
Role of surface adsorption in tuning the properties of black phosphorus
W. P.
Xu
and
H.
Xu
 *
*
Department of Physics, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: xu.h@sustc.edu.cn
First published on 30th November 2017
Abstract
H2O and O2 are believed to be key factors that influence the structural stability of black phosphorus (BP) in ambient conditions. In this work, the interactions of H2O and/or O2 with BP are investigated using first-principles calculations. The results indicate that water molecules prefer to adsorb on the BP surface and form a six-member water ring. The dissociation barrier of O2 is significantly reduced in the presence of H2O, which dramatically promotes the degradation of BP. Moreover, the introduction of O2 also facilitates the adsorption of water on the surface. The effects of H2O and/or O2 on the quasiparticle band gap and exciton binding energy of BP are also investigated. The results suggest that water adsorption has only a slight influence on the electronic properties and exciton binding energy, while O2 adsorption causes obvious changes in the properties of BP, which results in a direct-to-indirect band gap transition in BP.
1. Introduction
Similar to graphite, the interlayer of black phosphorus (BP) is connected by weak van der Waals interactions. Therefore, it is feasible to prepare few-layer BP samples by exfoliation. Single layer BP, which is also known as phosphorene, is a promising candidate for next-generation photoelectric devices because of its unique electronic structure, high carrier mobility, current on–off ratio and large exciton binding energy (EBE).1–12 However, an inevitable issue that is encountered in various applications is how to avoid the oxidation of BP. The lone pairs of electrons on the exfoliated BP make it easy to oxidize, which leads to the irreversible degradation of BP into PxOy compounds when it is exposed to the ambient environment.13–19The interactions of O2 and/or water with BP have recently received considerable academic attention. Theoretically, Ziletti et al.20 found that O2 interacts strongly with the BP surface in the dissociative form with an energy release of 2.08 eV per O atom. The reaction barrier in the process of O2 dissociation is only 0.15 eV, and the dissociated O2 serves as anchors to improve the adsorption ability of water by forming hydrogen bonds.20 In addition, the calculations performed by Wang et al.21 indicated that O2 can spontaneously dissociate on a BP surface at room temperature, while water only weakly binds to pristine BP. Experimentally, the synergetic effect of O2 and H2O in the oxidation of BP has been confirmed by X-ray photoelectron spectroscopy (XPS) measurements.22 Moreover, the site-selective oxidation of BP after exposure to high-purity O2 or H2O has been systematically studied using XPS and transmission electron microscopy (TEM) observations,23 and an oxidation mechanism involving a multistep reaction has been proposed. Despite intensive efforts, the interplay between water molecules on the BP surface and the synergetic effect of O2 and H2O during BP oxidation at the atomic level remain unclear. More importantly, it is desirable to investigate the influence of H2O and/or O2 adsorption on the electronic properties of BP, especially on the quasiparticle (QP) band gap and EBEs.
2. Methods
First-principles calculations were performed using the Vienna Ab initio Simulation Package.24,25 The projector augmented wave (PAW)26 method and Perdew–Burke–Ernzerhof (PBE)27 functional were employed. The wave functions were expanded in a plane wave basis that was truncated at an energy cutoff of 450 eV. The one-shot GW approach (known as G0W0)28 was applied to yield accurate band gaps, and the QP band structures were calculated using a set of maximally localized Wannier functions.29 The excitonic effects were studied by employing the Bethe–Salpeter equation (BSE),30,31 in which the ten highest valence bands and the ten lowest conduction bands were included. In the GW-BSE calculations, the response functions of energy cutoff were set to be 200 eV. The QP band structures and optical absorption spectra were obtained using a spatial separation of 22 Å between the neighboring slabs. The semi-empirical DFT-D3 correction was included to evaluate the van der Waals (vdW) interactions.32 All the parameter that were used have been carefully checked to ensure the convergence of the calculations.3. Results and discussion
We first studied the adsorption behavior of H2O molecules on the BP surface. To avoid the artificial interactions between repeating slabs, we performed calculations using a p(6 × 6) supercell. The calculated results show that both H atoms in the H2O molecule point toward the surface P atoms as shown in Fig. 1(a). The adsorption energy per H2O is −0.086 eV, and it is further reduced to −0.185 eV by considering the vdW interactions, which is in excellent agreement with prior results.20,21,33 To study the interactions between water molecules, more H2O molecules were included. The results clearly indicated that water molecules are likely to attract one another through hydrogen bonding and form a ring structure when there are at least three H2O molecules (see Fig. 1(b)). In this ring configuration, each water molecule forms one hydrogen bond. In addition, there is one hydrogen atom in each H2O molecule that points toward the surface P atoms. This type of configuration can reduce the total energy of the system. The corresponding average adsorption energies per H2O with and without the vdW interactions are −0.283 eV and −0.392 eV, respectively. Considering the vdW effect on the adsorption energy, we found that the average adsorption energy per H2O is reduced by approximately 0.1 eV when the vdW correction is considered. Therefore, the vdW correction cannot be neglected, and it was included in all subsequent calculations. The influence of the vdW interactions on the average adsorption energy was determined and is listed in Table 1. | ||
| Fig. 1 The adsorption of (a) a single H2O molecule, (b) three H2O molecules and (c) six H2O molecules on the p(6 × 6) BP surface. | ||
When the number of H2O molecules further increases, the H2O molecules still tend to form a ring structure with more intermolecular hydrogen bonds. In the H2O ring, most non-hydrogen-bonded H atoms point toward the BP surface. However, several H atoms point upwards, which could reduce the repulsive forces between H atoms and further reduce the total energy. The six-member ring is an example: six hydrogen atoms in the H2O molecules form a ring through hydrogen bonds, four hydrogen atoms in the H2O molecules point toward the BP surface, and the remaining two H atoms of diagonal H2O molecules point upwards (see Fig. 1(c)). The average adsorption energy per H2O of a six-member H2O ring is −0.498 eV. As shown in Table 1, our results indicate that the adsorption energy reaches its minimum (−0.498 eV) when the six-member ring forms, which suggests that the optimal number for H2O adsorption on a BP surface is six. Furthermore, we also investigated the stable configuration at temperatures of 60 and 100 K using molecular dynamics (MD) calculations. The results also show that the average adsorption energy is the lowest when H2O molecules adopt a ring-like structure, which is consistent with the above findings.
| nH2O | 1 | 2 | 3 | 455 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| E 1 | −0.080 | −0.196 | −0.283 | −0.356 | −0.389 | −0.391 | −0.388 |
| E 2(vdw) | −0.185 | −0.306 | −0.392 | −0.470 | −0.490 | −0.498 | −0.496 |
| ΔE | 0.095 | 0.110 | 0.111 | 0.114 | 0.101 | 0.107 | 0.108 |
In ambient conditions, H2O molecules always coexist with O2 molecules on the BP surface. In the following, we investigated the possible adsorption structure of the water molecules on the BP surface in the presence of O2. The dissociation of O2 on the BP surface has been intensively studied in the literature.20 To verify the dissociation process, we also calculated the O2 dissociation on the BP surface. The calculated O–P bond length is 1.505 Å, and the adsorption energy per O atom is −2.10 eV. The calculated adsorption energy per O atom for O2 dissociation at the ortho-site (see Fig. 2(a)) is 80 meV lower than at the meta-site (see Fig. 2(b)), which is consistent with prior results.20 The two O atoms at the ortho-site with large separation experience a weak repulsive force, which results in a lower adsorption energy.
 | ||
| Fig. 2 A single O2 molecule that is dissociated at (a) the ortho-site and (b) the meta-site of the p(6 × 6) BP surface. | ||
During O2 dissociation on the BP surface, the dissociated O2 initially bonds to two meta-site P atoms.20 Although it is not the energetically favorable configuration, it can form on the BP surface because of the larger diffusion barrier of the oxygen atom (∼2 eV) to form the ortho-site configuration. Moreover, the calculated average adsorption energy per H2O is lower for the meta-site configuration (see Fig. 3). Thus, we only considered the meta-site configuration below. The results show that each H atom in H2O points to its neighboring O atom, forming two hydrogen bonds, as shown in Fig. 4(a). Compared to the H2O adsorption on a pristine BP surface, the introduction of O2 is predicted to increase the hydrogen bonding and further reduces the average adsorption energy of the H2O molecules. The calculated adsorption energy per H2O with O2 dissociation (−0.389 eV) is 0.204 eV lower than on the pristine BP surface, which indicates the hydrophilic properties of the oxidized BP. Similarly, as shown in Fig. 4(b), three H2O molecules form a ring structure that is bonded by three pairs of hydrogen bonds. Two of the unpaired H atoms between the neighboring H2O molecules point to the adsorbed O atoms and the rest of the H atoms point upwards. The ring structure again forms when more H2O molecules are adsorbed on the BP surface, such as the five-member ring that is shown in Fig. 4(c). The calculated average adsorption energy per H2O with different numbers of H2O molecules that is shown in Fig. 3 indicates that the five-member water ring has the lowest value of −0.532 eV. Therefore, the optimal number for H2O adsorption on a BP surface with dissociated O2 is five.
 | ||
| Fig. 3 The average adsorption energy of different water molecules with the dissociation of an O2 molecule at the meta-site and ortho-site on the p(6 × 6) BP surface. | ||
 | ||
| Fig. 4 The adsorption of (a) a single H2O molecule, (b) three H2O molecules, and (c) five H2O molecules on the p(6 × 6) BP surface with the dissociation of O2. | ||
Next, we performed calculations to reveal the influence of H2O on the dissociation barrier of O2. In the absence of water, the dissociation barrier of O2 on a p(3 × 5) BP surface is approximately 0.2 eV, as shown in Fig. 5(a), which is consistent with prior results.20 In the presence of one H2O molecule, both H atoms of the H2O molecule point toward the dissociated O2 to form two hydrogen bonds, which promote the decomposition of O2. The dissociation barrier of O2 is found to be almost 0 when H2O is introduced as shown in Fig. 5(b), indicating a spontaneous reaction. In other words, the presence of H2O molecules can dramatically accelerate the decomposition of O2 molecules on the BP surface. Simultaneously, the presence of O2 gives rise to an enhancement of water adsorption on a BP surface. Thus, O2 and H2O molecules have a strong synergetic effect in the oxidation of BP. Therefore, the degradation of BP not only starts from edges, but also occurs on the BP surface in wet conditions.
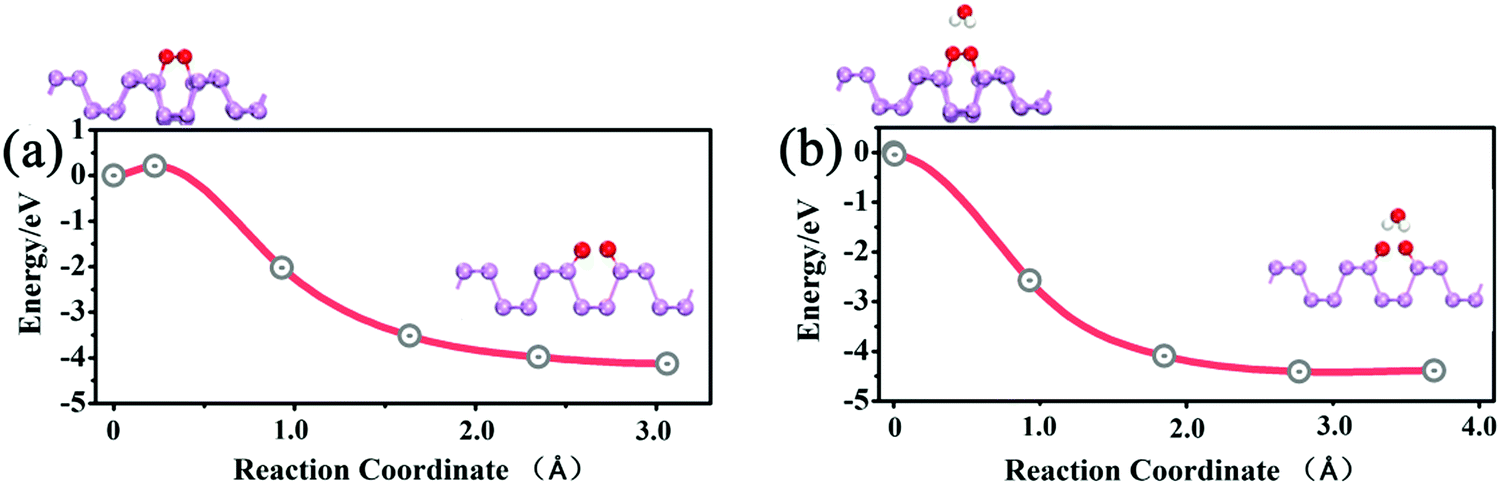 | ||
| Fig. 5 (a) The O2 dissociation process on the p(3 × 5) BP surface. (b) The O2 dissociation process on the p(3 × 5) BP surface in the presence of one adsorbed H2O molecule. | ||
Because BP is easily damaged by H2O and O2 molecules, it is important to know whether the electronic properties suffer from oxidation. Because of the expensive computational cost, a p(2 × 2) supercell was used, and the models that were used are shown in Fig. 6. The calculated QP band gap and GW-BSE optical band gap of the pristine BP are 1.958 eV (see Fig. 7(a)) and 1.252 eV (see Fig. 7(e)), respectively. Therefore, the calculated EBE is 0.706 eV, which is consistent with the literature.34–41 As illustrated in Fig. 7(a), BP shows direct band gap semiconductor features, where both the conduction-band minimum (CBM) and valence-band maximum (VBM) are located at the Γ point. The projected density of states (PDOS) indicates that both VBM and CBM are mainly contributed by pz orbitals, which is in good agreement with prior results.42
 | ||
| Fig. 6 The p(2 × 2) surfaces used to calculate the EBE. (a) The bare surface. (b) The dissociation adsorption of a single O2 molecule, (c) the adsorption of a single H2O molecule, and (d) the co-adsorption of H2O and O2. | ||
 | ||
| Fig. 7 The calculated G0W0 band structures, PDOS, and BSE spectra. The G0W0 band structures and PDOS of (a) the pristine BP, (b) a single H2O molecule, (c) a single O2 molecule, and (d) the co-adsorption of H2O and O2 on the BP surface. BSE spectra of (e) the pristine BP, (f) a single H2O molecule, (g) a single O2 molecule, and (h) the co-adsorption of H2O and O2 on the BP surface. The charge distribution of the VBM and CBM are plotted as insets in (a) and (c). The isosurface values are set to 0.0045 e Å−3. | ||
Because the influence of O2 and/or H2O adsorption on the QP band structure and optical spectra within the GW-BSE have not yet been reported, it is particularly important to perform such calculations. When a single H2O molecule is adsorbed on a p(2 × 2) supercell, the band structure is almost unchanged, and CBM and VBM are still located at the Γ point. The orbitals from the adsorbed H2O are located at deep levels. The calculated QP band gap and GW-BSE optical band gap are 1.972 eV and 1.282 eV, as reported in Fig. 7(b) and (f), respectively. The calculated EBE is 0.690 eV, which is almost the same as the EBE of the pristine BP. Different from water, the adsorption of O2 on a BP surface has a significant influence on the electronic properties as shown in Fig. 7(c). The VBM is still at the Γ point, and it is also originated from the pz orbitals of P atoms. Oxygen adsorption has a significant impact on the CBM. The CBM deviates from the Γ point, which locates along Γ–M direction. The CBM is dominated by the px and py orbitals of P atoms bonding to oxygen, while 2p orbitals of O atoms also contribute to CBM. The formation of O–P bond is accompanied by a strong orbital hybridization, and it causes a decrement of the underlying P–P bond lengths, leading to an enhancement of the splitting between the pz bonding and antibonding orbitals of P atoms. As the pz antibonding orbitals of P atoms are raised in energy and the contributions from the px and py orbitals of P atoms increase, it results in a direct-to-indirect band gap transition. The QP and GW-BSE band gaps at the Γ point are 2.482 eV and 1.956 eV, respectively. The calculated EBE becomes 0.526 eV upon the adsorption of O2. In addition, we find that the co-adsorption of H2O and O2 on the BP surface is similar to O2 dissociation, which also leads to a direct–indirect band gap transition. As shown in Fig. 7(d) and (h), the QP and GW-BSE band gaps at the Γ point are 2.474 eV and 1.926 eV, respectively, resulting in an EBE of 0.548 eV. The above analysis explains why H2O adsorption has scarcely any influence on BP and why O2 adsorption and their co-adsorption change the electronic properties of BP.
4. Conclusions
In summary, the influence of surface adsorption in the structural instability and electronic properties of a BP surface is systematically investigated. We find that water molecules prefer to form a six-member water ring, while a five-member water ring becomes the most energetically favorable after the introduction of O2. Moreover, the dissociated O2 can strengthen the adsorption ability of water. In addition, the dissociation barrier of O2 can be drastically reduced in the presence of H2O, which could explain the rapid decomposition of BP in ambient conditions. Interestingly, the effect of H2O adsorption on the electronic properties and EBE is negligible. In contrast, O2 adsorption and the co-adsorption of O2 and H2O have a remarkable effect on the electronic properties of BP, and cause a direct–indirect band gap transition, which significantly depresses photo-absorption efficiency.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, Grant No. 11674148 and 11334004), the Guangdong Natural Science Funds for Distinguished Young Scholars (No. 2017B030306008) and the Basic Research Program of Science, Technology, and Innovation Commission of Shenzhen Municipality (Grant No. JCYJ20160531190054083)References
- V. Tran, R. Soklaski, Y. Liang and L. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235319 CrossRef.
- S.-L. Yau, T. P. Moffat, A. J. Bard, Z. Zhang and M. M. Lerner, Chem. Phys. Lett., 1992, 198, 383–388 CrossRef CAS.
- L. Li, Y. Yu, G. J. Ye, Q. Ge, X. Ou, H. Wu, D. Feng, X. H. Chen and Y. Zhang, Nat. Nanotechnol., 2014, 9, 372–377 CrossRef CAS PubMed.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- J. Qiao, X. Kong, Z.-X. Hu, F. Yang and W. Ji, Nat. Commun., 2014, 5, 4475 CAS.
- S. P. Koenig, R. A. Doganov, H. Schmidt, A. H. Castro Neto and B. Özyilmaz, Appl. Phys. Lett., 2014, 104, 103106 CrossRef.
- F. Xia, H. Wang and Y. Jia, Nat. Commun., 2014, 5, 4458 CAS.
- X. Wang, A. M. Jones, K. L. Seyler, V. Tran, Y. Jia, H. Zhao, H. Wang, L. Yang, X. Xu and F. Xia, Nat. Nanotechnol., 2015, 10, 517–521 CrossRef CAS PubMed.
- C.-G. Andres, V. Leonardo, P. Elsa, O. I. Joshua, K. L. Narasimha-Acharya, I. B. Sofya, J. G. Dirk, B. Michele, A. S. Gary, J. V. Alvarez, W. Z. Henny, J. J. Palacios and S. J. v. d. Z. Herre, 2D Mater., 2014, 1, 025001 CrossRef.
- H. Liu, Y. Du, Y. Deng and P. D. Ye, Chem. Soc. Rev., 2015, 44, 2732–2743 RSC.
- L. Kou, C. Chen and S. C. Smith, J. Phys. Chem. Lett., 2015, 6, 2794–2805 CrossRef CAS PubMed.
- A. S. Rodin, A. Carvalho and A. H. Castro Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 075429 CrossRef.
- A. Ziletti, A. Carvalho, P. E. Trevisanutto, D. K. Campbell, D. F. Coker and A. H. Castro Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 085407 CrossRef.
- M. T. Edmonds, A. Tadich, A. Carvalho, A. Ziletti, K. M. O’Donnell, S. P. Koenig, D. F. Coker, B. Özyilmaz, A. H. C. Neto and M. S. Fuhrer, ACS Appl. Mater. Interfaces, 2015, 7, 14557–14562 CAS.
- D. Grasseschi, D. A. Bahamon, F. C. B. Maia, A. H. C. Neto, R. O. Freitas and C. J. S. d. Matos, 2D Mater., 2017, 4, 035028 CrossRef.
- A. Favron, E. Gaufrès, F. Fossard, A.-L. Phaneuf-Lheureux, N. Y. W. Tang, P. L. Lèvesque, A. Loiseau, R. Leonelli, S. Francoeur and R. Martel, Nat. Mater., 2015, 14, 826–832 CrossRef CAS PubMed.
- J. D. Wood, S. A. Wells, D. Jariwala, K.-S. Chen, E. Cho, V. K. Sangwan, X. Liu, L. J. Lauhon, T. J. Marks and M. C. Hersam, Nano Lett., 2014, 14, 6964–6970 CrossRef CAS PubMed.
- S. Gamage, Z. Li, V. S. Yakovlev, C. Lewis, H. Wang, S. B. Cronin and Y. Abate, Adv. Mater. Interfaces, 2016, 3, 1600121 CrossRef.
- Y. Huang, J. Qiao, K. He, S. Bliznakov, E. Sutter, X. Chen, D. Luo, F. Meng, D. Su, J. Decker, W. Ji, R. S. Ruoff and P. Sutter, Chem. Mater., 2016, 28, 8330–8339 CrossRef CAS.
- A. Ziletti, A. Carvalho, D. K. Campbell, D. F. Coker and A. H. Castro Neto, Phys. Rev. Lett., 2015, 114, 046801 CrossRef CAS PubMed.
- G. X. Wang, J. S. William, P. Ravindra and P. K. Shashi, 2D Mater., 2016, 3, 025011 CrossRef.
- W. Luo, Y. Z. Dmitry, A. M. Cory, Y. Du, L. Yang, Y. Wu and D. Y. Peide, Nanotechnology, 2016, 27, 434002 CrossRef PubMed.
- K. L. Kuntz, R. A. Wells, J. Hu, T. Yang, B. Dong, H. Guo, A. H. Woomer, D. L. Druffel, A. Alabanza, D. Tománek and S. C. Warren, ACS Appl. Mater. Interfaces, 2017, 9, 9126–9135 CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- M. S. Hybertsen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 5390–5413 CrossRef CAS.
- A. A. Mostofi, J. R. Yates, Y.-S. Lee, I. Souza, D. Vanderbilt and N. Marzari, Comput. Phys. Commun., 2008, 178, 685–699 CrossRef CAS.
- N. E. Bickers, D. J. Scalapino and S. R. White, Phys. Rev. Lett., 1989, 62, 961–964 CrossRef PubMed.
- J. P. A. Charlesworth, R. W. Godby and R. J. Needs, Phys. Rev. Lett., 1993, 70, 1685–1688 CrossRef CAS PubMed.
- N. Marom, A. Tkatchenko, M. Rossi, V. V. Gobre, O. Hod, M. Scheffler and L. Kronik, J. Chem. Theory Comput., 2011, 7, 3944–3951 CrossRef CAS PubMed.
- L. S. Wanderlã, S. S. Everson and R. H. Miwa, J. Phys.: Condens. Matter, 2017, 29, 075002 CrossRef PubMed.
- J.-H. Choi, P. Cui, H. Lan and Z. Zhang, Phys. Rev. Lett., 2015, 115, 066403 CrossRef PubMed.
- L. Seixas, A. S. Rodin, A. Carvalho and A. H. Castro Neto, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 115437 CrossRef.
- E. Prada, J. V. Alvarez, K. L. Narasimha-Acharya, F. J. Bailen and J. J. Palacios, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 245421 CrossRef.
- T. Vy, R. Fei and Y. Li, 2D Mater., 2015, 2, 044014 CrossRef.
- P. Cudazzo, L. Sponza, C. Giorgetti, L. Reining, F. Sottile and M. Gatti, Phys. Rev. Lett., 2016, 116, 066803 CrossRef PubMed.
- Z. Nourbakhsh and R. Asgari, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 94, 035437 CrossRef.
- C. E. P. Villegas, A. S. Rodin, A. Carvalho and A. R. Rocha, Phys. Chem. Chem. Phys., 2016, 18, 27829–27836 RSC.
- Z. Jiang, Z. Liu, Y. Li and W. Duan, Phys. Rev. Lett., 2017, 118, 266401 CrossRef PubMed.
- X. Han, H. M. Stewart, S. A. Shevlin, C. R. A. Catlow and Z. X. Guo, Nano Lett., 2014, 14, 4607–4614 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2018 |