
Spectroscopy of prospective interstellar ions and radicals isolated in para-hydrogen matrices
 †*a
Yuan-Pern
Lee
*ab
†*a
Yuan-Pern
Lee
*ab
Abstract
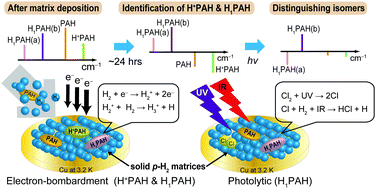
para-Hydrogen (p-H2) serves as a new host in matrix-isolation experiments for an investigation of species of astrochemical interest. Protonated and mono-hydrogenated species are produced upon electron bombardment during deposition of p-H2 containing a precursor in a small proportion. The applications of this novel technique to generate protonated polycyclic aromatic hydrocarbons (H+PAH), protonated polycyclic nitrogen heterocycles (H+PANH), and their neutral counterparts, which are important in the identification of interstellar unidentified infrared emission bands, demonstrate its superiority over other methods. The clean production with little fragmentation, ease of distinction between protonated and neutral species, narrow lines and reliable relative infrared intensities of the lines, and broad coverage of the spectral range associated with this method enable us to assign the isomers unambiguously. The application of this method to the protonation of small molecules is more complicated partly because of the feasible fragmentation and reactions, and partly because of the possible proton sharing between the species of interest and H2, but, with isotopic experiments and secondary photolysis, definitive assignments are practicable. Furthermore, the true relative infrared intensities are critical to a comparison of experimental results with data from theoretical calculations. The spectra of a proton-shared species in solid p-H2 might provide insight into a search for spectra of proton-bound species in interstellar media. Investigations of hydrogenated species involving the photolysis of Cl2 or precursors of OH complement those using electron bombardment and provide an improved ratio of signal to noise. With careful grouping of observed lines after secondary photolysis and a comparison with theoretical predictions, various isomers of these species have been determined. This photolytic technique has been applied in an investigation of hydrogenated PAH and PANH, and the hydrogenation reactions of small molecules, which are important in interstellar ice and the evolution of life. The electronic transitions of molecules in solid p-H2 have been little investigated. The matrix shift of the origins of transitions and the spectral width seem to be much smaller than those of noble-gas matrices; these features might facilitate a direct comparison of matrix spectra with diffuse interstellar bands, but further data are required to assess this possibility. The advantages and disadvantages of applying these techniques of p-H2 matrix isolation to astrochemical research and their future perspectives are discussed.
- This article is part of the themed collections: PCCP Perspectives, 2018 PCCP HOT Articles and Theory, experiment, and simulations in laboratory astrochemistry
 Please wait while we load your content...
Please wait while we load your content...

