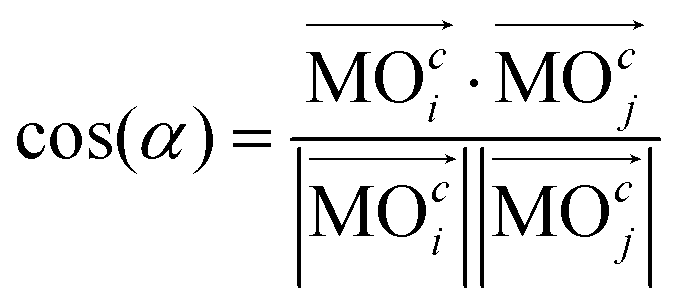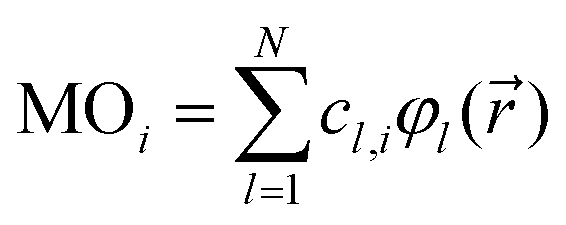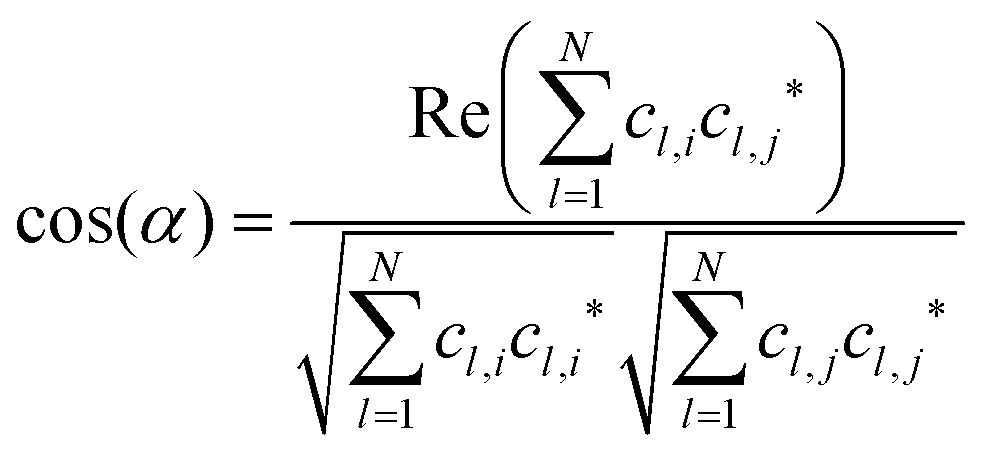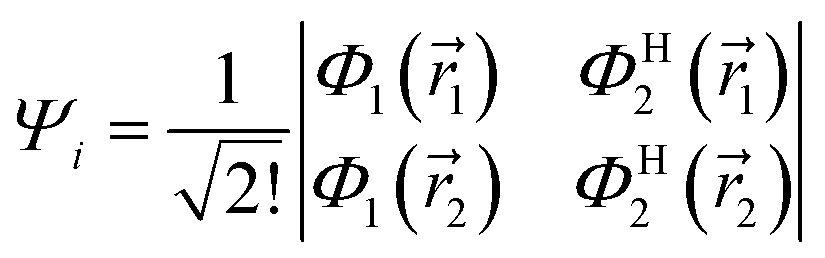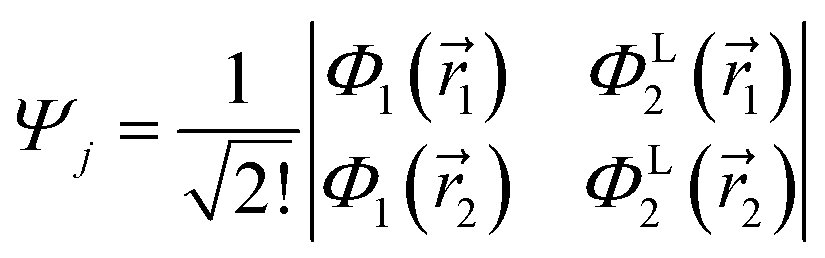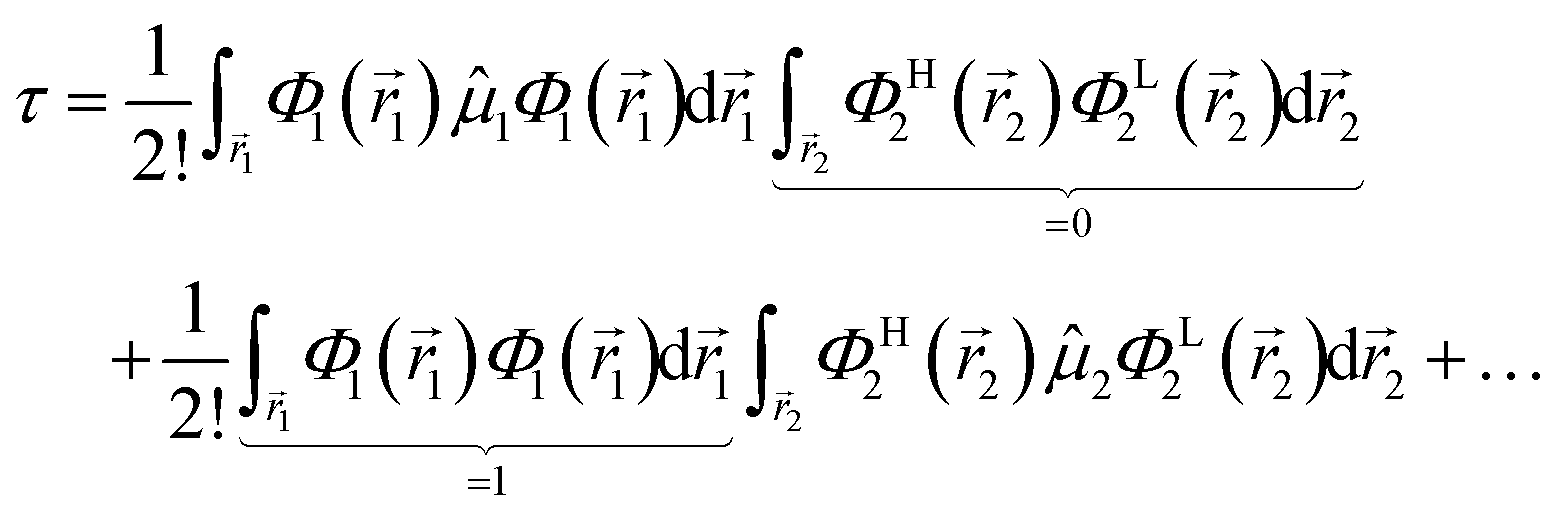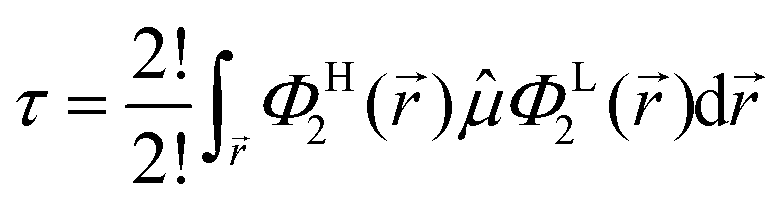Structural determinants in the bulk heterojunction†
Received
17th December 2017
, Accepted 30th January 2018
First published on 30th January 2018
Abstract
Photovoltaics is one of the key areas in renewable energy research with remarkable progress made every year. Here we consider the case of a photoactive material and study its structural composition and the resulting consequences for the fundamental processes driving solar energy conversion. A multiscale approach is used to characterize essential molecular properties of the light-absorbing layer. A selection of bulk-representative pairs of donor/acceptor molecules is extracted from the molecular dynamics simulation of the bulk heterojunction and analyzed at increasing levels of detail. Significantly increased ground state energies together with an array of additional structural characteristics are identified that all point towards an auxiliary role of the material's structural organization in mediating charge-transfer and -separation. Mechanistic studies of the type presented here can provide important insights into fundamental principles governing solar energy conversion in next-generation photovoltaic devices.
1 Introduction
At the molecular level the arrival of a photon at the photoactive layer of a solar cell must be considered an extremely rare event. For example, assuming incident solar radiation of power 1000 W m−2,1,2 a particular photosensitive molecule of nanometer sized dimensions could encounter a photon only every 0.4 milliseconds. It follows that efficient photovoltaic devices cannot afford missing any of these incoming photons and thus need to be designed in an appropriate way. This appears to be particularly important in the field of organic photovoltaics and imposes considerable constraints on the morphological details of the bulk heterojunction (BHJ),3–5i.e. the layer where photoexcitation,6–8 electron transfer6–9 and charge separation6,7,10,11 take place.
Organic photovoltaics offers numerous advantages over conventional technology, e.g. flexible devices, low cost manufacturing, sizeable power conversion efficiencies, thin film deposition techniques, semi-transparency etc.12,13 However, additional development is needed in order to fully exploit all these opportunities.14 As outlined by Ratner and coworkers15 too much emphasis is currently placed on power conversion efficiency while deeper knowledge is required in the microscopic details of the active layer and its physicochemical properties. Central domain of interest is the BHJ at sub-nanometer resolution16 where key challenges remain in processing technologies,17–22 reliability and chemical design of donor/acceptor pairs23–27 and control of the process of charge separation and recombination.1,6,9–11,15,28–30
Experimental characterization of the charge-transport properties of molecules has always been a challenge.31 Auxiliary analysis based on theoretical considerations32–39 can provide additional insight into specific details of the BHJ. Of particular interest are multiscale approaches33,40 where several length- and time-scales are examined in hierarchical order with different methods applied at increasing level of detail. This way the relationship between molecular nanostructure and characteristic electronic/optical properties of the BHJ can be explored.22,41,42 Here, we follow such an approach starting with the classical condensed matter representation of the BHJ and subsequently refine our focus onto atomistic and electronic degrees of freedom. We chose to investigate terthiophene–fullerene (3T–C60),26,27 a typical dyad with covalently linked donor/acceptor moieties, because of the anticipated simplification in structural complexity.
This paper is organized as follows: Section 2 describes individual components of the applied multiscale approach with a focus on technical details, in Section 3 we discuss obtained results following the multiscale approach in a top-down sense, meaning from macroscopic to molecular/electronic properties and in Section 4 we briefly summarize obtained key findings.
2 Experimental/computational
2.1 Molecular dynamics (MD)
2.1.1 Set up of structures and geometries.
Terthiophene–fullerene (3T–C60)26,27 was constructed with the help of MOLDEN43 and minimized with GAUSSIAN44 (version 03) using PM345 model chemistry. A new residue type was defined using ANTECHAMBER46 based on the GAFF47 force field. Quick qualitative optimization without the assignment of atomic partial charges was followed by a second parameterization step using BCC charges.48 An orthorhombic box containing 216 units of 3T–C60 was set up following previous protocols49,50 where a number density was chosen that prevented individual units from initial cross-contacts.
2.1.2 Minimization, equilibration, annealing.
500 steps of steepest descent minimization were performed with program SANDER from the AMBER package51 before switching to conjugate gradient minimization for another 9500 steps, both times without restraints and a cutoff of 12 Å. The minimized structure was heated to 298 K within 100 ps of equilibration MD based on a Berendsen thermostat in the NTV ensemble.52 Particle mesh Ewald summation was used throughout (cut off radius of 10 Å). Bonds involving H-atoms were considered by the SHAKE algorithm53 and a time step of 2 fs was used. Another 100 ps of equilibration MD was carried out at 298 K and constant pressure of 1 atm to fix box dimensions. This was followed by 1000 ps of equilibration MD in the NpT ensemble using an Anderson thermostat,54,55 a cutoff of 12 Å and a time step of 2 fs. To remove all potentially introduced inter-particle arrangements the system was heated to 1300 K simulation temperature within 1000 ps of MD simulation at otherwise similar conditions to the room temperature equilibration. Another equilibration MD was run for 10 ns at this elevated simulation temperature of 1300 K, again in the NpT ensemble using an Anderson thermostat, a cutoff of 12 Å and a time step of 1.5 fs. This was followed by a cooling simulation to the target simulation temperature of 300 K within 1000 ps, NpT ensemble (Anderson), 12 Å cutoff and a time step of 1.5 fs.
2.1.3 Production MD.
The equilibrated system was taken up for production MD within the NpT ensemble again using an Anderson thermostat,54,55 a cutoff of 12 Å and a time step of 1.5 fs. Overall sampling time was 110 ns within 3 individual runs covering 10 ns, 50 ns and again 50 ns.
2.1.4 Diffusion coefficients.
Diffusion coefficients were computed according to the Einstein–Smoluchowski relation56,57 where PTRAJ58 was used to extract mean square displacements (MSD) from the MD trajectory (module diffusion). The data was fit with GNUPLOT59 and slopes were determined from the fit then multiplied by 10/6 to yield diffusion constants in units of 10−5 cm2 s−1.
2.1.5 Radial distribution functions.
Structural ensemble snapshots were extracted from the MD trajectory and used to compute radial distribution functions, g(r). The 216 3T–C60 units of each of the extracted ensemble snapshots were divided into a central core unit and the remaining 215 other units forming the periphery. A subset of 17 specific units of 3T–C60 with distance to the box origin smaller than 28 Å was selected for the central core position, i.e. the r = 0 point in g(r). On the order of 770 structural snapshots were taken into account, hence averages were based on approximately 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 evaluations. A generalized form of g(r) was applied where a subset of atoms could be defined inside a particular 3T–C60 unit to form a single point of reference. Thus atoms 1 to 32 defined the donor moiety (D = 3T) whereas atoms 33 to 92 described the acceptor part (A = C60). Truncation effects were avoided by a 26-fold replication of the central simulation cell in all possible orthorhombic neighbour positions. Because box dimensions did change in the NpT ensemble, these 26× replications needed to be geometrically re-adjusted for every single structural snapshot. Radial distribution functions were then computed in the standard way60 sampling radii in the range of 0 Å to 30 Å in incremental “bins” of 0.1 Å. Once characteristic properties of g(r) had been determined the evaluation was repeated now screening for subsets of 3T–C60 units most suitable to reproduce essential signatures of g(r). At first the focus was on identifying groups composed of seven 3T–C60 units with an inner core unit surrounded by 6 neighbours separated by approximately 9.75 Å where the selection was done in the “least squares” sense. Next specific C60–3T and 3T–3T distances were taken into account in a similar way again following g(r) signatures. A shortlist of 9 structural scaffolds was thus prepared and visually examined and a final group of 13 D/A pairs was extracted to become subject to in-depth analysis at higher levels of theory. Graphical control was exercised by superimposing the inner core unit of the extracted 9 subsets with the help of PTRAJ58 (module rms reference) and visualizing the extent of variation in 3T-chain geometry with VMD.61
000 evaluations. A generalized form of g(r) was applied where a subset of atoms could be defined inside a particular 3T–C60 unit to form a single point of reference. Thus atoms 1 to 32 defined the donor moiety (D = 3T) whereas atoms 33 to 92 described the acceptor part (A = C60). Truncation effects were avoided by a 26-fold replication of the central simulation cell in all possible orthorhombic neighbour positions. Because box dimensions did change in the NpT ensemble, these 26× replications needed to be geometrically re-adjusted for every single structural snapshot. Radial distribution functions were then computed in the standard way60 sampling radii in the range of 0 Å to 30 Å in incremental “bins” of 0.1 Å. Once characteristic properties of g(r) had been determined the evaluation was repeated now screening for subsets of 3T–C60 units most suitable to reproduce essential signatures of g(r). At first the focus was on identifying groups composed of seven 3T–C60 units with an inner core unit surrounded by 6 neighbours separated by approximately 9.75 Å where the selection was done in the “least squares” sense. Next specific C60–3T and 3T–3T distances were taken into account in a similar way again following g(r) signatures. A shortlist of 9 structural scaffolds was thus prepared and visually examined and a final group of 13 D/A pairs was extracted to become subject to in-depth analysis at higher levels of theory. Graphical control was exercised by superimposing the inner core unit of the extracted 9 subsets with the help of PTRAJ58 (module rms reference) and visualizing the extent of variation in 3T-chain geometry with VMD.61
2.1.6 Reduction to D/A pairs.
All chemical linkers connecting D- with A-moieties inside a particular 3T–C60 unit were removed in all the 9 samples of bulk-representing 3T–C60 scaffolds and unsaturated valencies were fixed with H-atoms. Individual D/A pairs were extracted from the scaffold based on physical proximity (see Movie 1, ESI†). In the majority of cases, D/A pairs were located on different units, i.e. inter-rather than intra-molecular electron transfer was taken into account. However, from time to time the closest D unit to a particular A unit was found to be the initially bound one, hence intra-molecular electron transfer was considered in these cases. Occasionally a particular scaffold included more than one suitable D/A geometry and respective coordinate sets were then termed case 1, case 2 etc. Naming convention was according to the unit number providing the A-part and a list of the studied D/A pairs is given in Table 1. For graphical comparison the complete set of all 13 D/A structures was superimposed with the help of PTRAJ58 (module rms reference) and color-coded with VMD.61
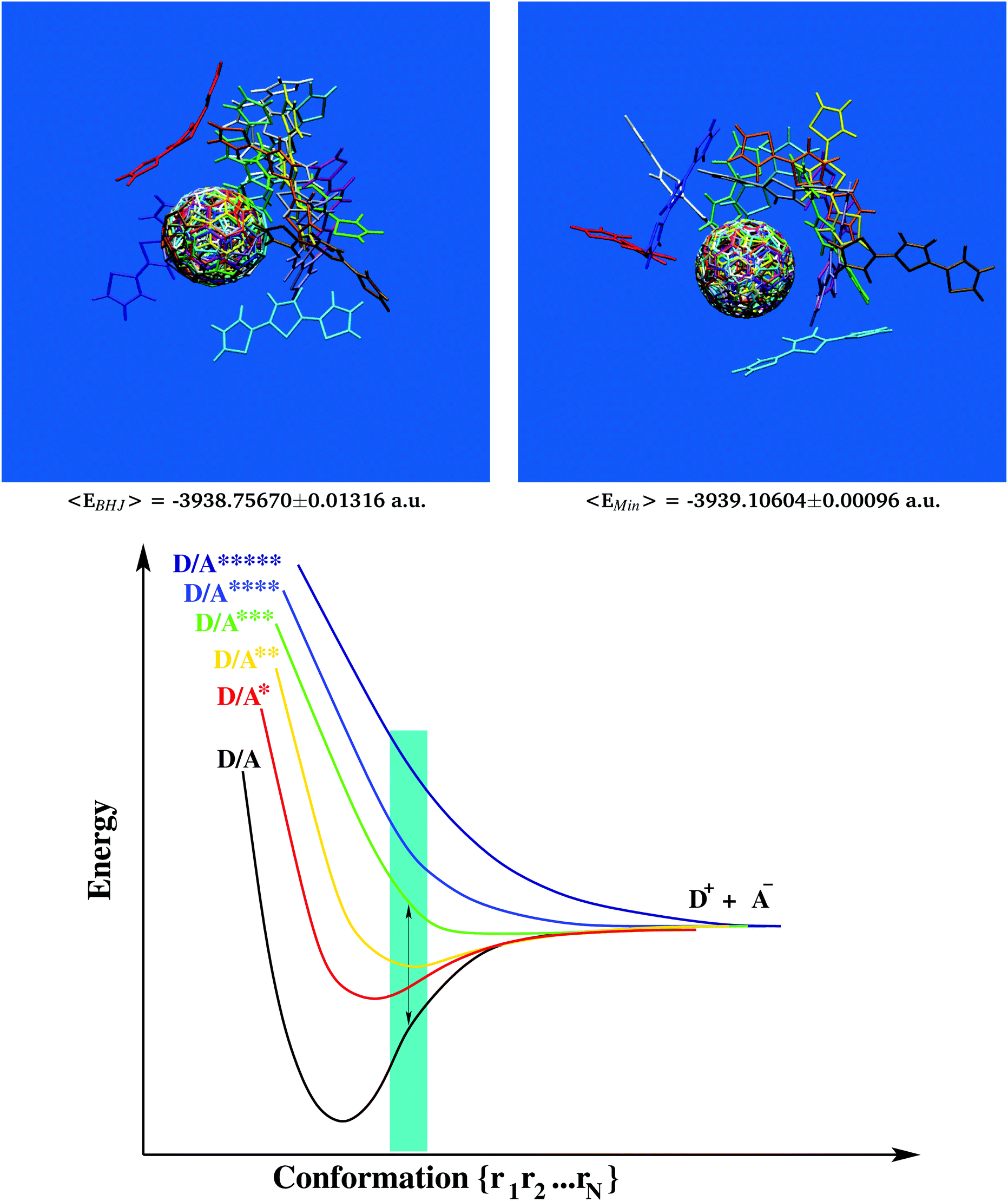
Table 1 Ground state energies of selected D/A pairs from the BHJ (model chemistry: PBEPBE/6-31g*). Average values, −3938.75670 [±0.01316] a.u., are significantly higher than corresponding ground state energies of minimized structures, −3939.10604 [±0.00096] a.u., with a mean gap of +0.34934 [±0.01273] a.u. which is approximately 5 times the photon energy (also see Fig. 2 and Movie 2, ESI, for a 360° view)
| Sample |
E
BHJ(D/A) [Hartree] |
E
Min(D/A) [Hartree] |
ΔE [Hartree] |
Colorb (Fig. 2) |
|
Penultimate geometry due to non-convergent minimization.
VMD61 jargon.
|
| 3T–C6056(case 1) |
−3938.7278 |
−3939.1041a |
+0.3763 |
White |
| 3T–C6056(case 2) |
−3938.7484 |
−3939.1081 |
+0.3597 |
Iceblue |
| 3T–C6076 |
−3938.7441 |
−3939.1053 |
+0.3612 |
Green3 |
| 3T–C6098 |
−3938.7511 |
−3939.1058 |
+0.3547 |
Ochre |
| 3T–C60156 |
−3938.7536 |
−3939.1054 |
+0.3518 |
Blue |
| 3T–C60162(case 1) |
−3938.7538 |
−3939.1052 |
+0.3514 |
Red |
| 3T–C60162(case 2) |
−3938.7845 |
−3939.1070 |
+0.3225 |
Green |
| 3T–C60167 |
−3938.7567 |
−3939.1062 |
+0.3495 |
Yellow |
| 3T–C60168 |
−3938.7572 |
−3939.1068 |
+0.3496 |
Cyan |
| 3T–C60171(case 1) |
−3938.7591 |
−3939.1063 |
+0.3472 |
Purple |
| 3T–C60171(case 2) |
−3938.7675 |
−3939.1057 |
+0.3382 |
Cyan2 |
| 3T–C60199(case 1) |
−3938.7618 |
−3939.1067 |
+0.3449 |
Orange2 |
| 3T–C60199(case 2) |
−3938.7715 |
−3939.1059 |
+0.3344 |
Silver |
2.2 Single point ab initio calculations
Single point DFT calculations were carried out for all 13 bulk-representative D/A pairs in unmodified original geometries as obtained from MD trajectories. The PBEPBE/6-31g* level of theory62–64 was applied and ground states were considered at overall neutral charge state. MO coefficients were also written out and coupled D/A systems were split into individual D- and A-parts which were considered for isolated ground state calculations too (at otherwise similar conditions to the fully coupled D/A systems). Next all the 13 D/A pairs were first minimized and subsequently considered for ground state energy calculations using identical levels of theory. One explicit sample, 3T–C60162(case 2), was minimized using fully intact 3T–C60 units in order to probe whether actual minimum structures in the gas phase were properly reproduced from reduced D/A complexes isolated here (Fig. S2, ESI†). All ab initio computations were carried out with GAUSSIAN65 and results are summarized in Table 1. All minimized structures were also superimposed with the help of PTRAJ58 and color coded with VMD61 to facilitate simple graphical comparison to the bulk-representative analogues. As an example, system 3T–C60171(case 1) was also studied in an extended form where additional explicit partial charges were considered in the form of a background charge distribution. Partial charges were those used during MD simulation and the inner subset of 7 D/A units was taken into account (see Fig. S16, ESI†). Similarly, background charges where also included when running electron transfer calculations (see below). In these latter cases, however, one direct D- or A-unit was included in ionic form with net charge +1 or −1. These took into account the charge-transfer complex established after photo-excitation and primary electron transfer. A second independent series of single point calculations was carried out for all 13 D/A pairs in bulk-representative as well as minimized geometry using the MM description to assess general force field performance (see Fig. S3, ESI†).
2.3 Similarity coefficients
Generalization of the angle between two vectors forms the basis of a criterion used to estimate the similarity between two molecular orbitals (MOs). In particular, the generalized scalar product,| | 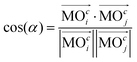 | (1) |
yields values close to 1 for a pair i, j of rather similar MOs, but indicates unrelatedness when values are approaching 0. As implied by the superscript c, the focus here is entirely on the vector of coefficients, cl, forming the specific linear combination of a particular MO,| | 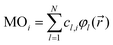 | (2) |
while all associated basis functions, φl(![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) ), will be ignored. |
), will be ignored. |![[a with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0061_20d1.gif) | in eqn (1) denotes “the norm/length of vector ” which should be 1 for MOs anyway owing to normalization. Because MOs are not unique with respect to a uniform sign change in all coefficients, cl, the expression in eqn (1) needs to consider two variants, one using the original set of coefficients, cl,j, for one of the two vectors considered, then a second variant with the same set of coefficients but all signs inverted, ±cl,j, where the larger of the two resulting cos(α)-values will be used for actual characterization of similarity. Apart from the extension to arbitrary dimensions, N, the other important generalization in eqn (1) is the applicability to vectors of complex coefficients.66 Here the relation in full detail becomes,
| in eqn (1) denotes “the norm/length of vector ” which should be 1 for MOs anyway owing to normalization. Because MOs are not unique with respect to a uniform sign change in all coefficients, cl, the expression in eqn (1) needs to consider two variants, one using the original set of coefficients, cl,j, for one of the two vectors considered, then a second variant with the same set of coefficients but all signs inverted, ±cl,j, where the larger of the two resulting cos(α)-values will be used for actual characterization of similarity. Apart from the extension to arbitrary dimensions, N, the other important generalization in eqn (1) is the applicability to vectors of complex coefficients.66 Here the relation in full detail becomes,| | 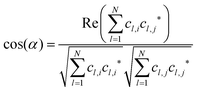 | (3) |
where Re() denotes the “real part of a complex number” and c* indicates the “complex conjugate of c”. Similarity coefficients have been used in two ways: (i) to compare a particular MO of the assembled D/A pair with its corresponding D- or A-orbital of the isolated donor- or acceptor-system considered stand-alone (and the missing part filled with zeros), (ii) to identify those MOs that become (un)occupied during electron transfer.
2.4 Transition dipole moments
The transition dipole moment67,68 is a convenient property to evaluate whether an electronic transition from an initial state Ψi to a final state Ψj is likely to occur as a result of interaction with electromagnetic radiation. Its expectation value| | τ = 〈Ψi|![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/char_e0b3.gif) |Ψj〉 |Ψj〉 | (4) |
may be written in terms of the electric dipole moment operator, ![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b3.gif) , a classical one-electron operator. Consider for example a molecule with two electrons where both the initial as well as the final state are approximated by single Slater determinants, in particular,
, a classical one-electron operator. Consider for example a molecule with two electrons where both the initial as well as the final state are approximated by single Slater determinants, in particular,| | 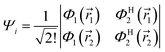 | (5) |
and| | 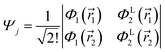 | (6) |
and Φ1,2 are MOs following eqn (2) in initially explicit electronic coordinates, 1, 2. The HOMO in Ψi is substituted with the LUMO in Ψj, hence the superscripts in ΦH/L2, and the usual orthogonality relations apply, i.e. 〈Φi|Φj〉 = δij. For this particular case, the dipole moment operator is a sum of two terms, = 1 + 2 acting on either of the two electrons and application of eqn (4) will result in a sum of eight terms of the following type,| | 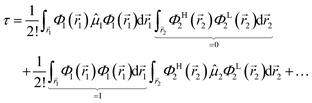 | (7) |
from which only a number of 2! will not vanish, which further turn out to be analogous expressions in different explicit coordinates and may thus further be combined to a single final relation (in general coordinates), i.e.| | 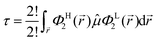 | (8) |
If we had considered another substitution in Ψi, for example to replace the HOMO with LUMO+1, we had ended up with exactly the same relation except that ΦL+12 had to be used on the r.h.s. of eqn (8). Consequently, by pairing the HOMO with a set of potential LUMOs and computing individual expectation values of the transition dipole moment, the propensity of the corresponding electronic transitions may be ranked. Here transitions between HOMO, LUMO, LUMO+1,…, LUMO+9 were considered for both the 13 bulk conformations as well as their counterparts in relaxed geometry. Program CMPTRQ69 was applied in a slightly modified version to compute expectation values of the transition dipole moment according to eqn (8).
2.5 Electron transfer calculations
The time dependent Schrödinger equation70 has been considered via the Cayley algorithm71–74 to trace the time-evolution of approximate D/A MOs in incremental time steps of duration Δt = 0.048 fs (for method details see ref. 75). The PBEPBE/6-31g* level of theory62–64 was applied throughout. Given the extent of the calculations, an efficient implementation such as the one utilizing the GPU75 was of considerable advantage. The focus was on detecting electron transfer (ET), i.e. the shift from occupied MO sections belonging to the donor to initially vacant sections (cl,i = 0 in eqn (2)) associated with the acceptor. This shift has been investigated here both in orthogonal and non-orthogonal MOs75 but the difference turned out to be negligible for all practical purposes. Every fifth time step ET has been probed and the similarity between runtime MOs and the initial set of uncoupled donor- or acceptor-MOs been analyzed (see Section 2.3). This way the time coordinate could be resolved and those MOs loosing/gaining an electron be identified in an unequivocal manner (see for example lower left/right panels in Fig. 4).
3 Results and discussion
3.1 The bulk heterojunction exhibits specific structural characteristics that support photo-excitation and transfer of electrons
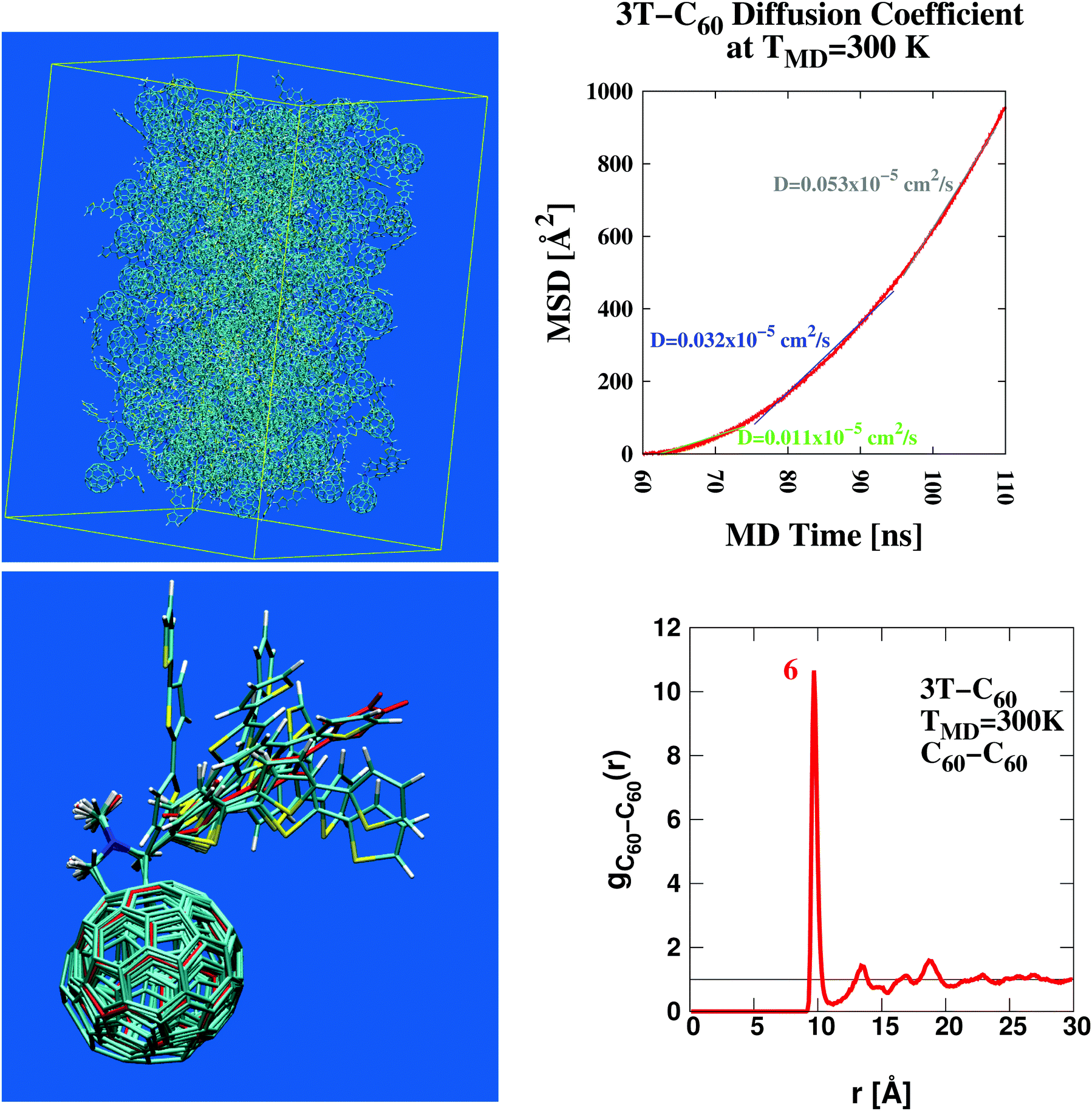
Using classical Molecular Dynamics simulation76 (MD) we study the microscopic organization of terthiophene–fullerene (3T–C60)26,27 which is considered a model system of BHJs with direct implications to various other materials used in current organic photovoltaics.2,77–80 A major design advantage with 3T–C60 is the chemical linkage between electron donor- (D) and acceptor- (A) moiety bringing together both these groups into close proximity in a well defined ratio of 1:1. This way the frequently stressed importance of intimate mixing of neat D- and A-phases81–85 is respected by design. A number of complicating factors can thus be avoided that would otherwise arise within the conventional model of BHJs established to date (see for example the schematic illustration given in Fig. 4 of ref. 85). These include, among others, structural heterogenity of unclear shape and dimension, spatial extent of individual D/A-phases, actual texture of the interface, charge carrier mobilities etc. Despite a relatively small anticipated power conversion efficiency of about 0.2% (as approximated from structurally related oligothiophene–C60 dyads86–89) this material should form an excellent basis to study fundamental processes of charge carrier creation as a function of BHJ morphology. An ensemble of 216 such dyads was simulated in a central box under periodic boundary conditions using AMBER51 (for a structural snapshot see Fig. 1, upper left panel). Such a detailed picture of the molecular organization of the photoactive layer is a hallmark of theoretical approaches such as the one pursued here. This way new insight into D/A operation can be gained from a realistic molecular perspective that otherwise appears difficult to achieve with purely experimental techniques. A self-diffusion coefficient of approximately 0.05 × 10−5 cm2 s−1 was derived from the MD trajectory (Fig. 1, upper right panel). Such a figure is indicative of a strongly viscous liquid similar to oil from sunflower seeds.90
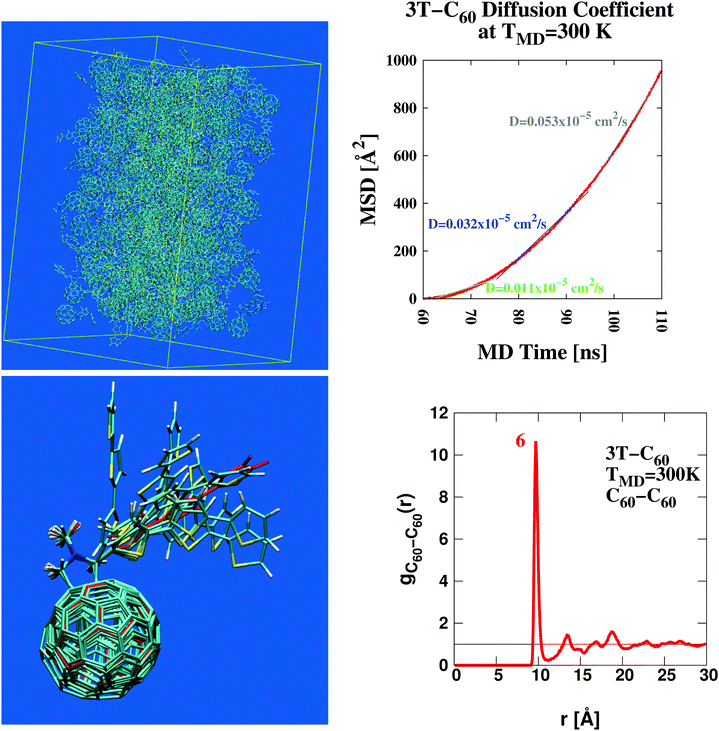 |
| | Fig. 1 Structural characterization of the photovoltaic material terthiophene–fullerene (3T–C60)26,27 from MD simulation describing an ensemble of 216 crosslinked dyads (upper left panel). Determination of the self diffusion coefficient reveals the signature of a strongly viscous liquid (upper right panel). Significant local order is seen from the radial distribution function, g(r) (lower right panel and Fig. S1, ESI†), where figures in red denote the integral over a particular peak, i.e. numbers of neighbours. Results obtained from g(r) can be used to extract bulk-like samples for in-depth analysis. A corresponding selection of 9 such samples displays considerable structural diversity as illustrated from molecular superposition of the central dyad in subgroups composed of 7 dyads (lower left panel). | |
Important insight into the molecular architecture of condensed matter is often gained from analyzing the radial distribution function,91g(r). Here we have computed a generalized form of g(r) where individual 3T–C60 units are represented by two geometric centres, one for the donor-part (3T), the other for the acceptor-part (C60). Corresponding charts are shown in Fig. 1 (lower right panel, A–A subset) and Fig. S1, ESI† (D–D and D–A subsets). Self-counts arising within the same unit were omitted. The most significant structural pattern thus obtained was (i) a complex composed of 7 D/A units with a central A-unit surrounded by 6 others all at a distance of approximately 9.75 Å, (ii) about 2 D-units approximately 6–9 Å away from a central A-part, (iii) only negligible correlation between individual D-units. Once major characteristics of the radial distribution function were identified, the evaluation was repeated now screening for particular scaffolds of 3T–C60 units most appropriate to represent key features in g(r). A selection of 9 such sets was made and central units were superimposed onto each other to assess the degree of local diversity between individual conformations (see Fig. 1 lower left panel). Especially with respect to the 3T chain a great variety of different arrangements was observed, hence the BHJ exhibits a rather rich and complex microstructure. The selected 9 sets were further refined and single bulk-representative D/A pairs isolated from the groups. An example of how such a selection would work in practice is given in Movie 1, ESI† (A-unit in red, D-unit in blue). At first all chemical linkers were neglected because—by design—they would not interfere with photo-excitation or primary electron transfer.89 Then single explicit pairs of D/A groups were extracted from the scaffold based on physical proximity (see Movie 1, ESI†). As shall be demonstrated later, this second step is still conserving all major electronic characteristics of the bulk-embedded D/A pair. All in all 13 D/A pairs were prepared and passed on for in-depth analysis (Table 1, leftmost column). Detecting common signals within this group of 13 samples would now appear to be of greatly enhanced significance because (i) all these 13 cases are bulk-representative, i.e. each of them represents frequently occurring conformations in the ensemble, (ii) structurally, there is still a great deal of variation involved, hence characteristic electronic signatures shared among all 13 samples should really highlight deeply anchored principles.
Most of the interesting properties of materials used in organic photovoltaics develop only inside the BHJ. Therefore, comparing bulk-like samples to structures predominant in the gas phase could be a route to discover essential requirements of photo-voltaic devices. Following this idea, all 13 bulk-representative D/A pairs were considered for ground state energy calculations at the PBEPBE/6-31g* level of theory62–64 which had been shown to produce reliable results at reasonable computational cost for photophysical and electronic properties of C60-based systems forming BHJs.92–94 Similarly, all 13 samples were also first minimized and then subjected to ground state energy calculations in their relaxed conformation (which is more relevant to the gas phase). Results are summarized in Table 1. Since for this quantitative comparison we had to consider identical D/A skeletons in either BHJ- or minimum-conformation, it was also of interest to analyze whether fully integral 3T–C60 dyads without removal of the linker would still exhibit similar minimum geometries, i.e. not suffer from any steric hindrance. A corresponding case study is shown in Fig. S2, ESI,† for sample 3T–C60162(case 2) revealing predominantly identical geometries hence confirming the validity of our selection/reduction procedure to simplify structural complexity.
All bulk conformations showed significantly increased ground state energies when compared to their corresponding structures in the relaxed state. Part of this discrepancy may stem from the non-perfect agreement between MM- and QM-surfaces of potential energy, although qualitatively both descriptions appear to be comparable (see Fig. S3, ESI†). Of particular note is the rather small standard deviation seen in the set of bulk conformation energies (±0.01316 Hartree, also included in Fig. 2) making the energy gap even more significant. The magnitude of the energy gap was comparable to about 5 times the photon energy.95 However, no obvious structural feature could be identified to explain this gap. For example, all 13 samples in bulk-like as well as relaxed conformation were structurally superimposed and overlays confronted to each other (see Fig. 2 upper panels and Movie 2, ESI,† for a 360° view). Apart from a sporadically appearing bulge in the 3T chain (compare for example red units in Fig. 2 and Movie 2, ESI†), little structural evidence could be provided that would allow a distinction between bulk and relaxed conformations. An important consequence of all of this is the necessary paradigm shift away from the classic picture of Franck–Condon excitation. Owing to the uphill elevation of ground state energies, primary excitation of electrons in the BHJ should follow a non-Franck–Condon pathway as graphically sketched in Fig. 2 (lower panel, off-minimum domain coloured in cyan96). Taking into account the anticipated high viscosity, we can assume that inside the BHJ individual 3T–C60 units will be considerably constrained, hence will have little conformational freedom to drift away from the BHJ-geometry. It is also for these BHJ-internal constraints that we believe that individual 3T–C60 units will be permanently “forced out” of their minimum geometries, hence the important role of morphology in BHJs eventually leading to the remarkable shift to non-Franck–Condon excitation. This has important implications for the mechanistic understanding of the BHJ such as for example the framework developed by Vandewal and coworkers11 where (i) hypothetical CT states would now no longer have to be shifted to the right on the nuclear coordinate axis, and (ii) the auxiliary construct of “vibrationally excited ground states” could be alleviated to a large extent.
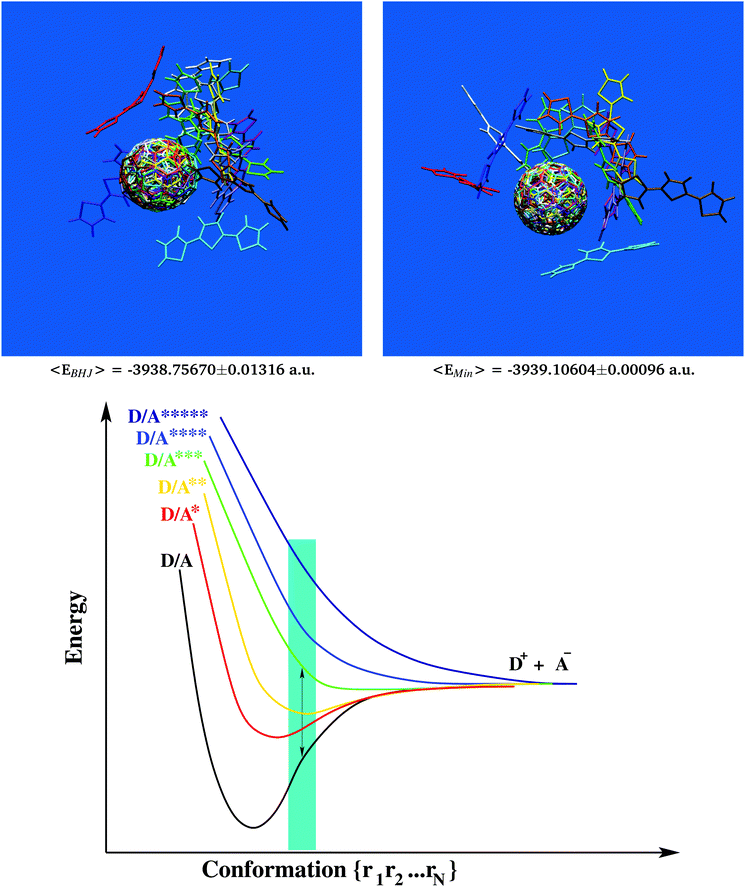 |
| | Fig. 2 Extraction of 13 characteristic bulk-like D/A pairs from the MD data following major signals of the radial distribution function. Structural superposition of D/A geometries (upper left panel) considered for in-depth analysis of electron transfer. The same set of D/A pairs displays significantly decreased ground state energies in relaxed conformation (upper right panel and Table 1). It follows that primary excitation must be of non-Franck–Condon type as graphically outlined by a simplified 2-dimensional potential energy chart for electron excitation and primary transfer in the BHJ (cyan area, lower panel). | |
3.2 Characteristic electronic properties of D/A pairs can help to explain primary charge-transfer and -separation in the BHJ
Next we computed transition dipole moments67,68 for all 13 samples in bulk-like as well as relaxed conformation and results (vector magnitudes) are summarized in Table 2 and Tables S1–S12, ESI.† Only single excitation Slater determinants were considered using the set of molecular orbitals (MO) obtained from calculation of ground state energies (see Section 3.1). Energy levels of involved key MOs are also shown in Fig. 3 and Fig. S4–S15, ESI.†
Table 2 Transition dipole moments in a.u. for the selected D/A pair 3T–C60171(case 1) in bulk conformation (above the bar values) and in relaxed conformation (below the bar values)
|
|
HOMO |
LUMO |
LUMO+1 |
LUMO+2 |
LUMO+3 |
LUMO+4 |
LUMO+5 |
LUMO+6 |
LUMO+7 |
LUMO+8 |
| LUMO |
0.262/0.075 |
|
|
|
|
|
|
|
|
|
| LUMO+1 |
0.157/0.053 |
0.053/0.016 |
|
|
|
|
|
|
|
|
| LUMO+2 |
0.076/0.100 |
0.533/0.011 |
0.312/0.008 |
|
|
|
|
|
|
|
| LUMO+3 |
0.061/0.141 |
2.729/3.492 |
3.509/3.249 |
1.492/1.251 |
|
|
|
|
|
|
| LUMO+4 |
0.065/0.044 |
2.067/1.828 |
2.171/3.052 |
3.445/3.395 |
0.588/0.021 |
|
|
|
|
|
| LUMO+5 |
0.151/0.124 |
2.893/2.961 |
2.185/2.080 |
2.838/3.347 |
0.414/0.021 |
0.747/0.012 |
|
|
|
|
| LUMO+6 |
3.187/3.362 |
0.477/0.133 |
0.610/0.203 |
0.285/0.160 |
0.534/0.509 |
0.410/0.234 |
0.632/0.471 |
|
|
|
| LUMO+7 |
1.002/0.074 |
1.388/0.111 |
1.443/0.062 |
0.459/0.082 |
1.749/0.053 |
1.070/0.017 |
0.842/0.010 |
3.911/0.282 |
|
|
| LUMO+8 |
0.052/0.055 |
0.066/0.013 |
0.222/0.006 |
0.310/0.023 |
0.228/0.025 |
0.272/0.003 |
0.046/0.008 |
0.276/0.232 |
0.100/0.174 |
|
| LUMO+9 |
0.051/0.146 |
0.566/0.013 |
0.398/0.021 |
0.369/0.019 |
0.709/0.028 |
0.545/0.015 |
0.264/0.014 |
0.537/0.519 |
1.403/0.070 |
0.788/0.019 |
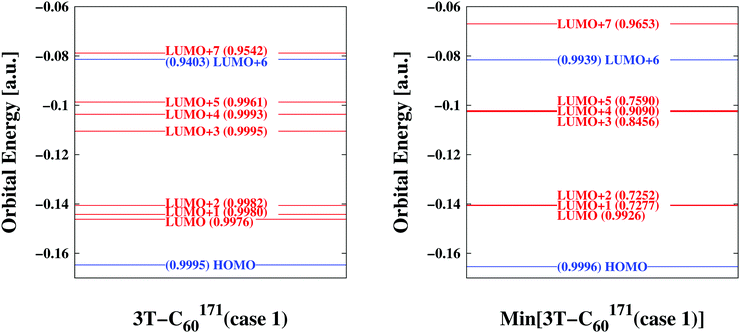 |
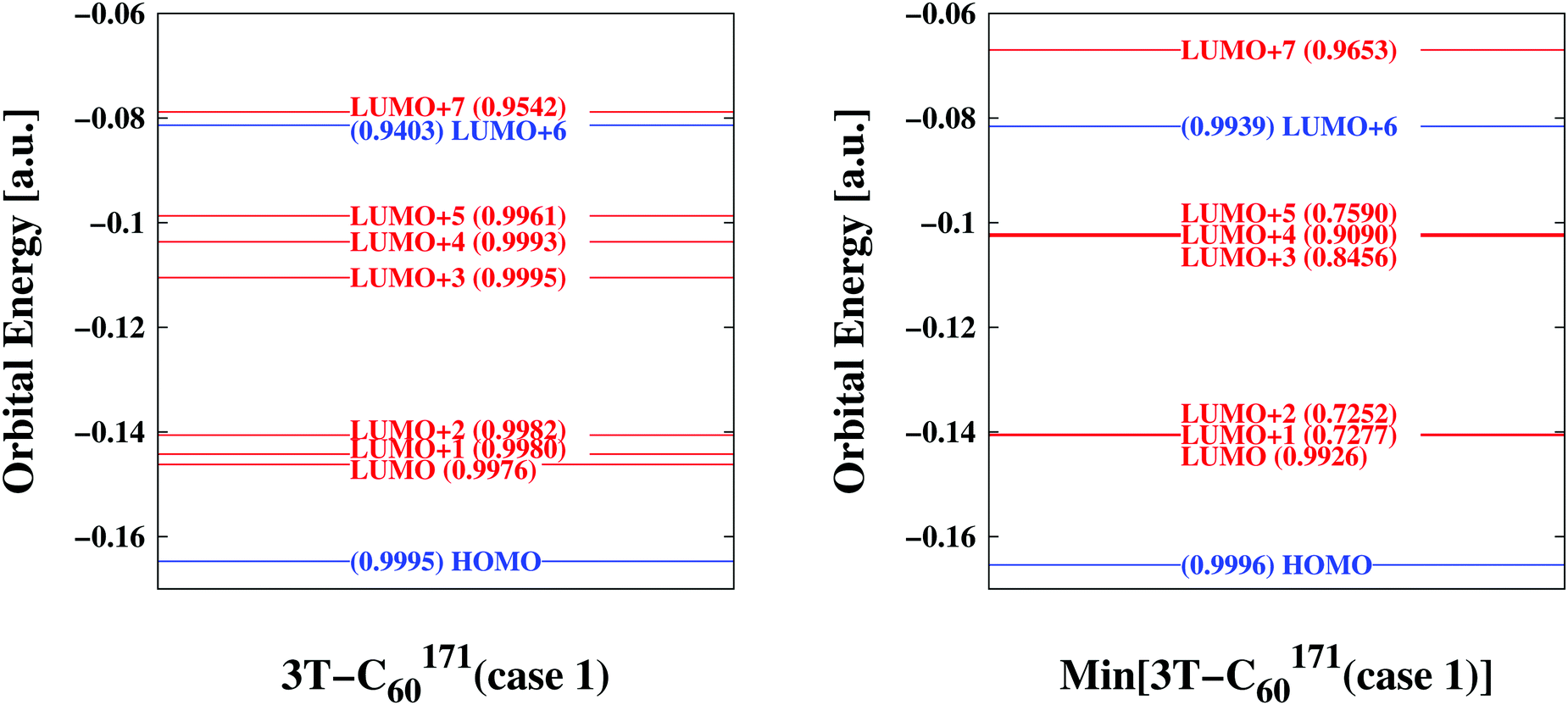
| | Fig. 3 Comparison of key orbitals involved in photo-excitation and primary electron transfer emphasizing differences/similarities between bulk conformation (left panel) and relaxed form (right panel)—sample 3T–C60171(case 1). Orbitals predominantly located on D are indicated in blue while those concentrated on A are given in red. A measure of similarity is also included in parentheses (1.0 indicating perfect agreement) where the resemblance is quantified between a particular orbital of the assembled D/A pair and its corresponding D- or A-orbital of the isolated D- or A-systems considered stand-alone. The bulk (i) distributes vacant energy levels more evenly (ii) makes accessible LUMO+7 for direct excitation into an A-orbital (also see Table 2) (iii) decouples electronic degrees of freedom (compare similarity coefficients). | |
At first we were interested in the fundamental transition of the HOMO electron. As can be seen in Fig. 3 (and Fig. S4–S15, ESI†) the HOMO is entirely located on the donor part of the D/A complex confirming its chemical mission of providing electrons. Transition dipole moments would suggest HOMO → LUMO+6 or HOMO → LUMO+7 transitions as the most likely to occur. Both would approximately fall into the energy range a single photon can provide assuming ordinary sunlight irradiation. However, only the latter is specific to the BHJ (compare above the bar values to below the bar values in column 2 of Table 2). LUMO+7 is an orbital entirely located on the A part, thus the bulk facilitates direct excitation from a D- into an A-orbital by lowering the corresponding energy level (compare levels of LUMO+7 in Fig. 3 and Fig. S4–S15, ESI†). Additional remarkable features of the BHJ are the much broader distribution of energy levels of LUMO to LUMO+5 and the significant degree of decoupling of electrons (compare for example similarity coefficients for LUMO+1, LUMO+2, LUMO+3 and LUMO+5 in Fig. 3 and Fig. S4–S15, ESI†). Thus, the combination of transition dipole moments with similarity coefficients offers novel insight into electronic degrees of freedom at the MO level of detail.
The most ineffective—yet rather probable—continuation of the electrons journey after having been excited into any of the LUMO levels, is to simply decay down to the ground state again, i.e. back to the HOMO level. This would, however, be accompanied by the emission of another photon of quasi-identical energy to the one that had been absorbed in the first place. Moreover, the re-emitted photon would appear locally and could promptly be re-absorbed by any of the surrounding A-units, thus the molecular architecture of the BHJ guarantees efficient harvesting of all the arriving photons either by primary or induced follow-up excitations. On the other hand, decaying down to any of the intermediate LUMO levels would again mean perfect charge separation because all of these LUMOs are entirely located on the A part (see similarity coefficients of LUMO to LUMO+5 in Fig. 3 and Fig. S4–S15, ESI†). Provided the coupling to vibrational states remains small, additional, less energetic photons would be co-released, hence additional re-absorptions could take place at adjacent A-units now promoting energetically smaller transitions according to the pattern given in Table 2. Along these lines, the aforementioned broadening of energy levels in the BHJ would be a welcome side-effect as it facilitates repercussions of transitions of smaller scale. All in all, the picture emerging is that of a rather rich and complex interplay of all various kinds of excitations and triggered emissions occurring all throughout the BHJ. The remarkable finding of CT absorptions at energies below the optical gap7,29,97 would seamlessly fit into such a conceptual understanding of the BHJ. It will be interesting to see whether the scheme presented here will also be compatible to the recently reported “hot” dissociation channels based on high-energy charge-separated (CS) states.98
Additional comparisons of key orbitals were made in order to verify the extraction procedure of bulk-representative D/A pairs outlined above (also see Movie 1, ESI†). To assess the neglected influence of neighbouring 3T–C60 units, the finally isolated D/A pair was considered embedded in the matrix of all its direct neighbours and corresponding MOs were derived from ground state energy calculations including a set of atomic partial charges from adjacent 3T–C60 units forming the matrix. Results for sample 3T–C60171(case 1) are shown in Fig. S16, ESI.† MO levels have uniformly shifted to higher energies and the gap between LUMO+7 and LUMO+6 has slightly increased but all major characteristics of the involved key orbitals have been upheld (compare to Fig. 3), thus the reduction to single isolated D/A pairs appears justified.
3.3 Continued charge separation is hyper-fast and barrierless
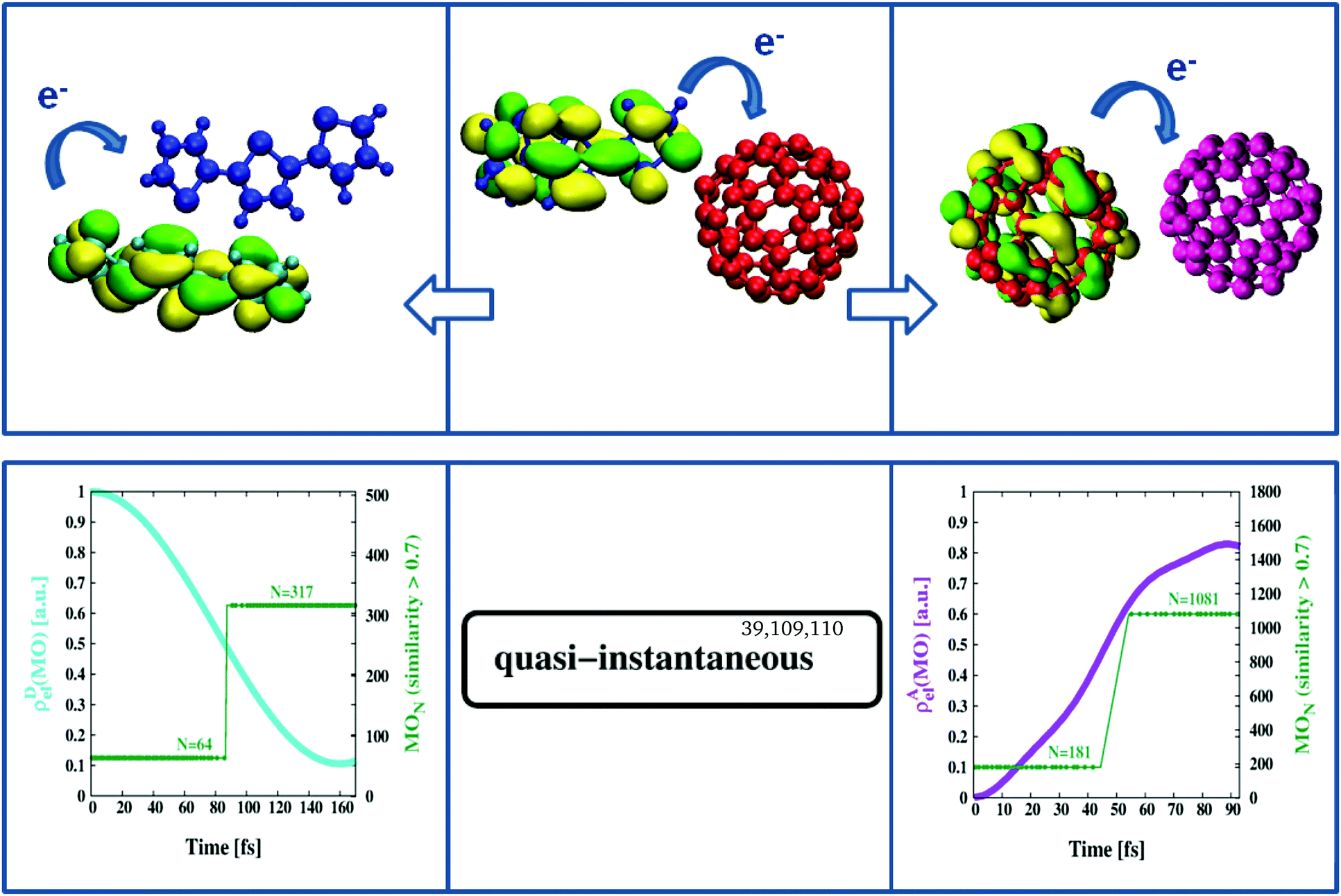
We have recently proposed an efficient method to study electron transfer on the computer.75 As the methodology is well established, we refer the interested reader to ref. 99. Briefly, MOs of two non-interacting systems, e.g. an isolated D- and corresponding A-structure in the geometry of the D/A complex but considered stand-alone, are extended with redundant coordinates (zeros only) to formally include the missing part (A or D) and examined for cross-reactivity by a combined Hamiltonian that describes both systems (A/D) in fully coupled detail. The focus is on changes in MO composition over time, in particular whether formally extended MO sections (just zeros at start-up) become occupied (non-zero) in the course of the calculation. The latter is taken as indication of electron transfer based on the type of coupling the combined Hamiltonian can provide. In a sense the approach is similar to a time-resolved version of MO-theory100 where the A- and D-structures formally replace the parts of atomic orbitals on both sides of the MO diagram and the calculation carries out the placement of electrons into MO levels. For example, in Fig. 4 we see the time evolution of certain key orbitals responsible for the continuation of charge separation once primary electron excitation and transfer has taken place. Thus we start from a negatively charged [C60]− (red in Fig. 4), and a positively charged [3T]+ (blue in Fig. 4), and combine them with potential interaction partners from the BHJ. These will be another C60-unit (purple in Fig. 4) and another 3T-unit (cyan in Fig. 4) both in neutral charge state and the geometrical arrangement typical of the BHJ. Combined Hamiltonians will then act on either pair to probe the tendency to pass on the extra charge. Here the strength of the present multiscale approach becomes particularly evident since it allows to respect the morphology of the BHJ in two ways, (i) by selecting D/A pairs representative of the BHJ and (ii) by including adjacent 3T–C60 units in the form of background charges characteristic of the bulk. Of particular note is the inclusion of the charge-transferred part to the background charges, i.e. a [3T]+ unit when [C60]− is considered for second stage electron transfer, and vice versa, a [C60]− unit when [3T]+ is considered for opposite promotion of the electron hole. This way the frequently mentioned strongly bound charge-transfer complex can be accounted for, at least within the limitations of the method. Since all MOs have been extended to include D- as well as A-sections, we can compute individual fractional electron densities, ρD,Ael(MO), specific to a particular MO that are entirely due to the D- or the A-part. Thus when tracking the HOMO in [C60]− (i.e. the extra electron of D in this particular case), ρAel(MO) will initially be zero and show an increasing trend only in case the partner C60 (purple, A in this particular case) is able to accept the extra electron (see purple graph in Fig. 4 lower right panel). Similarity coefficients can then be used to identify the actual MO the extra electron has been transferred to. For example, from Fig. 4 (lower right panel) we learn that the HOMO of D (MO number 181) slowly morphs into MO number 1081 which is the LUMO of A (each C60 is described by 900 basis functions and the computation is for the β density which includes the extra electron). This process is hyper-fast (terminates within approximately 100 fs) and occurs barrierless.101 Similarly, following the HOMO in 3T implies that ρDel(MO) should change from 1 to 0 in case the acceptor-unit ([3T]+ in this particular case) is able to incorporate the electron. In terms of MOs this shifts “the hole” from the LUMO of A (MO number 317) to the HOMO of D (MO number 64) again barrierless within approximately 200 fs (see cyan graph in Fig. 4 lower left panel). This must still be considered very fast, but is considerably slower than the opposite transfer of an electron from [C60]− to another C60, hence might be limiting photoconversion efficiency as observed in related materials.86,88,102,103 In general, the dynamics reported here appears to agree well with previously published transfer rates obtained from spectroscopic measurements.39,104–108 The more severe limitation, however, is a less intertwined network of individual 3T chains lacking universal overlap to promote the transfer of the “electron hole”, hence a purely structural constraint. While finding suitable candidates of C60–C60 pairs likely to pass on an electron was always possible in numerous ways, identifying a suitable pair of 3T units properly aligned for “electron hole transport” to potentially occur required elaborate procedures of manual screening. Thus the remarkable difference in RDF signals between A–A and D–D pairs did manifest itself even here, in the domain of electrons and molecular orbitals (compare Fig. 1 lower right panel with Fig. S1, ESI†). The obvious consequence would be that many of the initiated pathways of “electron hole transport” will eventually get stuck and not reach the terminal layer of the device electrode.103 Again, the combination of time-resolved computations of electron transfer together with the periodic consideration of similarity coefficients has been demonstrated to be a powerful technique that offers specific insight into fundamental processes characteristic of a particular type of photoactive material.
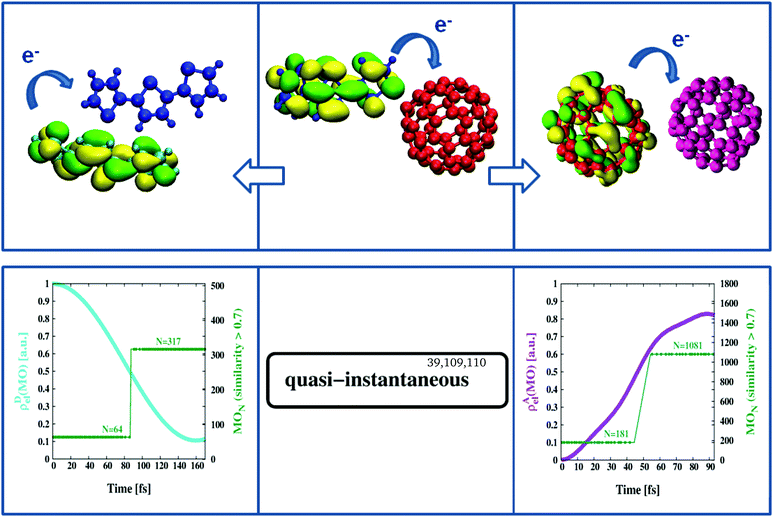 |
| | Fig. 4 Second stage charge separation. Once primary electron transfer has taken place (middle panel) the negatively charged [C60]− (red) can pass on its excess electron to a second C60 unit (purple) within approximately 100 fs (right panels). At the same time the void in the positively charged [3T]+ (blue) can be refilled from a second, neutral, 3T unit (cyan) within about 200 fs (left panels). | |
BHJs are frequently described as semiconductors using Fermi–Dirac statistics. All these approaches relate the open-circuit voltage, i.e. the maximum voltage available from a solar cell, to the HOMO/LUMO gap with empirical corrections accounting for various effects.1,6,9,10,13,28,30 Despite its widespread use in performance assessment, the restricted focus on bandgap tuning has been reported critical for optimizing power conversion efficiency.13,15,28 Sensitive issues are experimental limitations in determining HOMO/LUMO offsets from cyclic voltammetry,6,13,15,28 the actual role of weakly/strongly bound charge transfer states,1,9,11,29,30 and the influence of HOMO/LUMO offsets between D and A moieties.1,2,9,29 The liquid state description presented here is an alternative way of looking at BHJs, where frequently occurring conformations of D/A pairs are examined in terms of electronic rearrangements characteristic of the bulk. Ideally, the initially extracted D/A complex would next be augmented with adjacent D- and A-units in a cluster-like expansion thereby increasingly approaching bulk properties at mesoscopic scale. Unfortunately the associated computational cost of explicit electronic structure calculations pursuing such a cluster expansion is quickly becoming prohibitive,98 hence only approximate treatments in the form of background charge distributions can be accomplished to date (see for example Fig. 4 and Fig. S16, ESI†). However, the detailed insight into key orbitals (e.g.Fig. 3), the systematic analysis of relevant electronic transitions (e.g.Table 2), the clear cut separation into primary electron transfer and second stage charge separation (e.g.Fig. 4) together with the systematic application of different methods appropriate for different length- and time-scales, appear to make such an approach a worthwhile endeavour.
4 Conclusion
We demonstrate significant local order for pairs of C60 in the structural organization of terthiophene–fullerene26,27 using Molecular Dynamics.76 A multiscale approach is used to characterize essential morphological features of the bulk heterojunction. The aim is to provide an accurate—still feasible—description of the relevant physics governing fundamental processes and molecular organization of the photoactive layer. For a bulk-representative set of donor/acceptor pairs we observe significantly increased ground state energies as opposed to their corresponding forms in relaxed conformation characteristic of the gas phase. The important conceptual consequence for the bulk heterojunction11,29,97 is that of non-Franck–Condon type of electron excitation and primary transfer. A range of characteristic changes affecting the electronic properties of bulk-like donor/acceptor pairs are identified, all of them pointing towards an auxiliary role of the bulk heterojunction in facilitating charge-transfer and -separation. Of particular importance is the promotion of electrons/holes to adjacent units, a process identified here to be hyper-fast and barrierless. A notable limitation is the depletion of network contacts for terthiophene-chains leading to a likely scenario of numerous pathways of “electron hole transport” ending in deadlock. The applied multiscale approach demonstrates its potential for examining the effective charge carrier dynamics resulting from the inner morphology of the active layer. This work also illustrates the critical need for additional structural and mechanistic studies of the molecular operation of bulk heterojunctions to foster our understanding and improve the design of next-generation photovoltaic devices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Felix Plasser (University of Vienna) for helpful discussions on evaluating MO similarity. The authors acknowledge the TU Wien University Library for financial support through its Open Access Funding Program.
References
- M. Scharber and N. S. Sariçiftçi, Prog. Polym. Sci., 2013, 38, 1929–1940 CrossRef CAS PubMed.
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- H. Hoppe and N. S. Sariçiftçi, J. Mater. Chem., 2006, 16, 45–61 RSC.
- X. Yang and J. Loos, Macromolecules, 2007, 40, 1353–1362 CrossRef CAS.
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS PubMed.
- P. Parkinson, J. Lloyd-Hughes, M. B. Johnston and L. M. Herz, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 115321 CrossRef.
- C. J. Brabec, G. Zerza, G. Cerullo, S. D. Silvestri, S. Luzzati, J. C. Hummelen and N. S. Sariçiftçi, Chem. Phys. Lett., 2001, 340, 232–236 CrossRef CAS.
- X. Gong, M. Tong, F. G. Brunetti, J. Seo, Y. Sun, D. Moses, F. Wudl and A. J. Heeger, Adv. Mater., 2011, 23, 2272–2277 CrossRef CAS PubMed.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
- K. Vandewal, S. Albrecht, E. T. Hoke, K. R. Graham, J. Widmer, J. D. Douglas, M. Schubert, W. R. Mateker, J. T. Bloking, G. F. Burkhard, A. Sellinger, J. M. J. Fréchet, A. Amassian, M. K. Riede, M. D. McGehee, D. Neher and A. Salleo, Nat. Mater., 2014, 13, 63–68 CrossRef CAS PubMed.
-
Y. Galagan and R. Andriessen, Organic Photovoltaics: Technologies and Manufacturing, Third Generation Photovoltaics, InTech, 2012 Search PubMed.
-
C. N. Hoth, P. Schilinsky, S. A. Choulis, S. Balasubramanian and C. J. Brabec, Applications of Organic and Printed Electronics, Springer, New York, 2013 Search PubMed.
- J. D. Servaites, S. Yeganeh, T. J. Marks and M. A. Ratner, Adv. Funct. Mater., 2010, 20, 97–104 CrossRef CAS.
- N. E. Jackson, B. M. Savoie, T. J. Marks, L. X. Chen and M. A. Ratner, J. Phys. Chem. Lett., 2015, 6, 77–84 CrossRef CAS PubMed.
- O. V. Mikhnenko, H. Azimi, M. Scharber, M. Morana, P. W. M. Blom and M. A. Loi, Energy Environ. Sci., 2012, 5, 6960–6965 CAS.
- T. A. Shastry, S. C. Clark, A. J. E. Rowberg, K. A. Luck, K.-S. Chen, T. J. Marks and M. C. Hersam, Adv. Energy Mater., 2016, 6, 1501466 CrossRef.
- B. Ray and M. A. Alam, Sol. Energy Mater. Sol. Cells, 2012, 99, 204–212 CrossRef CAS.
- L. A. Perez, K. W. Chou, J. A. Love, T. S. van der Poll, D.-M. Smilgies, T.-Q. Nguyen, E. J. Kramer, A. Amassian and G. C. Bazan, Adv. Mater., 2013, 25, 6380–6384 CrossRef CAS PubMed.
- N. D. Treat and M. L. Chabinyc, Annu. Rev. Phys. Chem., 2014, 65, 59–81 CrossRef CAS PubMed.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. McCulloch, C.-S. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- U. Zhokhavets, G. Gobsch, H. Hoppe and N. S. Sariçiftçi, Synth. Met., 2004, 143, 113–117 CrossRef CAS.
- D. A. R. Barkhouse, R. Debnath, I. J. Kramer, D. Zhitomirsky, A. G. Pattantyus-Abraham, L. Levina, L. Etgar, M. Grätzel and E. H. Sargent, Adv. Mater., 2011, 23, 3134–3138 CrossRef CAS PubMed.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- Y. Zheng and J. Xue, Polym. Rev., 2010, 50, 420–453 CrossRef CAS.
- K. Lewandowska, W. Bednarski, G. Milczarek, S. Waplak, A. Graja, E. Y. Park, T.-D. Kim and K.-S. Lee, Synth. Met., 2011, 161, 1640–1645 CrossRef CAS.
- B. Barszcz, B. Laskowska, A. Graja, E. Y. Park, T.-D. Kim and K.-S. Lee, Chem. Phys. Lett., 2009, 479, 224–228 CrossRef CAS.
- G. Garcia-Belmonte, A. Guerrero and J. Bisquet, J. Phys. Chem. Lett., 2013, 4, 877–886 CrossRef CAS PubMed.
- M. A. Loi, S. Toffanin, M. Muccini, M. Forster, U. Scherf and M. Scharber, Adv. Funct. Mater., 2007, 17, 2111–2116 CrossRef CAS.
- C. Deibel, T. Strobel and V. Dyakonov, Adv. Mater., 2010, 22, 4097–4111 CrossRef CAS PubMed.
- M. A. Ratner, Nat. Chem., 2015, 7, 181 CrossRef PubMed.
- E. Jankowski, H. S. Marsh and A. Jayaraman, Macromolecules, 2013, 46, 5755–5785 CrossRef.
- V. Rühle, J. Kirkpatrick and D. Andrienko, J. Chem. Phys., 2010, 132, 134103 CrossRef PubMed.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 12057–12067 CrossRef CAS PubMed.
- T. Liu and A. Troisi, Adv. Mater., 2013, 25, 1038–1041 CrossRef CAS PubMed.
- D. L. Cheung and A. Troisi, J. Phys. Chem. C, 2010, 114, 20479–20488 CAS.
- S. Klaiman, E. V. Gromov and L. S. Cederbaum, Phys. Chem. Chem. Phys., 2014, 16, 13287–13293 RSC.
- F. Gajdos, H. Oberhofer, M. Dupuis and J. Blumberger, J. Phys. Chem. Lett., 2013, 4, 1012–1017 CrossRef CAS PubMed.
- S. M. Falke, C. A. Rozzi, D. Brida, M. Maiuri, M. Amato, E. Sommer, A. D. Sio, A. Rubio, G. Cerullo, E. Molinari and C. Lienau, Science, 2014, 344, 1001–1005 CrossRef CAS PubMed.
-
Multiscale Modelling of Organic and Hybrid Photovoltaics, ed. D. Beljonne and J. Cornil, Springer, Berlin, Heidelberg, 2014 Search PubMed.
- P. J. Brown, D. S. Thomas, A. Köhler, J. S. Wilson, J.-S. Kim, C. M. Ramsdale, H. Sirringhaus and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 064203 CrossRef.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. B. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- J. J. P. Stewart, J. Comput. Chem., 1989, 10, 209–220 CrossRef CAS.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- A. Jakalian, B. L. Bush, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2000, 21, 132–146 CrossRef CAS.
- S. Höfinger and F. Zerbetto, J. Phys. Chem. A, 2003, 107, 11253–11257 CrossRef.
- S. Höfinger and F. Zerbetto, Theor. Chem. Acc., 2004, 112, 240–246 CrossRef.
- D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo, K. M. Merz-Jr., A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. D. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- W. F. van Gunsteren and H. J. C. Berendsen, Mol. Phys., 1977, 34, 1311–1327 CrossRef CAS.
- H. C. Andersen, J. Chem. Phys., 1980, 72, 2384–2393 CrossRef CAS.
- S. C. Harvey, R. K.-Z. Tan and T. E. Cheatham III, J. Comput. Chem., 1998, 19, 726–740 CrossRef CAS.
- A. Einstein, Ann. Phys., 1905, 322, 549–560 CrossRef.
- M. Smoluchowski, Ann. Phys., 1906, 326, 756–780 CrossRef.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
-
H. B. Bröker, J. Campbell, R. Cunningham, D. Denholm, G. Elber, R. Fearick, C. Grammes, L. Hart, L. Heckingu, P. Juhász, T. Koenig, D. Kotz, E. Kubaitis, R. Lang, T. Lecomte, A. Lehmann, A. Mai, B. Märkisch, E. A. Merritt, P. Mikulík, C. Steger, S. Takeno, T. Tkacik, J. V. der Woude, J. R. V. Zandt, A. Woo and J. Zellner, gnuplot 4.6 – An Interactive Plotting Program, http://sourceforge.net/projects/gnuplot, 2014.
-
M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Clarendon Press, Oxford, 1989 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.02, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
-
http://https://en.wikipedia.org/wiki/Dot_product#Complex_vectors
.
-
http://https://en.wikipedia.org/wiki/Transition_dipole_moment
.
- F. Plasser, M. Wormit and A. Dreuw, J. Chem. Phys., 2014, 141, 024106 CrossRef PubMed.
- S. Höfinger and M. Wendland, Int. J. Quantum Chem., 2002, 86, 199–217 CrossRef.
- E. Schrödinger, Ann. Phys., 1926, 79, 361–376 CrossRef.
- J. S. Graves and R. E. Allen, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13627–13633 CrossRef.
- N. Watanabe and M. Tsukada, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 2914–2923 CrossRef CAS.
- H. W. van der Hart, M. A. Lysaght and P. G. Burke, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 76, 043405 CrossRef.
- A. Acocella, G. A. Jones and F. Zerbetto, J. Phys. Chem. A, 2006, 110, 5164–5172 CrossRef CAS PubMed.
- S. Höfinger, A. Acocella, S. C. Pop, T. Narumi, K. Yasuoka, T. Beu and F. Zerbetto, J. Comput. Chem., 2012, 33, 2351–2356 CrossRef PubMed.
-
M. Karplus, Nobel Lecture, http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2013/karplus-lecture.pdf, 2013.
- R. A. J. Janssen, N. S. Sariçiftçi and A. J. Heeger, J. Chem. Phys., 1994, 100, 8641 CrossRef CAS.
- P. A. van Hal, E. H. A. Beckers, S. C. J. Meskers, R. A. J. Janssen, B. Jousselme, P. Blanchard and J. Roncali, Chem. – Eur. J., 2002, 8, 5415–5429 CrossRef CAS.
- W. J. D. Beenken, Chem. Phys., 2009, 357, 144–150 CrossRef CAS.
- N. Banerji, S. Cowan, M. Leclerc, E. Vauthey and A. J. Heeger, J. Am. Chem. Soc., 2010, 132, 17459–17470 CrossRef CAS PubMed.
-
A. J. Palkar and T. He, Donor–acceptor DYAD compounds in photovoltaics, http://https://www.lens.org/lens/patent/US_9006567_B2/fulltext, 2015.
- A. Cravino and N. S. Sariçiftçi, J. Mater. Chem., 2002, 12, 1931–1943 RSC.
- D. M. Guldi, C. Luo and A. Swartz, J. Org. Chem., 2002, 67, 1141–1152 CrossRef CAS PubMed.
- J.-F. Nierengarten, Sol. Energy Mater. Sol. Cells, 2004, 83, 187–199 CrossRef CAS.
- T. L. Benanti and D. Venkataraman, Photosynth. Res., 2006, 87, 73–81 CrossRef CAS PubMed.
- N. Negishi, K. Yamada, K. Takimiya, Y. Aso, T. Otsubo and Y. Harima, Chem. Lett., 2003, 32, 404–405 CrossRef CAS.
- Y. Murata, M. Suzuki and K. Komatsu, Org. Biomol. Chem., 2003, 1, 2624–2625 CAS.
- T. Nishizawa, K. Tajima and K. Hashimoto, Nanotechnology, 2008, 19, 424017 CrossRef PubMed.
- T. Otsubo, Y. Aso and K. Takimiya, J. Mater. Chem., 2002, 12, 2565–2575 RSC.
- E. E. Perez, A. A. Carelli and G. H. Crapiste, J. Food Eng., 2011, 105, 180–185 CrossRef CAS.
- P. Wernet, D. Nordlund, U. Bergmann, M. Cavalleri, M. Odelius, H. Ogasawara, L. A. Näslund, T. K. Hirsch, L. Ojamäe, P. Glatzel, L. G. M. Pettersson and A. Nilsson, Science, 2004, 304, 995–999 CrossRef CAS PubMed.
- H. Wang, Y. He, Y. Li and H. Su, J. Phys. Chem. A, 2012, 116, 255–262 CrossRef CAS PubMed.
- X. Qian, P. Umari and N. Marzari, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 245105 CrossRef.
- K. Saïl, G. Bassou, M. H. Gafour and F. Miloua, J. Exp. Theor. Phys., 2015, 121, 1015–1026 CrossRef.
-
http://www.pveducation.org/pvcdrom/properties-of-sunlight/energy-of-photon
.
- In principle the domain of vertical transitions (cyan) could also be shifted to the left of the minimum region on the conformation axis. However this would imply an increased penetration of electron clouds, thus should become visible in a drop in similarity coefficients in Fig. 3 where, however, the opposite is seen. Additional considerations for essential methods requirements are outlined in Fig. S3, ESI†.
- K. Vandewal, K. Tvingstedt, A. Gadisa, O. Inganäs and J. V. Manca, Nat. Mater., 2009, 8, 903–909 CrossRef PubMed.
- B. M. Savoie, A. Rao, A. A. Bakulin, S. Gelinas, B. Movaghar, R. H. Friend, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 2014, 136, 2876–2884 CrossRef CAS PubMed.
- A. Acocella, G. A. Jones and F. Zerbetto, J. Phys. Chem. B, 2010, 114, 4101–4106 CrossRef CAS PubMed.
-
R. S. Mulliken, Nobel Lecture, http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1966/mulliken-lecture.pdf, 1966.
- D. A. Vithanage, A. B. Matheson, V. Pranculis, G. J. Hedley, S. J. Pearson, V. Gulbinas, I. D. W. Samuel and A. Ruseckas, J. Phys. Chem. C, 2017, 121, 14060–14065 CAS.
- P. V. Kamat, J. A. Christians and J. G. Radich, Langmuir, 2014, 30, 5716–5725 CrossRef CAS PubMed.
- D. J. Coutinho, G. C. Faria, D. T. Balogh and R. M. Faria, Sol. Energy Mater. Sol. Cells, 2015, 143, 503–509 CrossRef CAS.
- S. Cook, R. Katoh and A. Furube, J. Phys. Chem. C, 2009, 113, 2547–2552 CAS.
- I.-W. Hwang, D. Moses and A. J. Heeger, J. Phys. Chem. C, 2008, 112, 4350–4354 CAS.
- J. Piris, T. E. Dykstra, A. A. Bakulin, P. H. M. van Loosdrecht, W. Knulst, M. T. Trinh, J. M. Schins and L. D. A. Siebbeles, J. Phys. Chem. C, 2009, 113, 14500–14506 CAS.
- R. A. Marsh, J. M. Hodgkiss, S. Albert-Seifried and R. H. Friend, Nano Lett., 2010, 10, 923–930 CrossRef CAS PubMed.
- S. Gélinas, A. Rao, A. Kumar, S. L. Smith, A. W. Chin, J. Clark, T. S. van der Poll, G. C. Bazan and R. H. Friend, Science, 2014, 343, 512–516 CrossRef PubMed.
- M. T. Hassan, T. T. Luu, A. Moulet, O. Raskazovskaya, P. Zhokhov, M. Garg, N. Karpowicz, A. M. Zheltikov, V. Pervak, F. Krausz and E. Goulielmakis, Nature, 2016, 530, 66–70 CrossRef PubMed.
- A. Sommer, E. M. Bothschafter, S. A. Sato, C. Jakubeit, T. Latka, O. Razskazovskaya, H. Fattahi, M. Jobst, W. Schweinberger, V. Shirvanyan, V. S. Yakovlev, R. Kienberger, K. Yabana, N. Karpowicz, M. Schultze and F. Krausz, Nature, 2016, 534, 86–90 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Radial distribution functions, g(r), for C60–3T pairs (A–D) and 3T–3T pairs (D–D); comparison of minimum structures for the reduced D/A form versus full integral form of entire 3T–C60 units; quality assessment of the employed MM description and illustration of MM/QM surfaces of potential energies; 12 additional samples of g(r)-derived D/A pairs in bulk as well as relaxed conformation analyzing transition dipole moments and related key orbitals; extended analysis of key orbitals in sample 3T–C60171(case 1) including direct neighbour units via a background charge distribution; two movie clips in avi format representing the selection of bulk-representative D/A pairs and a 360° view of superimposed structures in bulk-like as well as relaxed conformation. See DOI: 10.1039/c7cp08435h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *bc,
Ernst
Haunschmid
b,
Sergiu C.
Pop
a,
Tetsu
Narumi
d,
Kenji
Yasuoka
e,
Masato
Yasui
f and
Francesco
Zerbetto
a
*bc,
Ernst
Haunschmid
b,
Sergiu C.
Pop
a,
Tetsu
Narumi
d,
Kenji
Yasuoka
e,
Masato
Yasui
f and
Francesco
Zerbetto
a