
Experimental and theoretical investigation of fluorescence solvatochromism of dialkoxyphenyl-pyrene molecules†

Abstract
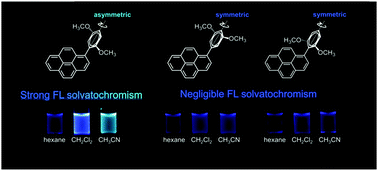
We investigated the fluorescence properties of dialkoxyphenyl-pyrene molecules experimentally as well as theoretically. Our experiments confirmed fluorescence solvatochromism in 2,5-dimethoxyphenyl-pyrene and, in contrast there was no significant solvent-effect on the emission properties of the isomers, 3,5- and 2,6-dimethoxyphenyl-pyrene. This clear difference in the solvent-dependence would reflect the difference in character of the excited-state between the isomers, which differ only in the substitution positions of the two methoxy groups. The positional effects of the di-substituted molecules are successfully explained theoretically by the topologies of the highest occupied molecular orbital of the phenyl group that are governed by the relative positions of the two substituents, though it is somewhat contradictory to the meta-effect for the mono-substituted molecules. Theoretical calculations were also used to analyze the character of the excited states; 2,5-dimethoxyphenyl-pyrene alone exhibited an intramolecular charge transfer character for the excited state, which was responsible for the solvatochromism effect. The dynamics of the excited states were analyzed using time-resolved fluorescence measurements, in which a characteristic increase of the fluorescence intensity was observed for 2,5-dialkoxyphenyl-pyrene; this observation was supported by the theoretical calculations as well.
- This article is part of the themed collection: Complex molecular systems: supramolecules, biomolecules and interfaces
 Please wait while we load your content...
Please wait while we load your content...

