
The effect of regioisomerism on the photophysical properties of alkylated-naphthalene liquids†

Abstract
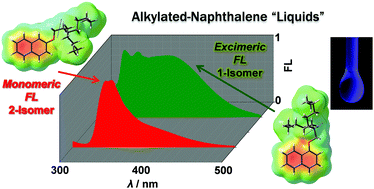
Novel regioisomeric alkylated-naphthalene liquids were designed and synthesized. In the solvent-free liquid state, 1-alkyloxy regioisomers showed excimeric luminescence, whereas 2-alkyloxy analogues exhibited monomer-rich luminescence features. Correlations among the molecular structures and the photophysical, calorimetric, and rheological properties are presented, demonstrating the impact of regioisomerism on the alkylated-chromophore liquid systems.
- This article is part of the themed collections: 2018 PCCP HOT Articles and Complex molecular systems: supramolecules, biomolecules and interfaces
 Please wait while we load your content...
Please wait while we load your content...

