
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic field effects dynamics of ethylammonium nitrate ionic liquid confined between glass plates†
Andrei
Filippov
 *ab and
Oleg N.
Antzutkin
ac
*ab and
Oleg N.
Antzutkin
ac
aChemistry of Interfaces, Luleå University of Technology, SE-97187 Luleå, Sweden. E-mail: Andrei.Filippov@ltu.se; Tel: +46 73-6782225
bInstitute of Physics, Kazan Federal University, 420008 Kazan, Russia
cDepartment of Physics, Warwick University, Coventry CV47AL, UK
First published on 31st January 2018
Abstract
Self-diffusion and NMR relaxation of the ethylammonium (EA) cation were studied in the protic ionic liquid, ethylammonium nitrate (EAN), confined between polar glass plates separated by a few μm distance and exposed to an external magnetic field of 9.4 T. The diffusion coefficient of EA (D) and the transverse NMR relaxation rate (1/T2) of –NH3 protons were increased immediately after placing the sample in the magnetic field by factors of ∼2 and ∼22, respectively, in comparison with those of bulk EAN. Further exposure of the sample to the magnetic field led to gradual changes in D, T1 and T2 towards their bulk values with a time constant of ∼70 min. Complete “recovery” of the sample to the “accelerated” D and “shortened” T2 values occurred at longer than 24 hours after the removal of the EAN sample from the magnet. Because the observed characteristic times of the change far exceed the times of molecular processes in EAN, we suggested that this phenomenon is related to reversible phase transformations occurring in confined EAN.
Introduction
Ionic liquids (ILs) are molten salts prepared from organic cations and either organic or inorganic anions.1,2 Their applications are continuously expanding, including their use, for instance, as electrolyte materials in lithium batteries3 and ultracapacitors,4 as media for chemical reactions and separation,2,5 and as lubricants.6 Ethylammonium nitrate (EAN) is a protic ionic liquid. This means that it has an exchangeable proton on the cation.1 EAN, first synthesized by Paul Walden in 1914,7 is the most frequently reported protic IL.1 It is used as a medium for chemical reactions, as a precipitating agent for protein separation,5 an electrically conductive solvent in electrochemistry,3 and in other applications. Like water, EAN has a three-dimensional hydrogen-bonding network and can be used as an amphiphilic self-assembly medium.8Ions of ILs can participate in a variety of interactions that may lead to the formation of ordered self-assembled structures.9 External conditions, such as temperature, pressure, electric field and confinement, may change the structural and dynamic parameters of ILs.9–13 Confinement in aligned carbon nanotubes,14 mesoporous carbon10 and silicon11,12 does increase the diffusion of ILs by a factor of 2–3 or more. The latter has been explained as a consequence of the decrease in the packing density of ions in pores with diameters comparable to the sizes of the ions.11 Recently, enhanced diffusion of ethylammonium (EA) cations has been observed for EAN confined between polar glass plates separated by ca. 4 μm,15 which has been related to the transformation of a sponge-like structure of bulk EAN. Magnetic field strength is also an important external variable that influences the properties of some organic substances, such as liquid crystals16 and organic-based semiconductors.17 No perceptible magnetic field effects have been observed for non-magnetic ionic liquids until now.9,18
In this work, we report the effect of applying a strong magnetic field to EAN confined between polar glass plates separated by ∼4 μm. This system demonstrated the accelerated translational diffusion of EA cations and the accelerated transverse NMR relaxation rates of EA –NH3 protons.15 After exposing this system to a strong magnetic field (9.4 T), we observed a slow (with a time constant of ca. 70 min) decrease in the diffusion coefficient of the EA cation and a gradual increase in T2 NMR relaxation times for EA –NH3 protons toward the bulk values of EAN. No such effect was observed earlier.
Experimental
Sample preparation
EAN (see Fig. 1) consists of an EA cation and a nitrate anion. EAN used in our experiment was synthesized, pretreated and characterized at Chemistry of Interfaces of Luleå University of Technology as described previously.15 | ||
| Fig. 1 Chemical structure of EAN, consisting of an ethylammonium (EA) cation (left) and a nitrate anion (right). | ||
NMR measurements for bulk EAN were performed by placing it in a standard 5 mm NMR glass tube. Confined EAN was prepared with thin glass plates arranged in a stack (see Fig. S1 in the ESI†). A chemical composition of plates (5 × 14 × 0.1 mm, Thermo Scientific Menzel-Gläser, Menzel GmbH, Germany) is presented in the ESI.† They were carefully cleaned before use (see the ESI†). Contact angle measurements with Milli-Q water provided a contact angle near 0° (see the ESI†). A stack of glass plates filled with EAN was prepared using a glove box in a dry nitrogen atmosphere. The samples were prepared by adding 2 μL of EAN to a first glass plate, placing a second glass plate on top, adding 2 μL to this glass plate, etc. until the thickness of the stack reached approximately 5 mm. Excess EAN from the sides of the stack was removed by wiping. The sample consisted of a stack of ca. 45 glass plates with EAN between them and placed in a rectangular sealed glass tube. The mean spacing between the glass plates was estimated by weighing the introduced EAN, which yielded d ∼ 3.8–4.5 μm.15 A detailed description of the sample preparation and characterization has been reported in our previous paper.15
EAN with deuterium-substituted –NH3 protons was prepared by mixing of the original EAN and D2O followed by water evaporation under high vacuum (∼10−3 mbar) for 48 h. The degree of substitution was determined from the intensity of the corresponding line in the 1H NMR spectrum.
Pulsed-field gradient diffusometry
1H NMR self-diffusion and T1 and T2 relaxation measurements of EAN, in bulk and confined between glass plates, were performed using a Bruker Ascend/Aeon WB 400 (Bruker BioSpin AG, Fällanden, Switzerland) NMR spectrometer with a working frequency of 400.27 MHz for 1H and at a magnetic field strength of 9.4 T with magnetic field homogeneity better than 4.9 × 10−7 T, using a Diff50 Pulsed-Field-Gradient (PFG) probe. An NMR solenoid 1H insert was used to macroscopically align the plates of the sample stack at 0 and 90 degrees with respect to the direction of the external magnetic field.The diffusional decays (DDs) were recorded using spin echo (td ≤ 5 ms) or stimulated echo (td > 5 ms) pulse sequences. For a single-component, the signal intensity, A, changes as:19,20
 | (1a) |
 | (1b) |
As has been shown in our previous work,15 there is no effect of restrictions on D in the direction along the plates (see Fig. S2 and S3 in the ESI†), while for diffusion normal to the plates the effect of the plates on D is noticeable at td longer than 3 ms (see Fig. S4–S6 in the ESI†). For diffusion in bulk and along the plates, the D values were obtained by fitting eqn (1) to the experimental decays. For the decays obtained in the direction normal to the plates at td = 3 ms, D was calculated from the equation:
 | (2) |
All measurements were performed at 293 K and were started immediately (in 1–2 minutes) after placing the sample in the NMR probe. The time required for a single NMR diffusion measurement was around 30 s, while that of a relaxation measurement was around 3 min. Data were processed using Bruker TopSpin 3.5 software.
To demonstrate the absence of effect on the background gradient g0 in such a heterogeneous system as glass plates with the confined ionic liquid, a 13-interval stimulated echo sequence with bipolar gradient pulses21 (SteBp2) was used. The latter pulse sequence removes the cross-terms arising from the applied pulsed gradients, g and g0, which are significant even if g0 values are small.
1H NMR relaxometry
1H T1 and T2 NMR relaxation time measurements were performed with inversion recovery (180°–τ–90°–fid) and CPMG (90°–τ–180°–τ–echo) pulse sequences, respectively.22 The duration of the 90° pulse was 7 μs. Phases of 90° and 180° pulses as well as the phase of the receiver in the CPMG pulse sequence were varied with a specific cycle, and the echo time was fixed to allow elimination of diffusion and J-coupling modulation effects.Results and discussion
Since all the measurements were performed at room temperature (293 K), the measurements were performed immediately after placing the sample in the magnet. The placement of bulk EAN in the magnetic field of the NMR spectrometer and systematic measurements of D, T1 and T2 over 24 hours showed no changes in these parameters (Table S1 in the ESI†). However, for EAN confined between glass plates, we observed the changes in D, and T1 and T2 of different chemical groups of EA, as shown in Fig. 2.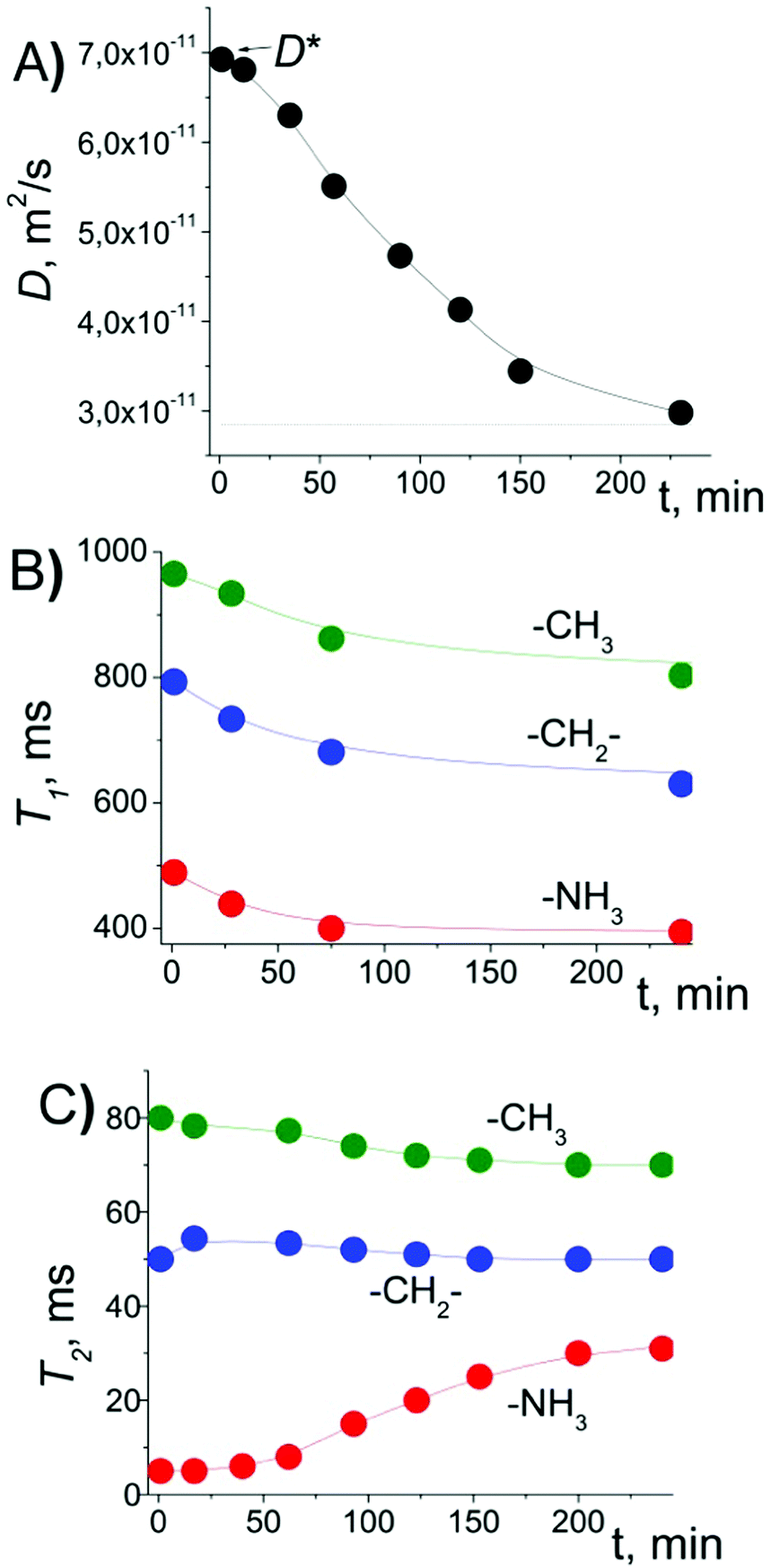 | ||
| Fig. 2 Changes in (A) diffusion coefficient of EA cations, (B) T1 NMR relaxation times and (C) T2 NMR relaxation times of protons of different chemical groups of the EA cation after placing EAN confined between polar glass plates at a magnetic field of 9.4 T. D was measured along the plates, td = 3 ms. | ||
Right after placement of the sample in the NMR probe, D was measured as D*, which was a factor of ∼2 larger than that in bulk (D0). The T1 relaxation recovery for protons of different groups demonstrated forms close to the exponential ones. T2 relaxation for the –NH3 group was exponential, while T2 relaxation of –CH2– and –CH3 groups was close to the sum of the two exponential functions (see Fig. S7 in the ESI†). In this case, the mean values of T2 were calculated as: T2 = (1/T21 + 1/T22)−1. The initial values of T2 (T2*, right after placement of the sample in the magnetic field) of different groups are decreased by factors of ∼6.2 (–CH2–), ∼6.4 (–CH3) and ∼22 (–NH3) relative to their bulk values (see Table S1 in the ESI†). Furthermore, we observed that D gradually decreased from D* to smaller values close to D0 (Fig. 2A). The same effect was observed either by applying unipolar pulse sequences (spin echo and stimulated echo) or by applying stimulated echo sequence with bipolar gradient pulses (SteBp2), Fig. S8 and S9 in the ESI.† Therefore, the effect is not related to the background gradient g0 of the magnetic field. This agrees well with linewidths of the NMR resonance lines, which should be broadened in the presence of the background gradient. In fact, the linewidths typical for bulk are maintained for EAN between glass plates.15
After placing the sample in a magnetic field, simultaneously with D, the T1 of different protons decreased by a factor of ∼1.25 (Fig. 2B). The mean values of T2 for –CH2– and –CH3 groups demonstrated small changes, while T2(–NH3) increased by a factor of ∼6 (Fig. 2C and Fig. S10 in the ESI†). A similar effect was observed for the sample oriented along and normal to the glass plates relative to the magnetic field (Fig. 3, black circles and stars).
 | ||
| Fig. 3 Changes in the diffusion coefficient of ionic liquids placed between polar glass plates: EA cations in EAN (black symbols); diffusion of EA cations in EAN with ∼30% protons of –NH3 groups replaced with deuterium (red circles). Dotted lines show the bulk values of D for the corresponding ionic liquids. Open and solid circles correspond to data from two independent experiments with the same parameters. | ||
From Fig. 2A, one can observe that the decrease in D and the increase in T2(–NH3) reached plateau after ca. 4 hours. The process can be characterized by the time during which the initial value changes by a factor of “e”, which was estimated to be nearly 70 min from Fig. 2A. The experiment was repeated several times with different time intervals, TE, between completion of the change in D and T2 values in the previous experiment and the beginning of the next experiment. In cases when TE was just few hours, the measured D value in the beginning of the new experiment was smaller than D*, and T2(–NH3) was larger than T2*(–NH3). But in cases when TE was on the order of a day or longer, D and T2 were completely “recovered” to D* and to T2*, respectively.
The observed acceleration of diffusion in the beginning of the experiment cannot be related to a “mechanical relaxation” of the sample in the strong magnetic field, which can lead to a flux in EAN. In this case, the measured (“apparent”) diffusion coefficient would be related to molecular displacements over longer distances; therefore, it would be higher than the true molecular diffusion coefficient. This would also lead to the increase of the apparent D with the increase of td. Here, there is no dependence of diffusion decays (and the measured diffusion coefficient) on the td (Fig. S3, solid symbols, ESI†). Therefore, measurements were performed under equilibrium conditions.
Additional experiments were performed to ensure that the effects observed for D and T2(–NH3) are due to the presence of a strong magnetic field, but not associated with other processes that may occur in the NMR spectrometer during diffusion and relaxation experiments. To investigate the effect of radio-frequency pulsing, a fully “recovered” sample was placed in the spectrometer probe for a rather long time, but no radio-frequency pulses were applied during this time interval. The first measurements of D and T2 were performed after 24 hours and yielded the same values as those from the experiments where continuous NMR measurements were considered. In another experiment, a sample that had “recovered” outside of the magnetic field of the spectrometer was placed between the poles of Neodymium permanent disk-shaped magnets with a magnetic field strength of ∼0.68 T for 24 hours before placing the sample in the NMR spectrometer for measurements. After placing the sample in the spectrometer, the measured value of D was between D* and D0. Therefore, one can conclude that the presence of a strong magnetic field is the main factor influencing the D and T2(–NH3) processes that occur with EAN confined between polar glass plates.
To reveal the main factors leading to the observed effect of alteration of diffusion coefficient in the presence of a magnetic field, few extra samples were prepared. For one of the samples, we used silanized nonpolar glass plates. The silanization protocol is described in the ESI.† In this case, D in the EAN layers between glass plates was close to D0 and no alteration occurred after placement of the sample in the magnetic field. For the preparation of another sample, we used nonprotic ionic liquid [P6,6,6,14][BOB]23 (Fig. S11 in the ESI†) and polar glass plates. Diffusion of the [P6,6,6,14]+ cation in the layers between the plates was just slightly lower than that in the bulk, and no effect of the magnetic field was observed. Therefore, the magnetic field has an effect only on the diffusion of protic IL placed between polar plates.
The state and dynamics of liquids between solid surfaces have been attracting scientific interest for many years. Earlier studies of interactions between charged plates led to the discovery of the Casimir effect.24,25 On a submicron scale, this force becomes the dominant force between uncharged conductors. Several recent experiments have reported data for the approach of two surfaces separated by ionic liquids which revealed relatively long-range forces (tens of nanometers) that appear to be electrostatic in origin.26 The original interpretation of these forces was controversial, and the origin of long-range forces in ionic liquids remains the subject of discussions. No forces acting in the μm range in an IL as well as no effects in the external magnetic field were reported. Earlier we discovered the acceleration of diffusion of EAN confined between polar glass plates and suggested that this is related to exchange processes of mobile –NH3 protons.15 To further explore this phenomenon, we substituted ca. 30% of these protons by deuterium by mixing of EAN and D2O followed by water evaporation under vacuum. It is well known that exchange rates of deuterium in chemical exchange reactions are considerably slower than those for protons, phenomena usually called the kinetic isotope effects. Indeed, for a sample containing EAN (with partially substituted protons by deuterons) confined between the glass plates, the rate of the decrease of D after placement of the sample in a magnetic field of 9.4 T was more than a factor of 10 slower as compared to non-deuterated EAN samples (Fig. 3, red circles). Therefore, the exchange rate of –NH3 protons plays a crucial role in the observed processes.
The timescale of the magnetic field phenomenon observed in EAN confined between polar plates is on the order of a few hours, which is much longer than the timescale of dynamic processes occurring with individual EAN ions confined between glass plates. Indeed, the longest of the dynamic processes is the self-diffusion of the ions, which covers the inter-planar distance during ca. 100 ms. For this reason, we suggested that the processes occurring with EAN between polar glass plates are the reversible phase transformations of EAN. It has been suggested that EAN, because of the competition of the strong hydrogen bonding and electrostatic interactions of EA cations and nitrate anions, and the hydrophobic interactions of –CH2– and –CH3 groups of EA cations, may form a bi-continuous sponge-like structure.9,27–29 Because this structure is characteristically a balance of forces, structural parameters can vary as a consequence of changes in the temperature, pressure and chemical modification of ions. Hindered diffusion is typical for this structure, as well as for the bi-continuous cubic phase, because of the curvilinear trajectories of ions along the micro-phase borders.30 It could be suggested that the balance of forces and structural parameters of the sponge-like phase may be modified in the presence of additional interactions of ions with the polar surfaces of glass plates. The change in structural parameters enhances the mobility of the ions.14,31 Another possibility is the transformation of the sponge-like phase to an isotropic phase. The phase transformation leads to enhancement of diffusive transport and accelerated –NH3 proton exchange. Application of a strong magnetic field additionally contributes to inter-ion interactions, which might be more efficient for moving ions, leading to further disturbance of the existing equilibrium. Thus, the application of a strong magnetic field to EAN confined between polar glass plates allows one to control the dynamics of EA ions.
Conclusions
In this work, we have experimentally shown that self-diffusion and NMR relaxation of EAN confined between polar glass plates reversibly alter after application of a strong magnetic field. The main factors that contributed to this effect are availability of protons in the protic EAN and the polarity of the surface. The exchange rate of –NH3 protons plays a crucial role in the observed processes. Because of the long timescale of this process, we suggested that the processes occurring with EAN between polar glass plates are the reversible phase transformations of EAN. Further structural studies are required to support this suggestion.Given that the dynamic properties of ionic liquids are significantly altered in confinement and in an external magnetic field, these results have strong implications for interface-intensive applications of ILs, such as their use as lubricants and in electrochemical and electro-magnetic systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. I. Furó (KTH, Stockholm) for valuable discussions concerning NMR diffusion and relaxation measurements and Prof. M. W. Rutland (KTH, Stockholm) for guiding in the preparation of polar and silanized glass plates and for contact angle measurements.References
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- C. Zhao, G. T. Burrell, A. A. J. Torriero, F. Separovic, N. F. Dunlop, D. R. MacFarlane and A. M. Bond, J. Phys. Chem. B, 2008, 113, 6923–6936 CrossRef PubMed.
- M. Lazzari, M. Mastragostino and F. Soavi, Electrochem. Commun., 2007, 9, 1567–1572 CrossRef CAS.
- J. A. Garlitz, C. A. Summers, R. A. Flowers II and G. E. O. Borgstahl, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 2037–2038 CrossRef CAS.
- F. U. Shah, S. Glavatskih and O. N. Antzutkin, Tribol. Lett., 2013, 51, 281–301 CrossRef CAS.
- P. Walden, Bull. Acad. Imp. Sci. St.-Petersbourg, 1914, 8, 405–422 Search PubMed.
- H. J. Jiang, P. A. FitzGerald, A. Dolan, R. Atkin and G. G. Warr, J. Phys. Chem. B, 2014, 118, 9983–9990 CrossRef CAS PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- S. M. Chathoth, E. Mamontov, P. F. Fulvio, X. Wang, G. A. Baker, S. Dai and D. J. Wesolowski, Europhys. Lett., 2013, 102, 16004 CrossRef.
- C. Iacob, J. R. Sangoro, P. Papadopoulus, T. Schubert, S. Naumov, R. Valiullin, J. Kärger and F. Kremer, Phys. Chem. Chem. Phys., 2010, 12, 13798–13803 RSC.
- A. Filippov, N. Azancheev, F. U. Shah, S. Glavatskih and O. N. Antzutkin, Microporous Mesoporous Mater., 2016, 230, 128–134 CrossRef CAS.
- K. Damodaran, Annu. Rep. NMR Spectrosc., 2016, 88, 215–244 CrossRef CAS.
- Q. Berrod, F. Ferdeghini, P. Judenstein, N. Genevaz, R. Ramos, A. Fournier, J. Dijon, J. Ollivier, S. Rols, D. Yu, R. A. Mole and J.-M. Zanotti, Nanoscale, 2016, 8, 7845–7848 RSC.
- A. Filippov, O. I. Gnezdilov, N. Hjalmarsson, O. N. Antzutkin, S. Glavatskih, I. Furó and M. W. Rutland, Phys. Chem. Chem. Phys., 2017, 19, 25853–25858 RSC.
- M. Wang, L. He, S. Zorba and Y. Yin, Nano Lett., 2014, 14, 3966–3971 CrossRef CAS PubMed.
- H. J. Jang, S. J. Pookpanratana, A. N. Brigeman, R. J. Kline, J. I. Basham, D. J. Gundlach, C. A. Hacker, O. A. Kirillov, O. D. Jurchescu and C. A. Richter, ACS Nano, 2014, 8, 7192–7201 CrossRef CAS PubMed.
- G. L. Burrell, I. M. Burgar, Q. Gong, N. F. Dunlop and F. Separovic, J. Phys. Chem. B, 2010, 114, 11436–11443 CrossRef CAS PubMed.
- P. T. Callaghan, Principles of Nuclear Magnetic Resonance Microscopy; Clarendon, Oxford, 1991 Search PubMed.
- J. E. Tanner, J. Chem. Phys., 1970, 52, 2523–2526 CrossRef CAS.
- R. M. Cotts, M. J. R. Hoch, T. Sun and J. T. Markert, J. Magn. Reson., 1989, 83, 252–256 CAS.
- J. A. Aguilar, M. Nilsson, G. Bodenhausen and G. A. Morris, Chem. Commun., 2012, 48, 811–813 RSC.
- F. U. Shah, S. Glavatskih, D. R. MacFarlane, A. Somers, M. Forsyth and O. N. Antzutkin, Phys. Chem. Chem. Phys., 2011, 13, 12865–12873 RSC.
- H. B. G. Casimir, Proc. K. Ned. Akad. Wet., 1948, 51, 793–795 Search PubMed.
- C. Genet, F. Intravaia, A. Lambrecht and S. Reynaud, Annales de la Fondation Louis de Broglie, 2004, 29, 331–348 Search PubMed.
- M. A. Gebbie, A. M. Smith, H. A. Dobbs, A. A. Lee, G. G. Warr, X. Banquy, M. Valtiner, M. W. Rutland, J. N. Israelachvili, S. Perkin and R. Atkin, Chem. Commun., 2017, 53, 1214–1224 RSC.
- R. Atkin and G. G. Warr, J. Phys. Chem. B, 2008, 112, 4164–4166 CrossRef CAS PubMed.
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162–5168 CAS.
- P. Niga, D. Wakeham, A. Nelson, G. G. Warr, M. Rutland and R. Atkin, Langmuir, 2010, 26, 8282–8288 CrossRef CAS PubMed.
- A. E. Frise, T. Ichikawa, M. Yoshio, H. Ohno, S. V. Dvinskikh, T. Kato and I. Furó, Chem. Commun., 2010, 46, 728–730 RSC.
- V. Aho, K. Mattila, T. Kühn, P. Kekäläinen, O. Pulkkinen, R. B. Minussi, M. Vihinen-Ranta and J. Timonen, Phys. Rev. E, 2016, 93, 043309 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp06554j |
| This journal is © the Owner Societies 2018 |