossRef CAS.
- V. Strehmel, H. Wetzel, A. Laschewsky, E. Moldenhaue and T. Klein, Polym. Adv. Technol., 2008, 19, 1383–1390 CrossRef CAS.
- R. H. Brown, A. J. Duncan, J.-H. Choi, J. K. Park, T. Wu, D. J. Leo, K. I. Winey, R. B. Moore and T. E. Long, Macromolecules, 2010, 43, 790–796 CrossRef CAS.
- L. M. Gan, P. Y. Chow, Z. Liu, M. Han and C. H. Quek, Chem. Commun., 2005, 4459–4461 RSC.
- S. Ueda, J. Kagimoto, T. Ichikawa, T. Kato and H. Ohno, Adv. Mater., 2011, 23, 3071–3074 CrossRef CAS PubMed.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef.
- M. G. Freire, A. F. M. Claudio, J. M. M. Araujo, J. A. P. Coutinho, I. M. Marrucho, J. N. C. Lopes and L. P. N. Rebelo, Chem. Soc. Rev., 2012, 41, 4966–4995 RSC.
- K. Fukumoto and H. Ohno, Angew. Chem., Int. Ed., 2007, 46, 1852–1855 CrossRef CAS PubMed.
- Y. Kohno, H. Arai, S. Saita and H. Ohno, Aust. J. Chem., 2011, 64, 1560–1567 CrossRef CAS.
- Y. Kohno and H. Ohno, Chem. Commun., 2012, 48, 7119–7130 RSC.
- Y. Kohno and H. Ohno, Phys. Chem. Chem. Phys., 2012, 14, 5063–5070 RSC.
- H. Ohno, K. Fujita and Y. Kohno, Phys. Chem. Chem. Phys., 2015, 17, 14454–14460 RSC.
- Y. Fukaya and H. Ohno, Phys. Chem. Chem. Phys., 2013, 15, 14941–14944 RSC.
- Y. Mieno, Y. Kohno, S. Saita and H. Ohno, Chem. – Eur. J., 2016, 22, 12262–12265 CrossRef CAS PubMed.
- S. Saita, Y. Mieno, Y. Kohno and H. Ohno, Chem. Commun., 2014, 50, 15450–15452 RSC.
- K. Fujita, D. R. MacFarlane and M. Forsyth, Chem. Commun., 2005, 4804–4806 RSC.
- K. Fujita and H. Ohno, Biopolymers, 2010, 93, 1093–1099 CrossRef CAS PubMed.
- K. Fujita, D. R. MacFarlane, M. Forsyth, M. Yoshizawa-Fujita, K. Murata, N. Nakamura and H. Ohno, Biomacromolecules, 2007, 8, 2080–2086 CrossRef CAS PubMed.
- Y. Ito, Y. Kohno, N. Nakamura and H. Ohno, Chem. Commun., 2012, 48, 11220–11222 RSC.
- Y. Ito, Y. Kohno, N. Nakamura and H. Ohno, Int. J. Mol. Sci., 2013, 14, 18350–18361 CrossRef PubMed.
- S. J. Singer and G. L. Nicolson, Science, 1972, 175, 720–773 CAS.
- Figure partly modified from the original one: http://genkiryokup.com/mainhp/category66/entry553.html.
| This journal is © the Owner Societies 2018 |