
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Salts with titanyl and vanadyl phthalocyanine radical anions. Molecular design and effect of cations on the structure and magnetic and optical properties†
Dmitri V.
Konarev
 *a,
Alexey V.
Kuzmin
b,
Salavat S.
Khasanov
b,
Mikhail S.
Batov
ac,
Akihiro
Otsuka
de,
Hideki
Yamochi
de,
Hiroshi
Kitagawa
d and
Rimma N.
Lyubovskaya
a
*a,
Alexey V.
Kuzmin
b,
Salavat S.
Khasanov
b,
Mikhail S.
Batov
ac,
Akihiro
Otsuka
de,
Hideki
Yamochi
de,
Hiroshi
Kitagawa
d and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, Moscow region, 142432 Russia. E-mail: konarev3@yandex.ru
bInstitute of Solid State Physics RAS, Chernogolovka, Moscow region, 142432 Russia
cMoscow State University, Leninskie Gory, 119991 Moscow, Russia
dDivision of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
eResearch Center for Low Temperature and Materials Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan
First published on 13th December 2017
Abstract
Radical anion salts of metal phthalocyanines have been providing a new family of compounds which can potentially possess promising optical, magnetic and conducting properties. Essential progress in this field has been reached only recently when series of salts of phthalocyanines with different metals and axial substituents were obtained. In this work we summarize our recent results about the radical anion salts of titanyl (TiIVOPc) and vanadyl (VIVOPc) phthalocyanines and nine new salts of these phthalocyanines are discussed. Different synthetic approaches for the preparation of these salts are presented, and the effect of the size and shape of counter cations on the crystal structures of the salts and consequently their magnetic and optical properties are considered. The {TiIVO(Pc˙3−)}˙− radical anions have an S = 1/2 spin on the Pc˙3− macrocycle, whereas the {VIVO(Pc˙3−)}˙− radical anions have two S = 1/2 spins on both the Pc˙3− macrocycle and the central VIV atom. Therefore, it is possible to compare the magnetic properties of isostructural compounds with different magnetic states of metal phthalocyanines. We also discuss the effect of reduction on the molecular structure and the optical and magnetic properties of negatively charged titanyl and vanadyl phthalocyanines.
Introduction
Metal-free and metal-containing phthalocyanines can possess promising optical, conducting and magnetic properties.1–9 As a result, some phthalocyanine derivatives are used as sensors and as materials for optical, photoelectronic and photovoltaic devices.1,2 To obtain conducting compounds phthalocyanines should be partially oxidized to introduce an unpaired electron into the phthalocyanine macrocycle. A columnar or layered arrangement of such macrocycles together with effective π–π stacking provides semiconductivity or quasi-one-dimensional metallic conductivity.3–7 The paramagnetic atoms in the center of the phthalocyanine macrocycle can induce a giant magnetoresistance.5–7 Molecular magnets containing manganese(III) phthalocyanine or substituted naphthalocyanine cations and tetracyanoethylene radical anions are also known.8,9 All these compounds were obtained by oxidation of metal phthalocyanines.Potentially, reduced metal-free and metal-containing phthalocyanines can also exhibit interesting magnetic or conducting properties. Theoretical calculations showed the possibility of metallic conductivity or even superconductivity at electron doping of non-transition metal phthalocyanines.10 Compounds with effective magnetic coupling of spins were obtained. For example, ferrimagnetic ordering of spins below 4.5 K was found in (Cp*2Cr+)[FeI(Pc2−)]−·4C6H4Cl2 which contains chains of alternating FeI (S = 1/2) and CrIII (S = 3/2) spins.11 Magnetic interactions are manifested between the Pc˙3− macrocycles in the case of their effective π–π interactions in dimers or layers.12,13 Reduction of metal phthalocyanines is realized at −0.4–−0.8 V vs. SCE in solutions.14,15 Therefore, all manipulations of metal phthalocyanine radical anions require anaerobic conditions. Probably, for this reason this field has been developed only recently and the number of reduced metal phthalocyanines obtained till now as crystals is still rather limited.16–28 Among them there are salts with cobalt(I) and iron(I) phthalocyanine anions16–20 as well as iron(I) hexadecachlorophthalocyanine anions.21,22 Their formation is accompanied by the reduction of metal atoms in the phthalocyanines. A series of salts with the [MPc]˙− radical anions (M = H2, CuII, NiII, SnII, PbII, TiIVO, VIVO, SnIVCl2, MgII)12,23–30 were also obtained. In this case the reduction is centered on the Pc macrocycle and is accompanied by the formation of paramagnetic Pc˙3− radical trianions. Several neutral metal phthalocyanine compounds are known which contain phthalocyanine radical trianions and even tetraanions: AlIII(Pc˙3−),31,32 GaIII(Pc˙3−),32 GeIV(Pc4−),33 and SnIVPh(Pc˙3−).34 In these compounds, the negative charge on Pc˙3− or Pc4− is compensated for by the positive +3 or +4 charge on the central metal atom.
Reduced metal phthalocyanines can show strong magnetic coupling of spins especially when they have only one axial ligand assisting the formation of π-stacking dimers in the salts.12,35 In this work we summarize our results on the preparation and study of radical anion salts of titanyl (TiIVOPc) and vanadyl (VIVOPc) phthalocyanines12,27,35 and present new salts of these phthalocyanines (Table 1). Different synthetic approaches for the preparation of these salts are presented. The effect of reduction on the molecular structure and properties of titanyl and vanadyl phthalocyanines is also discussed. In this work we discuss how the crystal structures of the salts can be affected by using cations of different size and shape. We found that it is possible to affect the overlapping degree of the Pc˙3− macrocycles in the dimers thereby affecting the magnetic and optical properties of the salts. Based on these data we show how it is possible to control the properties of the compounds by methods of molecular design.
| Cation | Neutral component | Phthalocyanine | Anion | Solvent | TiIVOPc | VIVOPc |
|---|---|---|---|---|---|---|
| Et4N+ – tetraethylammonium; Bu4N+ – tetrabutylammonium; Pent4N+ – tetrapentylammonium; Hex4N+ – tetrahexylammonium; MDABCO+ – N-methyldiazabicyclooctanium; i-Pr2Im+ – 1,3-di(iso-propyl)imidazolium; Me4P+ – tetramethylphosphonium; TPC – triptycene; cryptand: 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane; C6H4Cl2 – o-dichlorobenzene. | ||||||
| (Et4N+) | {MIVO(Pc˙3−)}˙− | C6H4Cl2 | 1 (ref. 12) | — | ||
| (Bu4N+) | {MIVO(Pc˙3−)}˙− | 2 (ref. 12) | 3 (ref. 12) | |||
| (Pent4N+) | {MIVO(Pc˙3−)}˙− | 4 (this work) | 5 (this work) | |||
| (Hex4N+) | {MIVO(Pc˙3−)}˙− | C6H4Cl2 | 6 (this work) | 7 (this work) | ||
| (MDABCO+) | {MIVO(Pc˙3−)}˙− | 8 (this work) | ||||
| (i-Pr2Im+) | {MIVO(Pc˙3−)}˙− | 0.75C6H4Cl2 | 9 (this work) | |||
| (Me4P+) | (TPC)0.5 | {MIVO(Pc˙3−)}˙− | C6H4Cl2 | 10 (ref. 35) | 11 (ref. 35) | |
| (MDABCO+) | (TPC) | {MIVO(Pc˙3−)}˙− | 12 (this work) | |||
| (MDABCO+)2 | {MIVO(Pc˙3−)}˙− | (I−) | 13 (this work) | 14 (this work) | ||
| {Cryptand(Na+)} | {MIVO(Pc˙3−)}˙− | C6H4Cl2 | 15 (ref. 27) | |||
Results and discussion
1. Synthesis
Metal phthalocyanines can be reduced by sodium fluorenone ketyl which can be prepared by direct interaction of fluorenone with metallic sodium in toluene.36 The first oxidation potential of this reductant of −1.3 V vs. Ag/AgCl (or −1.345 vs. SCE) in CH2Cl2 (ref. 37) allows the generation of radical anions of nearly all metal phthalocyanines.14,15 The sodium metal cation of the reductant is substituted by different organic cations at the reduction and these cations become the counterions of reduced metal phthalocyanine (see reaction 1). The reaction is carried out in pure o-dichlorobenzene (or in some cases in the presence of benzonitrile). Sodium halides are insoluble in these solvents; they precipitate from the solution to yield pure phthalocyanine salt.| M(Pc2−) + (Na+)(fluorenone˙−) + (cation+)(Hal−) → (Cation+){M(Pc˙3−)}˙− + fluorenone + (Na+)(Hal−)↓ | (1) |
It is important to use solvents which cannot solvate alkali metal cations, allowing their precipitation from the solution as chloride, bromide or iodide salts. The presence of cations of two types in the synthesis such as solvated alkali metal and organic cations generally does not allow the preparation of good quality single crystals. We used solvents purified of traces of oxygen and water by distillation over CaH2 or sodium benzophenone under argon and then degassing prior to use. All manipulations for the synthesis of the salts were carried out in a glove box with a controlled atmosphere and content of water and oxygen less than 1 ppm. It should be noted that although pristine metal phthalocyanines are very sparingly soluble in organic solvents, their radical anion salts especially with large organic cations are highly soluble in o-dichlorobenzene. Slow mixing of their solutions with n-hexane in the diffusion tube for 1–2 months allowed the preparation of good quality single crystals which precipitated on the walls of the tube. Crystals were studied by X-ray diffraction. After thorough separation of the crystals from any admixtures under a microscope and determination of possible phases according to the unit cell parameters, we analyzed their optical and magnetic properties. The latter properties were studied for polycrystalline samples sealed under an ambient pressure of argon or under a 10−5 torr vacuum in quartz tubes of 2 mm diameter by EPR and SQUID techniques.
There are different synthetic approaches for the preparation of the salts with small organic cations as illustrated by the synthesis of metal phthalocyanine salts with N-methyldiazabicyclooctinium cations (MDABCO+). Reduction of titanyl phthalocyanine in the presence of exactly one molar equivalent of (MDABCO+)(I−) (with a slight excess of sodium fluorenone ketyl as a reductant) followed by precipitation of the salt by n-hexane yields the crystals of (MDABCO+){TiIVO(Pc˙3−)}˙− (8). Since MDABCO+ is a small cation, addition of about 20% of benzonitrile is needed from the total volume of o-dichlorobenzene. Similarly, benzonitrile was added for the preparation of the salt with Et4N+ (ref. 12) and other salts containing MDABCO+.
For the synthesis of the radical anion salts it is possible to use a 2–3-fold molar excess of (MDABCO+)(I−) relative to metal phthalocyanine. The (MDABCO+)2{MIVO(Pc˙3−)}˙−(I−) salts (M = Ti for 13 and M = V for 14) containing two MDABCO+ cations and one I− anion per metal phthalocyanine were obtained by such a procedure. If o-dichlorobenzene molecules are involved in the salts, they contain solvent molecules instead of the second cation as in (Et4N+){TiIVO(Pc˙3−)}˙−·C6H4Cl2 (1) and (i-Pr2Im+){VIVO(Pc˙3−)}˙−·0.75C6H4Cl2 (13). In this case it is not possible to prepare salts with two cations. It should be noted that titanyl and vanadyl phthalocyanines form only 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salts with a large Bu4N+ cation even with an excess of (Bu4N+)(Br−) in the reaction mixture despite of planar phthalocyanines (H2Pc, CuIIPc, NiIIPc) form salts with two Bu4N+ cations and one Br− anions.12
:1 salts with a large Bu4N+ cation even with an excess of (Bu4N+)(Br−) in the reaction mixture despite of planar phthalocyanines (H2Pc, CuIIPc, NiIIPc) form salts with two Bu4N+ cations and one Br− anions.12
In the studies on fullerene salts, the authors derived the concept of multi-component approach in which a neutral component is added along with the source of the counterion to define the crystal packing at the salt.41 This method is successfully applied also in the anionic phthalocyanine chemistry. The aromatic hydrocarbon triptycene (TPC) can form layers with hexagonal vacancies which can accommodate small cations like MDABCO+ or Me4P+ to form the salt (MDABCO+)(TPC){TiIVO(Pc˙3−)}˙− (12) or (Me4P+)(TPC)0.5{MIVO(Pc˙3−)}˙−·C6H4Cl2 (M = Ti (10) and V (11)). In this case metal phthalocyanines are also packed in layers alternating with the cationic layers containing neutral TPC molecules. As a result, this method is suitable for the preparation of layered structures. Only small cations can be used together with TPC to form multi-component complexes. Even the Et4N+ cations are too large to be incorporated into the TPC layers and synthesis of the salt with Et4N+ and TPC yields (Et4N+){MIVO(Pc˙3−)}˙−·C6H4Cl2 (1) only.
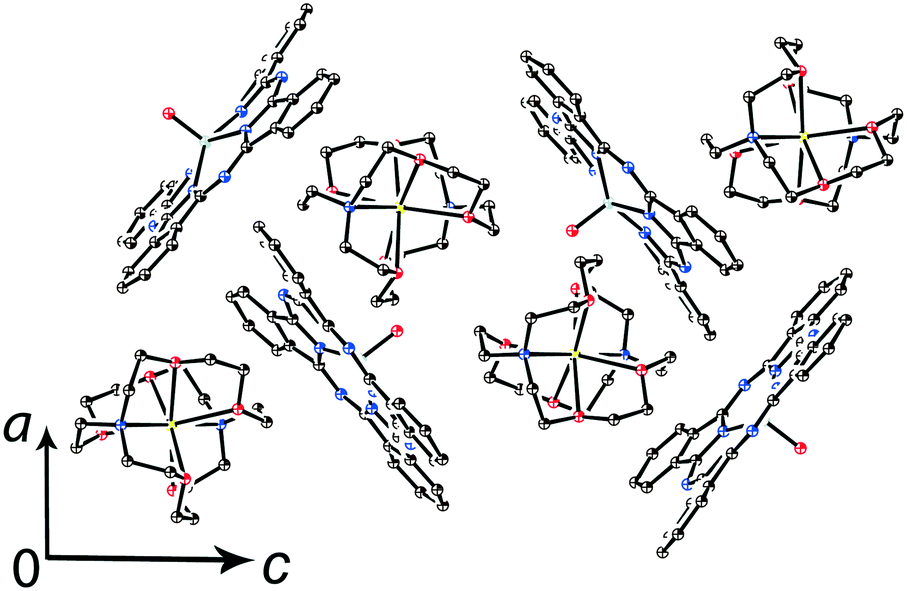
Components that cocrystallized with titanyl and vanadyl phthalocyanine radical anions in salts 1–14 are shown in Fig. 1. It is seen that the size of the cations varies from small (MDABCO+ and Me4P+) to large (Hex4N+ and cryptand[2,2,2](Na+)) (the latter cation is not shown in Fig. 1). The variation of the cations size allows one to study how the size of cations affects the crystal structure of the salts and packing of the macrocycles in the π-stacking [{MIVO(Pc˙3−)}˙−]2 dimers.
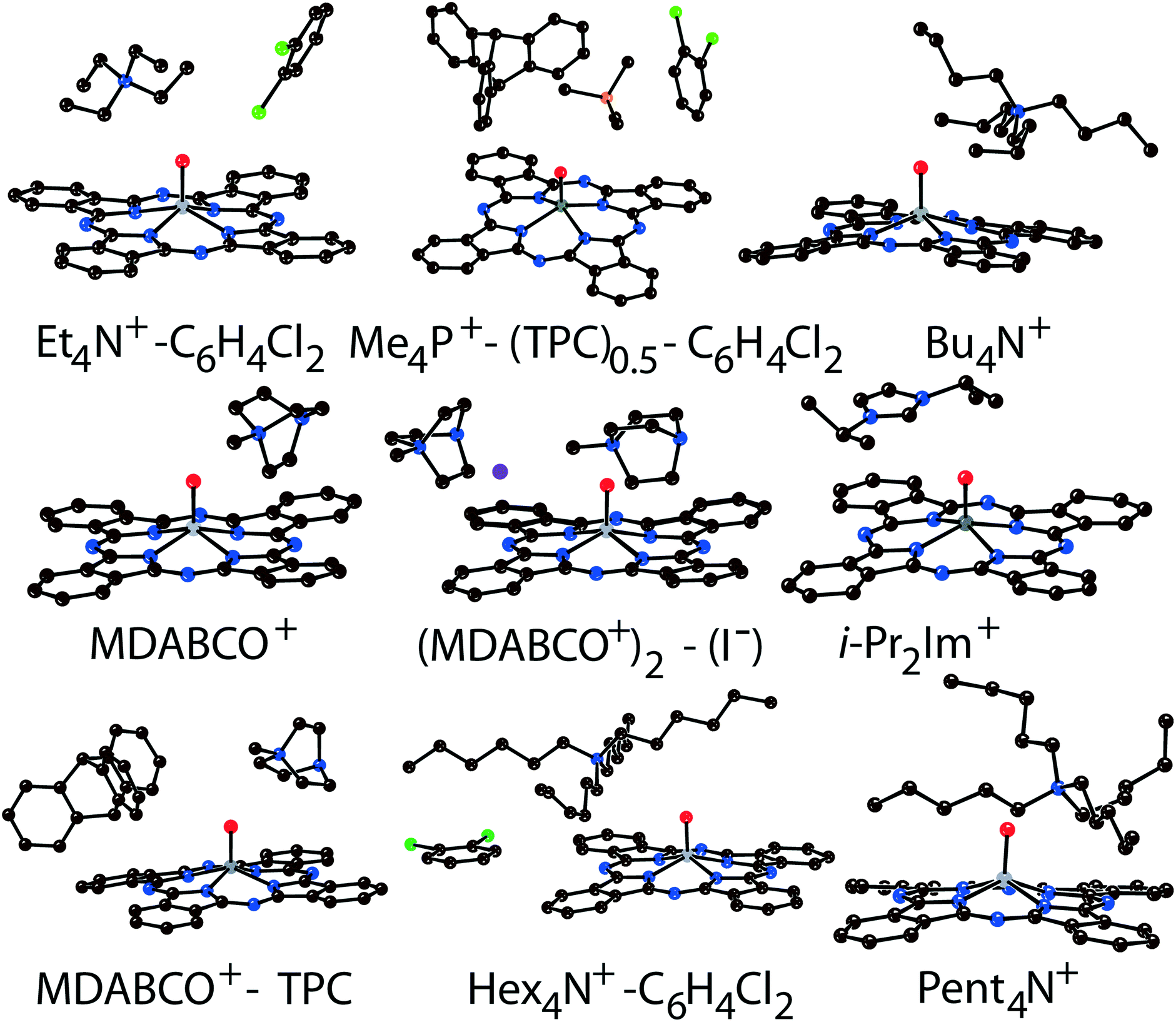 | ||
| Fig. 1 Molecular structures of the components in salts 1–14. Carbon atoms are reported in brown, nitrogen atoms in blue, oxygen atoms in red, metal atoms in grey, iodine atoms in violet, chlorine atoms in green, and phosphorus atom in orange. | ||
2. Crystal structures
The structure formed by titanyl phthalocyanine radical anions in (Et4N+){TiIVO(Pc˙3−)}˙−·C6H4Cl2 (1)12 is shown in Fig. 2. It is a layered structure in which phthalocyanine layers alternate with the Et4N+–C6H4Cl2 layers. Both the Et4N+ cations and solvent C6H4Cl2 molecules have a small size and a total of four such units are positioned over each phthalocyanine plane (Fig. 2b). The presence of only one axial substituent in {TiIVO(Pc˙3−)}˙− allows the formation of π–π stacking [{TiIVO(Pc˙3−)}˙−]2 dimers within the phthalocyanine layers. The Pc planes are arranged in these dimers very close to each other with an interplanar distance of only 3.13 Å, and 18 short van der Waals C,N⋯C,N contacts are formed between the phthalocyanines. There are several side-by-side C⋯C contacts in the layers between phthalocyanines belonging to neighboring dimers. These contacts are shown by green dashed lines in Fig. 2a. Thus, weak interactions between the dimers having a square arrangement in the layers are observed. The intermolecular overlap integral for the singly occupied molecular orbitals (SOMOs) of {TiIVO(Pc˙3−)}˙− in 1 was calculated by the extended Hückel method.12 The SOMO–SOMO overlap integral of 14.3 × 10−3 is large to show essential π–π interactions between the Pc˙3− macrocycles in 1. This is due to the small shift of phthalocyanines relative to each other in the dimers and a short interplanar distance between them. Indeed, macrocycles are positioned in such a way that the titanium atom of one phthalocyanine is positioned over the pyrrole nitrogen atom of the neighboring phthalocyanine (the shift of these macrocycles is 2.005 Å).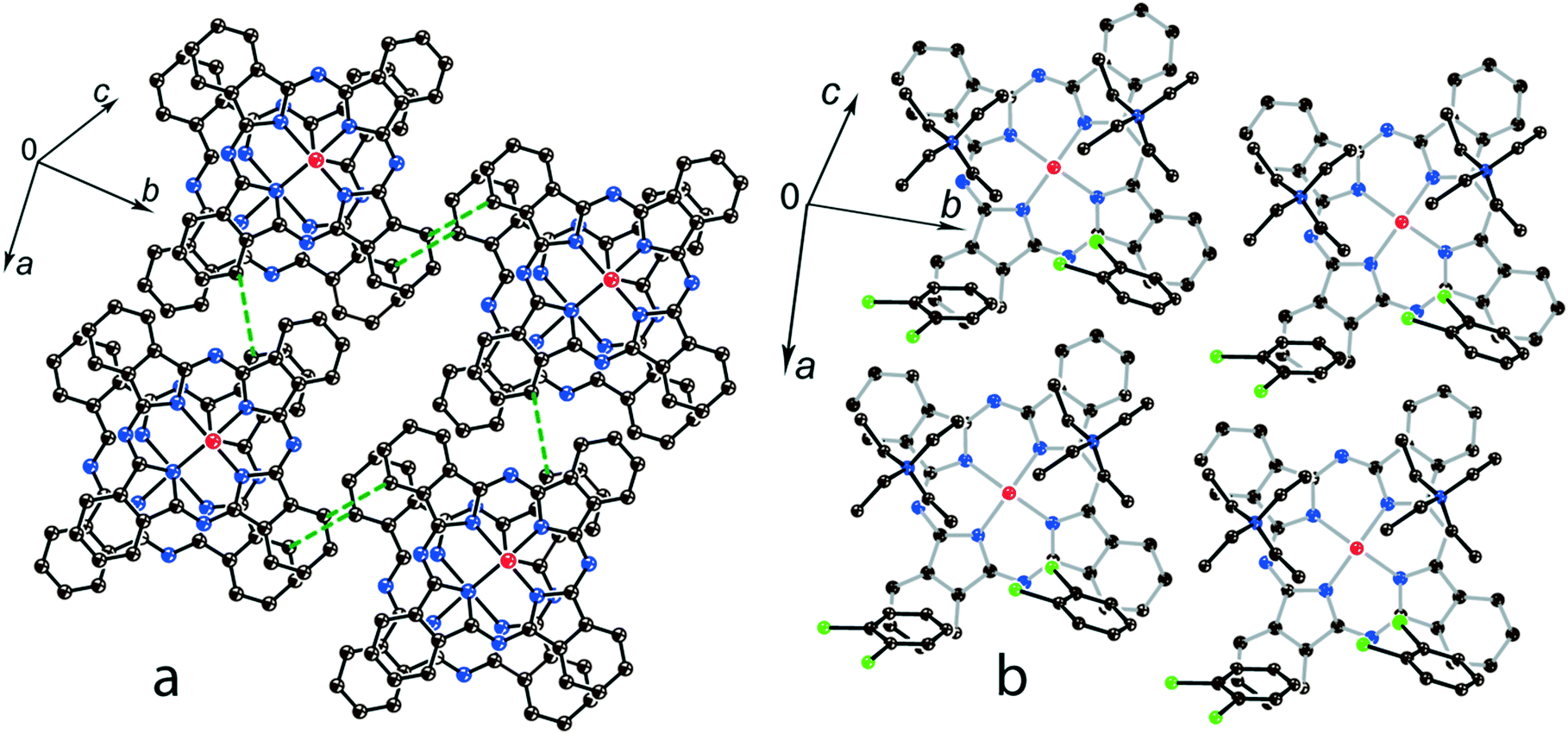 | ||
| Fig. 2 (a) View of the phthalocyanine layer composed of the π-stacking [{TiIVO(Pc˙3−)}˙−]2 dimers in 1.12 Short van der Waals C,N⋯C,N contacts are shown by green dashed lines. (b) Projection of the Et4N+–C6H4Cl2 layer on the phthalocyanine layer. Only one phthalocyanine from the dimer is shown (bonds are shown in grey). | ||
Crystal structures of multi-component salts (Me4P+)(TPC)0.5{MIVO(Pc˙3−)}˙−·C6H4Cl2 (M = Ti (10) and V (11)) were described previously.35 They also contain layers formed by π-stacking [{MIVO(Pc˙3−)}˙−]2 dimers alternating with Me4P+–TPC–C6H4Cl2 layers along the c axis. The shift of the Pc˙3− macrocycles in the dimers of 10 and 11 is larger than that in 1, providing essentially smaller SOMO–SOMO overlap integrals of 2.2 × 10−3 and 1.7 × 10−3, respectively.35
In previously described12 crystal structures of 2 and 3, the size of the Bu4N+ cations is essentially larger than that of Et4N+. As a result, only two Bu4N+ cations without solvent molecules are arranged near each Pc˙3− macrocycle, forming cationic layers (Fig. 3b). In this case, phthalocyanines are also packed in layers consisting of π-stacking [{MIVO(Pc˙3−)}˙−]2 dimers. However, the increase in cation size provides an essential shift (5.12 Å) of two Pc˙3− macrocycles relative to each other in the dimers. This effect can be explained by the fact that the increase in size of the cationic layer proportionally increases the size of the phthalocyanine layers. As a result, phthalocyanine macrocycles should be shifted relative to each other in the dimers to fit a larger area of the layers. The interplanar distance in the dimers increases up to 3.19 Å for both 2 and 3. The increase in cation size essentially decreases the π–π overlapping between the Pc˙3− macrocycles since the intermolecular SOMO–SOMO overlap integrals are only 0.1 × 10−3 and 0.3 × 10−3 for 2 and 3, respectively.12 Therefore, a several orders of magnitude decrease of the overlap integrals is observed for 2 and 3 compared with 1.
 | ||
| Fig. 3 (a) View of the phthalocyanine layer composed of the π-stacking [{TiIVO(Pc˙3−)}˙−]2 dimers in 2.12 Short van der Waals C,N⋯C,N contacts are shown by green dashed lines. (b) Projection of the Bu4N+ layer on the phthalocyanine layer. Only one phthalocyanine from the dimer is shown (bonds are shown in grey). | ||
The structures of 4–7 have not been published previously. The Pent4N+ cations form layers parallel to the bc plane in the isostructural (Pent4N+){TiIVO(Pc˙3−)}˙− (4) and (Pent4N+){VIVO(Pc˙3−)}˙− (5) salts. Two large Pent4N+ cations are arranged near each phthalocyanine plane, and the π-stacking [{MIVO(Pc˙3−)}˙−]2 dimers are also packed in layers (Fig. 4a). The extension of the alkyl substituents of the cations by one CH2 unit affects the packing of the macrocycles in the dimers. The interplanar distance in the dimers of 4 and 5 is increased up to 3.22–3.24 Å. The Pc˙3− macrocycles are shifted by 5.61 Å relative to each other in 4 and 5, providing only slight overlapping of the Pc˙3− macrocycles. These data show that the use of the Pent4N+ cations instead of Bu4N+ impairs the conditions for π–π interactions between Pc˙3− in the dimers. The size of the Pent4N+ cations can be considered as a boundary for the structures of metal phthalocyanines with one axial ligand at which the formation of π-stacking [{MIVO(Pc˙3−)}˙−]2 dimers is still possible.
 | ||
| Fig. 4 (a) View of the phthalocyanine layer composed of the π-stacking [{TiIVO(Pc˙3−)}˙−]2 dimers in 4. Short van der Waals C,N⋯C,N contacts are shown by green dashed lines. (b) Projection of the Pent4N+ layer on the phthalocyanine layer. Only one phthalocyanine from the dimer is shown (bonds are shown in grey). | ||
The study of the structures of (Hex4N+){TiIVO(Pc˙3−)}˙−·C6H4Cl2 (6) and (Hex4N+){VIVO(Pc˙3−)}˙−·C6H4Cl2 (7) shows that the increase in the cation size by one more CH2 unit does not allow the formation of π-stacking [{MIVO(Pc˙3−)}˙−]2 dimers. Instead, the {TiIVO(Pc˙3−)}˙− radical anions form one-dimensional chains with one side-by-side C⋯C contact (Fig. 5). These chains are completely isolated in a crystal by long alkyl substituents of the Hex4N+ cations.
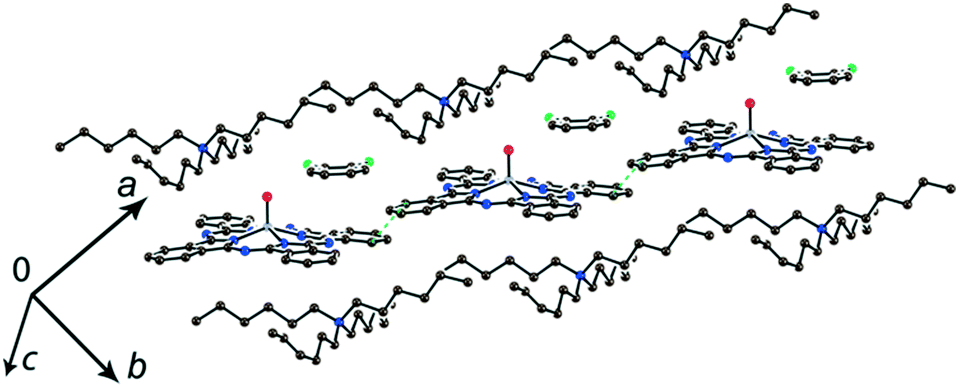 | ||
| Fig. 5 View of the chains from {TiIVO(Pc˙3−)}˙− in (Hex4N+){TiIVO(Pc˙3−)}˙−·C6H4Cl2 (6). | ||
Further increase of cation size does not allow the formation of one-dimensional phthalocyanine chains. Crystals of the {MIVO(Pc˙3−)}˙− salts with tetraoctylammonium cations were not obtained. However, our previous study shows that this cation assists in the formation of structures with a perpendicular arrangement of the Pc planes and large channels occupied by cations and solvent molecules.23 The structure of {cryptand[2,2,2](Na+)}{VIVO(Pc˙3−)}˙−·C6H4Cl2 (15) with large nearly spherical cryptand[2,2,2](Na+) cations also shows a perpendicular arrangement of the Pc˙3− macrocycles with channels occupied by cations and solvent molecules (Fig. 6).27 Any π–π interactions between the Pc planes are absent in such structures in spite of the presence of several side-by-side C⋯C contacts between Pc˙3−. Therefore, large cations do not yield salts with noticeable π–π interactions between Pc˙3−. Layered structures with π-stacking [{MIVO(Pc˙3−)}˙−]2 dimers are formed by using cations of relatively small or moderate size (smaller than Hex4N+). It is seen that the size of the cations defines the shift of the Pc˙3− macrocycles in the dimers affecting the overlap integrals between SOMO orbitals of {MIVO(Pc˙3−)}˙− and, correspondingly, the physical properties of the salts.
 | ||
| Fig. 6 View of the crystal structure of {cryptand(Na+)}{VIVO(Pc˙3−)}˙−·C6H4Cl2 (15) along the b axis and channels occupied by solvent C6H4Cl2 molecules (not shown for clarity) and the cryptand(Na+) cations.27 | ||
3. Magnetic properties
Titanyl and vanadyl phthalocyanine radical anions have different magnetic states. Titanyl phthalocyanine contains diamagnetic TiIV, and the magnetic properties of {TiIVO(Pc˙3−)}˙− are defined by the S = 1/2 spin delocalized over Pc˙3−. This system can be considered as an organic π-radical having an unpaired S = 1/2 spin only on the π-orbitals of the macrocycle. Vanadyl phthalocyanine radical anions, {VIVO(Pc˙3−)}˙−, have two S = 1/2 spins per radical anion since one S = 1/2 spin is delocalized over Pc˙3− and the other S = 1/2 spin is localized mainly on the VIV d-orbitals. Therefore, it is possible to study how the presence of paramagnetic metal atoms in the center of the macrocycle affects the magnetic properties of isostructural salts. Previously it was shown that paramagnetic metal centers like FeIII in some cases affect the conductivity of the {MIII(CN)2(Pc)}− salts with partially oxidized Pc macrocycles due to effective interactions of d-electrons of FeIII with conducting π-electrons of the macrocycles. This provides an anisotropic magnetoresistance according to the direction of magnetic field.5–7Data of magnetic measurements for the discussed salts are listed in Table 2. For all the salts with titanyl phthalocyanine radical anions, effective magnetic moments are in the 1.71–1.80 μB range at 300 K (Table 2). A theoretically calculated value for the system with one non-interacting S = 1/2 spin is equal to 1.73 μB and the observed magnetic moments of the salts are close to this value.
| Cation | TiIVOPc | VIVOPc |
|---|---|---|
| Et4N+ | 1, J/kB = −255 K, —, μeff. = 1.72 μB, 14.3 × 10−3, dimers12 | — |
| Me4P+(TPC)0.5 | 10, J/kB = −123 K, Θ = −131 K, μeff. = 1.71 μB, 2.2 × 10−3, dimers35 | 11, J/kB,inter = −105 K, Θ = −86 K, J/kB,intra = −15.2 K, μeff. = 2.19 μB, 1.7 × 10−3, dimers35 |
| Bu4N+ | 2, —, Θ = −57 K, μeff. = 1.72 μB, 0.1 × 10−3, dimers12 | 3, —, Θ = −9.6 K, μeff. = 2.37 μB, 0.3 × 10−3, dimers12 |
| Pent4N+ | 4, —, Θ = −1 K, μeff. = 1.80 μB, dimers | 5, Θ = −1.4 K, J/kB,intra = +5.6 K, μeff. = 2.38 μB, dimers |
| Hex4N+ | 6, —, Θ = − 3 K, μeff. = 1.78 μB, no dimers | 7, Θ = −2.3 K, J/kB,intra = +3.3 K, μeff. = 2.41 μB, no dimers |
| Cryptand(Na+) | — | 15, Θ = −1.3 K, J/kB,intra = +7.7 K, μeff. = 2.22 μB, no dimers27 |
For salt 1 with the smallest Et4N+ cations the decrease of molar magnetic susceptibility is observed below 300 K and dimers are transferred to the diamagnetic singlet state below 150 K (ref. 12) (Fig. 7a). Such a behavior can be explained by triplet–singlet transitions in the dimers at very strong antiferromagnetic coupling between spins.12 However, when we try to fit the experimental data by the Heisenberg model for pairs of isolated antiferromagnetically interacting spins,40 this model does not describe well the magnetic behavior of 1. At the same time the observed magnetic behavior can be fitted well by the Heisenberg model for quasi-two-dimensional square layers38 with a very large exchange interaction of J/kB = −255 K (Fig. 7a). Since the [{TiIVO(Pc˙3−)}˙−]2 dimers have a square arrangement in 1 and there is a weak overlapping between Pc˙3− macrocycles of the neighboring dimers (Fig. 2), we suppose the antiferromagnetic ordering of spins within phthalocyanine layers to be possible. Taking into account that magnetic coupling is realized only between π-electrons of Pc˙3−, the observed exchange interaction is very large for such a type of interactions.
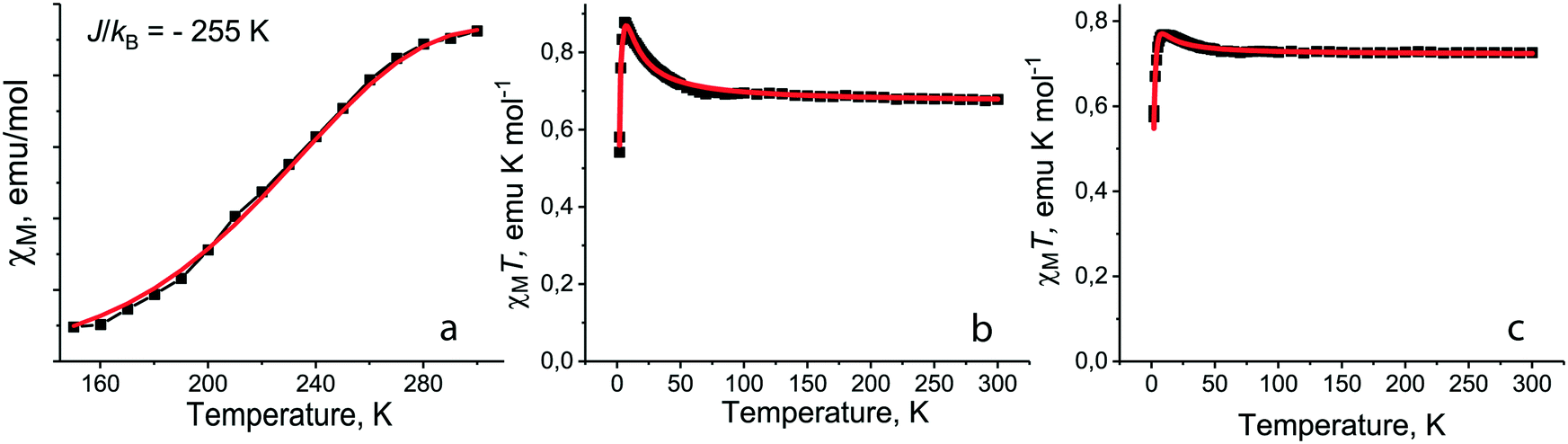 | ||
| Fig. 7 (a) Temperature dependence of molar magnetic susceptibility of 1 in the 150–300 K range (black squares) and fitting the data by the Heisenberg model for quasi-square layered arrangement of spins38 with J/kB = −255 K (red curve); fitting the experimental χMT data (black squares) by the modified S–T model39 for 5 (b) with Jintra/kB = +5.6 K, θ = −1.4 K, g = 2 (fix), f = 0.996 (red curve) and for 7 (c) with Jintra/kB = +3.3 K, θ = −2.3 K, g = 2 (fix), f = 0.999 (red curve). | ||
Introduction of neutral TPC molecules into the cationic Me4P+ layers in (Me4P+)(TPC)0.5{TiIVO(Pc˙3−)}˙−·C6H4Cl2 (10) increases their size and additionally shifts the Pc˙3− macrocycles in the dimers.35 The magnetic behavior of 10 is described well by the Heisenberg model for pairs of isolated antiferromagnetically coupled spins with an exchange interaction of −123 K. This interaction is still rather large, providing transition of the dimers to the diamagnetic state below T < 130 K, but it is nearly two times smaller than that in 1. The use of the model for pairs is supported by the isolation of dimers in 10 since SOMO–SOMO overlap integrals with the neighboring dimers within the layers are smaller than 0.1 × 10−3.35
Large Bu4N+ cations in (Bu4N+){TiIVO(Pc˙3−)}˙− (2) provide an even stronger shift of the Pc˙3− macrocycles in the dimers (Fig. 3) and a small SOMO–SOMO overlap integral showing weak π–π interactions in 2. As a result, no transition of the dimers to the diamagnetic singlet state is observed and spins are only antiferromagnetically coupled but with a rather large Weiss temperature of −57 K.12
The magnetic properties of salts 4–7 are presented in the ESI† (Fig. S15–S18). The Pent4N+ cations in (Pent4N+){TiIVO(Pc˙3−)}˙− (4) are larger than the Bu4N+ ones. The overlapping between the Pc˙3− macrocycles is still preserved in [{TiIVO(Pc˙3−)}˙−]2 but becomes weaker due to an additional shift of the macrocycles (Fig. 4). This salt shows a nearly paramagnetic behavior with a Weiss temperature close to zero (Θ = −1 K, Fig. S15†) which shows the absence of magnetic coupling between spins in the dimers. It is seen that the use of Pent4N+ cations suppresses magnetic coupling between spins within the dimers due to the essential shift of the macrocycles. Thus, a simple variation of cation size in such systems allows regulation of magnetic coupling between spins ranging from very strong up to nearly the absence of any magnetic coupling.
Salt (Hex4N+){TiIVO(Pc˙3−)}˙−·C6H4Cl2 (6) has a different packing of {TiIVO(Pc˙3−)}˙− which are arranged in the one-dimensional chains (Fig. 5). In this case only weak antiferromagnetic coupling between spins is realized with a Weiss temperature of −3 K (Table 2 and Fig. S16†). Similar weak magnetic interactions were found for different salts with the radical anions of copper(II), nickel(II), tin(II), lead(II) and metal-free phthalocyanines having nearly isolated packing of the Pc˙3− (or H2Pc˙−) macrocycles.12 The largest magnetic coupling among these salts with a Weiss temperature of −17 K was found for (Bu4N+)2{PbII(Pc˙3−)}˙3−(Br−).12
Several salts (3, 5, 7, 11, and 15) synthesized with the {VIVO(Pc˙3−)}˙− radical anions are isostructural to the {TiIVO(Pc˙3−)}˙− salts (Table 2). These salts have effective magnetic moments from 2.19 to 2.41 μB at 300 K in accordance with the presence of two S = 1/2 spins per one radical anion (Table 2). The calculated value for the system with two noninteracting S = 1/2 spins is equal to 2.45 μB. To understand the magnetic behavior of individual {VIVO(Pc˙3−)}˙− radical anions we consider the magnetic behavior of salts with isolated Pc˙3− packing: (Pent4N+){VIVO(Pc˙3−)}˙− (5), (Hex4N+){VIVO(Pc˙3−)}˙−·C6H4Cl2 (7) and {cryptand[2,2,2](Na+)}{VIVO(Pc˙3−)}˙−·C6H4Cl2 (15). All of them show similar magnetic behavior with a slight increase of the magnetic moment below 60 K and its abrupt decrease at low temperatures (T < 10 K) (Fig. 7b and c for salts 5 and 7, respectively). Such a magnetic behavior can be described well by a modified singlet–triplet (S–T) model39 with a rather strong ferromagnetic intramolecular exchange interaction between the Pc˙3− and VIV spins. This intramolecular exchange interaction (J/kB) ranges from +3.3 up to +7.7 K, while weak antiferromagnetic intermolecular coupling between the Pc˙3− macrocycles is found with a Weiss temperature (Θ) from −1.3 up to −2.3 K. In the case of 5, 7 and 15 intermolecular coupling is weak due to the absence of π–π interactions between Pc˙3−. When more effective π–π interactions are realized in 3 and 11, antiferromagnetic intermolecular exchange interactions can attain essentially larger values (up to J/kB = −105 K for 11).35 Since this value is nearly 10 times larger than those for the ferromagnetic intramolecular interactions, the total magnetic behavior of these salts is antiferromagnetic. Ferromagnetic intramolecular interactions and antiferromagnetic intermolecular interactions suppress each other. As a result, a comparative analysis of magnetic properties of salts 2, 3 and 10, 11 shows that the total antiferromagnetic coupling is noticeably weaker for the {VIVO(Pc˙3−)}˙− salts in comparison with the {TiIVO(Pc˙3−)}˙− salts (Table 2).
The EPR spectra of newly prepared compounds are shown in Fig. S19–S29† and the parameters of the signals are listed in Table 3. All salts with the {TiIVO(Pc˙3−)}˙− radial anions manifest EPR signals containing a main component with g = 1.9994–2.0008 and a linewidth (ΔH) of 1.50–3.67 mT. Only in case of salt 4 was a two-component spectrum found with the components of nearly equal intensity and linewidth (Fig. S19†). In spite of the S = 1/2 spin is delocalized over the Pc˙3− macrocycles in {TiIVO(Pc˙3−)}˙−, the parameters of the EPR signal in salts 1, 2, 4, 6, 10 and 13 are noticeably different from those of the salts with the metal-free phthalocyanine (H2Pc˙−) radical anions (g = 2.0030–2.0035 and ΔH = 0.1–0.3 mT).12,18 Therefore, the central metal TiIV atom, in spite of diamagnetism, affects the parameters of the EPR signal from the macrocycle. A temperature decrease generally narrows the EPR signals and increases their asymmetry since they are split into two or three lines at low temperatures (Table 3). Salts containing the {VIVO(Pc˙3−)}˙− radical anions show essentially broader EPR signals with g = 1.9878–1.9954 and ΔH = 7.80–25.30 mT. Since these species contain two paramagnetic centers (VIV and Pc˙3− with an S = 1/2 spin state), the observed signal originates from both centers having an exchange interaction. As a result, the observed signal has approximately intermediate parameters between those characteristic of VIV and Pc˙3−. In fact, for example, the room-temperature spectrum of pristine [VIVOPc2−]0 contains a broad main component (ΔH = 24.9 mT) from VIV with g = 1.9858 (ref. 12), whereas the EPR spectra of the salts with {TiIVO(Pc˙3−)}˙− containing Pc˙3− manifest EPR signals with the main component at g = 1.9994–2.0008 and ΔH = 1.50–3.67 mT (Table 3). Generally, a temperature decrease results in narrowing of the EPR signals from the salts and asymmetry increase as in case of the {TiIVO(Pc˙3−)}˙− salts. However, in several cases (11 and 14) essential broadening and shift to the higher g-factors are observed for the main components. This can be explained (as was shown previously for 11)35 by a strong antiferromagnetic coupling of spins delocalized over Pc˙3−. The contribution from Pc˙3− disappears at low temperatures due to the antiparallel arrangement of these spins, providing the manifestation of contribution only from weaker interacting VIV spins which have broad EPR signals at low temperatures.
| N | Room temperature g-Factor (linewidth (mT), relative intensity (%)) | Low temperature g-Factor (linewidth (mT), relative intensity (%)) |
|---|---|---|
| Salts with {TiIVO(Pc˙3−)}˙− | ||
| 1 (ref. 12) | 2 lines | 2 lines, 4.2 K |
| 2.0006 (3.54, 97%) | 1.9991 (1.0, main) | |
| 2.0025 (0.78, 3%) | 2.0023 (0.52) | |
| 2 (ref. 12) | 1 line | 2 lines, 4.2 K |
| 2.0003 (1.50) | 2.0010 (0.57, 50%) | |
| 1.9981 (1.00 50%) | ||
| 4 | 2 line | 3 lines, 50 K |
| 2.0033 (0.77, 50%) | 2.0037 (0.32, 20%) | |
| 1.9994 (1.17, 50%) | 2.0008 (0.62, 50%) | |
| 1.9963 (0.55, 30%) | ||
| 6 | 1 line | 2 lines, 50 K |
| 2.0008 (3.58) | 1.9952 (1.37, 80%) | |
| 2.0017 (0.74, 20%) | ||
| 10 (ref. 35) | 2 lines | — |
| 2.0001 (3.67, 93.2%) | ||
| 2.0031 (1.82, 6.8%) | ||
| 13 | 1 line | 2 lines, 4.2 K |
| 2.0007 (2.62) | 2.0024 (0.54, 45%) | |
| 1.9981 (1.25, 55%) | ||
| Salts with {VIVO(Pc˙3−)}˙− | ||
| 3 (ref. 12) | 1 line | 2 lines, 4.2 K |
| 1.9918 (7.8) | 1.9886 (7.12, 80%)1.9928 (4.04, 20%) | |
| 5 | 1 line | 2 lines, 50 K |
| 1.9902 (10.2) | 1.9849 (7.86, 10%) | |
| 1.9868 (29.7, 90%) | ||
| 7 | 1 line | 1 line |
| 1.9919 (9.17) | 1.9893 (3.74) | |
| 11 (ref. 35) | 2 lines | 2 lines, 6.9 K |
| 1.9954 (25.32, 90%) | 1.9832 (32.2, 40%) | |
| 2.0002 (10.49, 10%) | 2.0111 (27.3, 60%) | |
| 14 | 1 line | 2 lines |
| 1.9878 (11.19) | 1.9851 (9.97, 30%) | |
| 2.1323 (55.23, 70%) | ||
| 15 (ref. 27) | 1 line | 1 line |
| 1.9923 (8.91) | 1.9912 (4.37) | |
4. Control of the crystal packing of the macrocycles.
It is possible to control the crystal packing of the macrocycles by using different synthetic approaches. In the presence of small cations the [{MIVO(Pc˙3−)}˙−]2 dimers are packed in layers when the area size occupied by couples of the cations and solvent molecules (a total of 4 units) are close to that of the phthalocyanine macrocycle. For example, in (Et4N+){TiIVO(Pc˙3−)}˙−·C6H4Cl2 (1) two positions are occupied by the Et4N+ cations and two positions are occupied by solvent C6H4Cl2 molecules as described above (Fig. 2b). Synthesis with an excess of (MDABCO+)(I−) results in the formation of titanyl and vanadyl phthalocyanine salts with four MDABCO+ cations per one [{MIVO(Pc˙3−)}˙−]2 dimer. In this case two I− anions are involved in the salt to compensate for an excess of positive charge of the cations in (MDABCO+)2{MIVO(Pc˙3−)}˙−(I−) (M = Ti for 13 and M = V for 14). As a result, a total of four MDABCO+ cations are arranged near the phthalocyanine plane in both salts but two I− anions are inserted between the cations (Fig. 8b). Such a packing yields the layered structure in which phthalocyanine layers composed of the [{MIVO(Pc˙3−)}˙−]2 dimers are formed (Fig. 8a). Since MDABCO+ cations have a small size like Et4N+, the arrangements of the macrocycles in the dimers of salt 1 and both salts 13 and 14 are similar. They have the same small shift of the Pc˙3− macrocycles relative to each other (2.006 (13) and 2.003 Å (14)) and slightly larger interplanar distances of 3.205 Å for 13 and 14. It is possible to suppose that similarly to salt 1, salts 13 and 14 can show a rather strong magnetic coupling between the Pc˙3− spins in the dimers.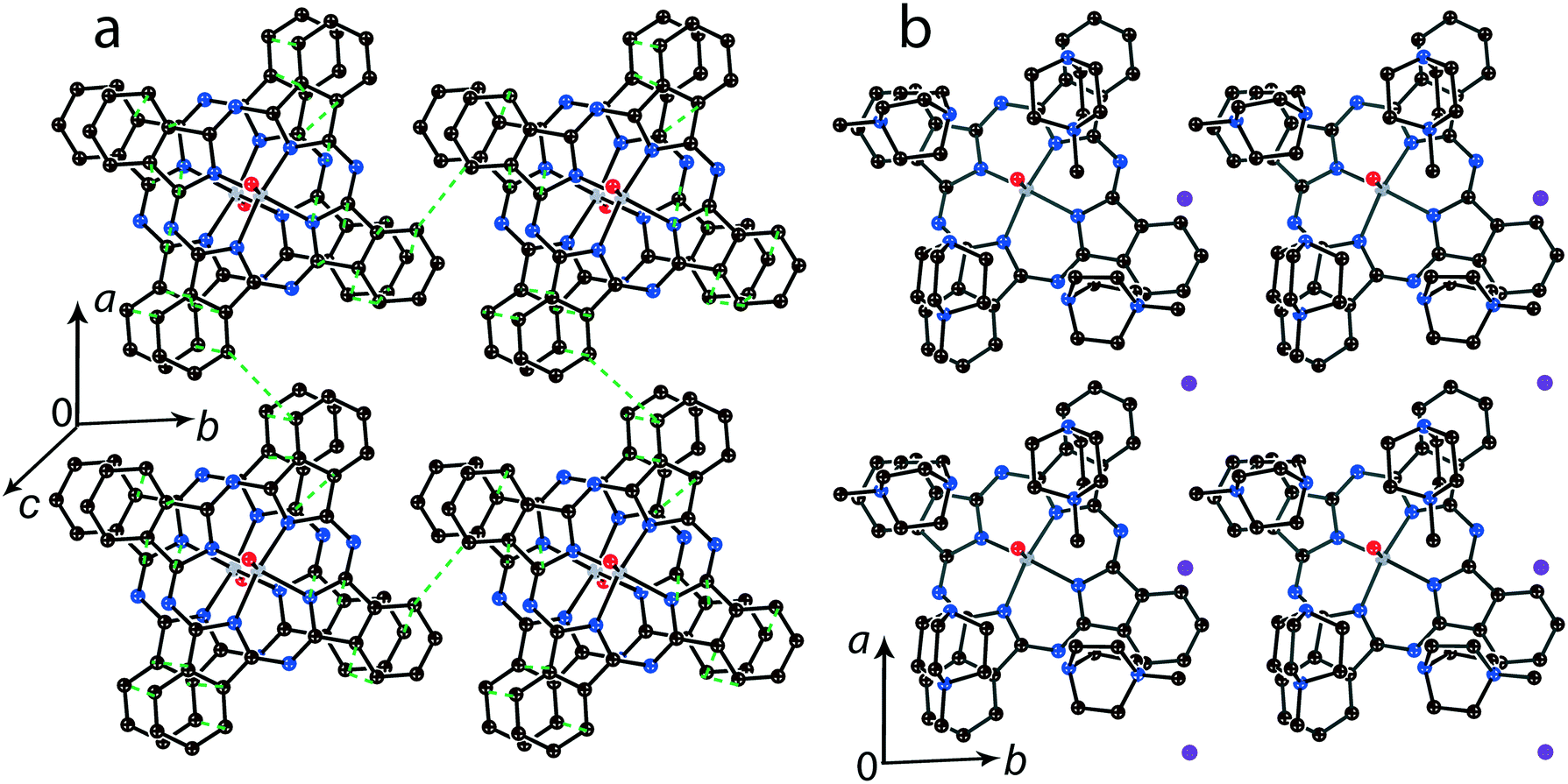 | ||
| Fig. 8 (a) View of the phthalocyanine layer composed of the π-stacking [{TiIVO(Pc˙3−)}˙−]2 dimers in 13. Short van der Waals C,N⋯C,N contacts are shown by green dashed lines. (b) Projection of the (MDABCO+)2 – (I−) layer on the phthalocyanine layer. Only one phthalocyanine from the dimer is shown (bonds are shown in grey). Salt 14 is isostructural to 13. | ||
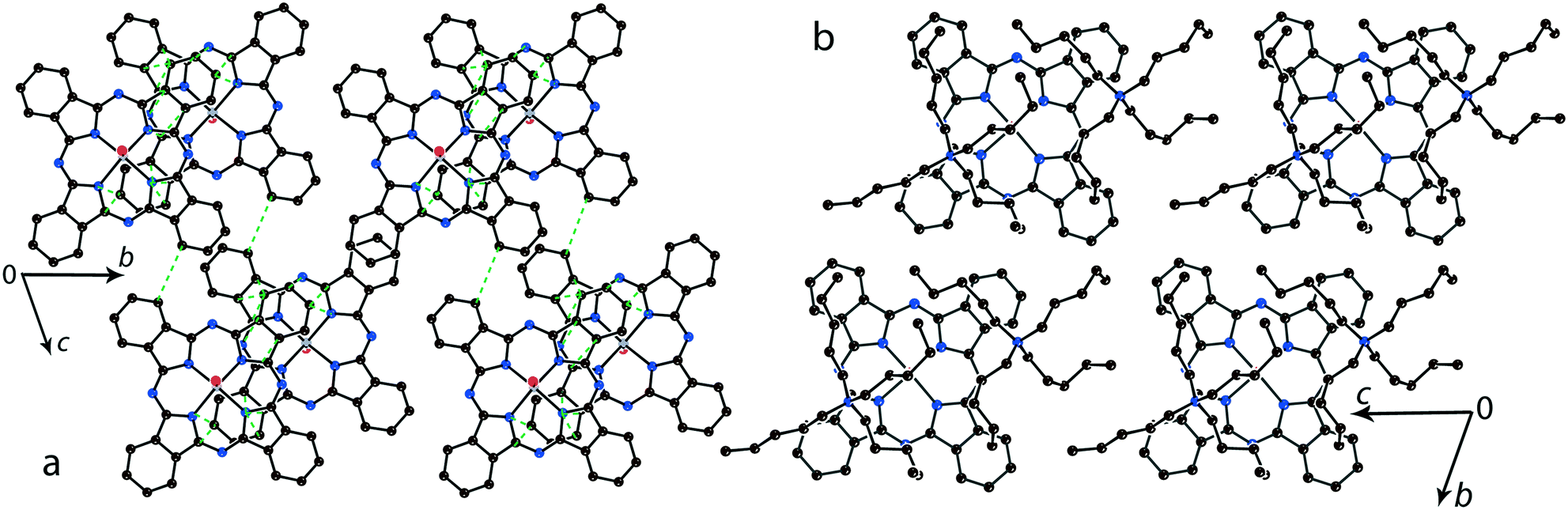
For the 1:1 salts with small organic cations like (MDABCO+){TiIVO(Pc˙3−)}˙− (8) and (i-Pr2Im+){VIVO(Pc˙3−)}˙− ·0.75C6H4Cl2 (9) another packing mode of the macrocycles is observed. In this case only two cations are positioned near each phthalocyanine plane, and these cations are alternated with the [{MIVO(Pc˙3−)}˙−]2 dimers in the chains as shown in Fig. 9. The structure is organized in such a way that two other positions above the phthalocyanine plane are occupied by phenylene substituents of two {MIVO(Pc˙3−)}˙− from the dimers belonging to the neighboring chains. In 8, such chains form layers parallel to the ab plane and the direction of the chains changes at the transition from one layer to another between [1 1 0] and [1−1 0]. The [{TiIVO(Pc˙3−)}˙−]2 dimers in 8 have a small shift of the Pc˙3− macrocycles (2.005 Å) like that observed for 1 and 13 and the interplanar distance between them is 3.231 Å. Thus, a rather strong magnetic coupling between spins can be realized within the isolated [{TiIVO(Pc˙3−)}˙−]2 dimers in 8.
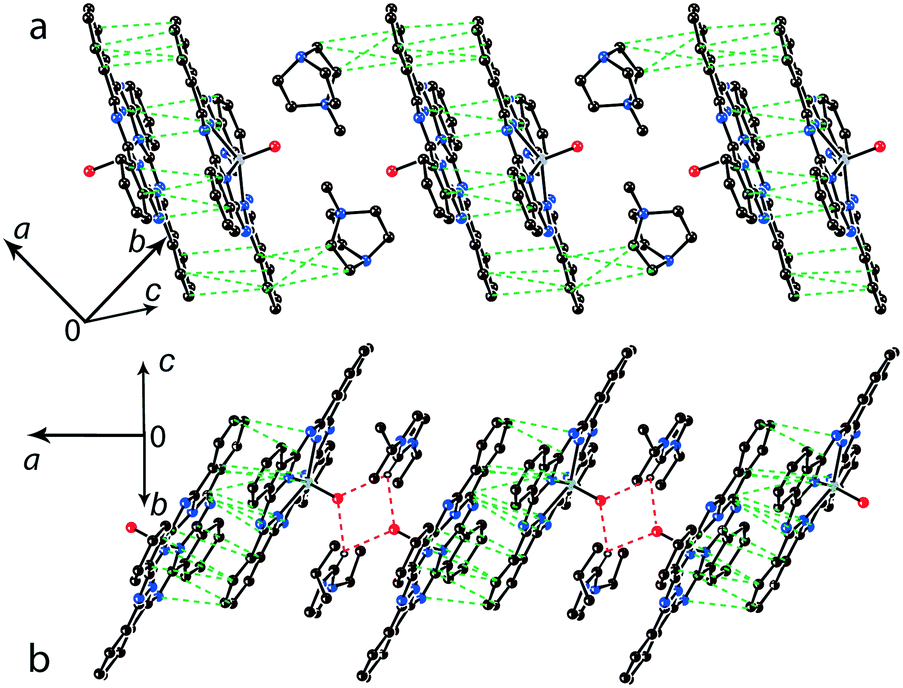 | ||
| Fig. 9 View of the chains composed of π-stacking [{TiIVO(Pc˙3−)}˙−]2 dimers and the MDABCO+ cations in 8 (a) and the [{VIVO(Pc˙3−)}˙−]2 dimers and i-Pr2Im+ cations in 9 (b). Van der Waals C,N⋯C,N contacts and hydrogen H(i-Pr2Im+)⋯O({VIVO(Pc˙3−)}˙−) bonds are shown by green and red dashed lines, respectively. | ||
Salt (i-Pr2Im+){VIVO(Pc˙3−)}˙−·0.75C6H4Cl2 (9) also contains alternating [{VIVO(Pc˙3−)}˙−]2 dimers and pairs of i-Pr2Im+ cations (Fig. 9b). This salt has some peculiarities due to the formation of multiple hydrogen bonds between hydrogen atoms of two i-Pr2Im+ cations and oxygen atoms of {VIVO(Pc˙3−)}˙− (Fig. 9b). The shortest bonds are formed between the H atom of the imidazole ring and two oxygen atoms of 2.487 and 2.525 Å lengths. Moreover, two hydrogen atoms of two methyl groups of i-Pr of each i-Pr2Im+ approach close to the oxygen atoms. Each oxygen atom forms a total of four such short contacts with H(i-Pr)⋯O distances of 2.58–2.88 Å (not shown in Fig. 9b). As a result, two oxygen atoms involved in the hydrogen bonding approach very close to each other at a 2.958 Å distance. Chains of alternating phthalocyanine dimers and cations in 9 are directed along the a axis. However, phthalocyanine planes have a different incline in the neighboring chains within the layer, forming a parquet structure. The dihedral angle between the 24-atom Pc planes of two {VIVO(Pc˙3−)}˙− from the neighboring chains is 57.88°. The shift of the Pc˙3− macrocycles in the [{VIVO(Pc˙3−)}˙−]2 dimers is equal to 5.60 Å. This shift is closer to that in (Pent4N+){VIVO(Pc˙3−)}˙− (5) which has a weak macrocycle overlapping and weak magnetic coupling of spins.
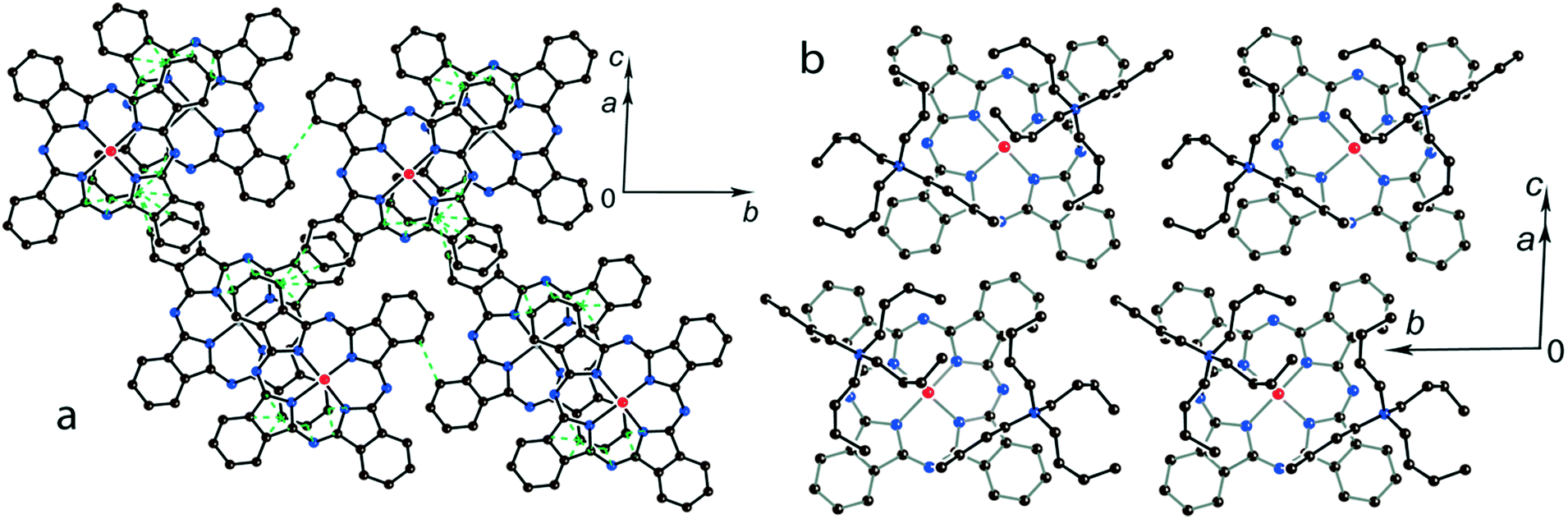
The multi-component approach allows (MDABCO+)(TPC) {TiIVO(Pc˙3−)}˙− (12) to be synthesized. In this salt, phthalocyanine layers composed of the [{TiIVO(Pc˙3−)}˙−]2 dimers alternate with the cationic MDABCO+ layers containing additional neutral TPC molecules (Fig. 10b). One-dimensional chains from the [{TiIVO(Pc˙3−)}˙−]2 dimers with close interphthalocyanine contacts can be outlined in the layers of 12 along the c axis (Fig. 11a). These chains are formed in such a way that the planes of phthalocyanines in each dimer are located obliquely relative to the cationic layers and dimers form stacks along the c axis with effective overlapping between phthalocyanines (Fig. 11a). The view along the phthalocyanine layers approximately along the a axis shows that the overlapping between the Pc˙3− macrocycles in this direction is realized as in other salts at the shift of Pc˙3− in the dimers (Fig. 11b). As a result, the packing of phthalocyanine layers in 12 is closer to a two-dimensional one (Fig. 10a). Previously, a multi-component layered fullerene complex (MDABCO+)(TPC)(C60˙−) including the MDABCO+ and TPC components was obtained.41 However, the MDABCO+-TPC layers in 12 have essentially looser packing compared with (MDABCO+)(TPC)(C60˙−). The distances between positively charged nitrogen atoms of MDABCO+ in the layers of 12 are 13.50 and 13.84 Å and these distances are essentially larger than 10 Å in the fullerene complex.41 This is due to the insertion of oxygen atoms of {TiIVO(Pc˙3−)}˙− into the cationic layers and the orientation of MDABCO+ cations lying sideways in 12. The shift of the Pc˙3− macrocycles in the [{TiIVO(Pc˙3−)}˙−]2 dimers of 12 is 4.29 Å and the interplanar distance is 3.251 Å. The observed shift is smaller than that in 2 (5.12 Å) but essentially larger than that in 1 (2.005 Å). It is seen that using different synthetic approaches and small cations of different sizes and shapes allows one to essentially modify the crystal structures of the salts, potentially affecting the magnetic and optical properties.
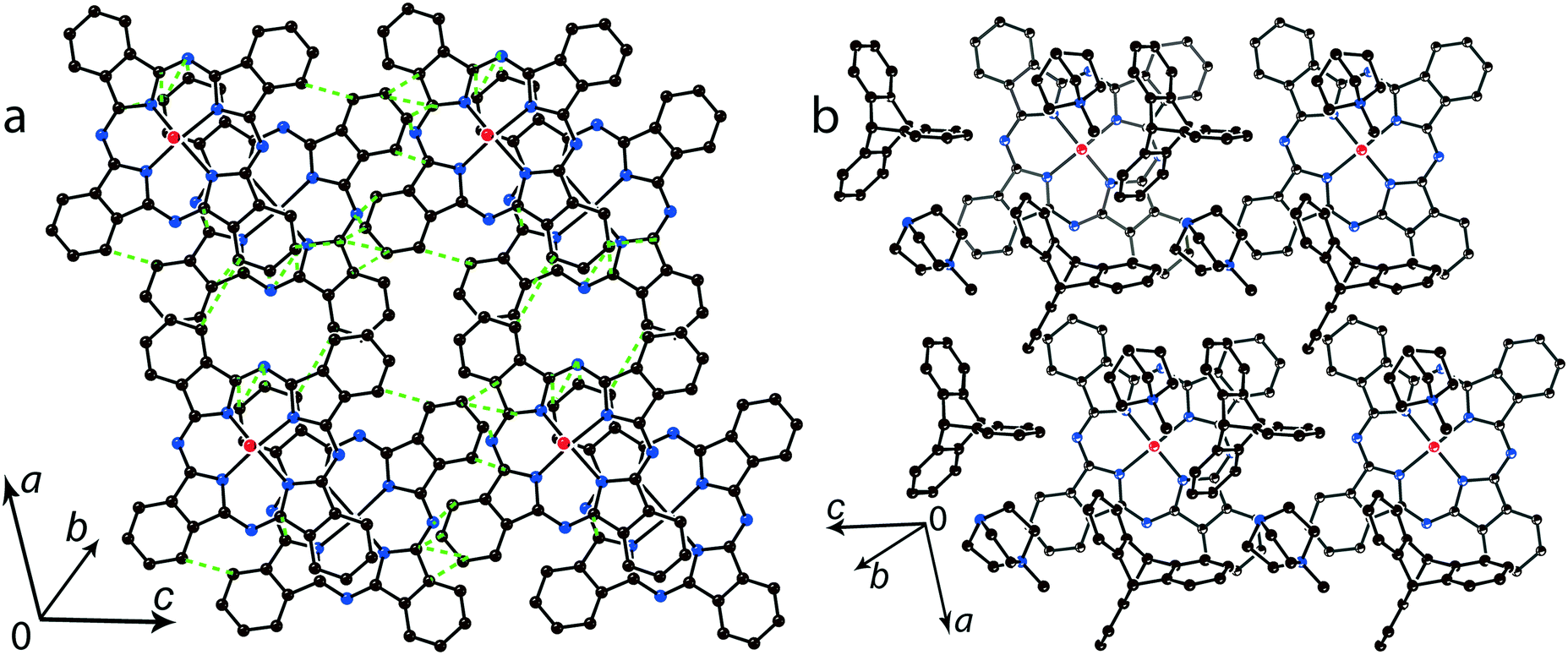 | ||
| Fig. 10 (a) View of the phthalocyanine layer composed of the π-stacking [{TiIVO(Pc˙3−)}˙−]2 dimers in 12. Short van der Waals C,N⋯C,N contacts are shown by green dashed lines. (b) Projection of the (MDABCO+)-TPC layer on the phthalocyanine layer. Only one phthalocyanine from the dimer is shown (bonds are shown in grey). | ||
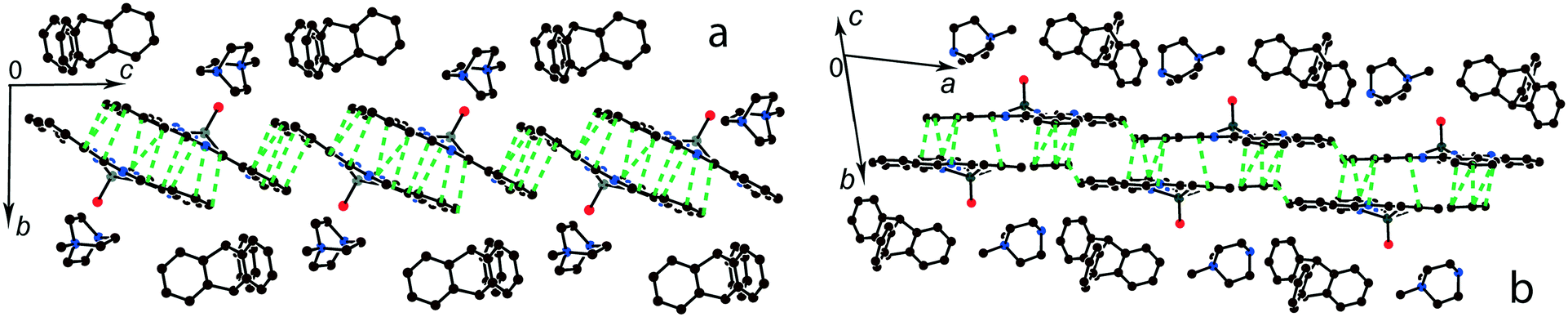 | ||
| Fig. 11 View along the phthalocyanine layers composed of the [{TiIVO(Pc˙3−)}˙−]2 dimers in 12: view along the a (a) and approximately along the c (b) axes. Short van der Waal contacts between phthalocyanines are shown by green dashed lines. | ||
To conclude on the overlapping of Pc˙3− in the phthalocyanine dimers, four types of π-π interactions between the 24-atom Pc planes can be considered (Fig. 12). The most effective overlapping with the shift of the Pc˙3− macrocycles by 2.005 Å is observed in 1 (Fig. 12a) as well as in 8 and 13. Partial overlapping with the shift of the Pc˙3− macrocycles by 5.12 Å is observed in 2 (Fig. 12b). Weak overlapping with the shift of the Pc˙3− macrocycles by 5.61 Å is found for 4 and 9 (Fig. 12c). Salt 12 has intermediate overlapping with the shift of the Pc˙3− macrocycles by 4.29 Å (Fig. 12d), but in this case a different packing mode of the dimers in the layers is observed. Isostructural salts with the [{VIVO(Pc˙3−)}˙−]2 dimers have a similar arrangement of the 24-atom Pc planes within the dimers.
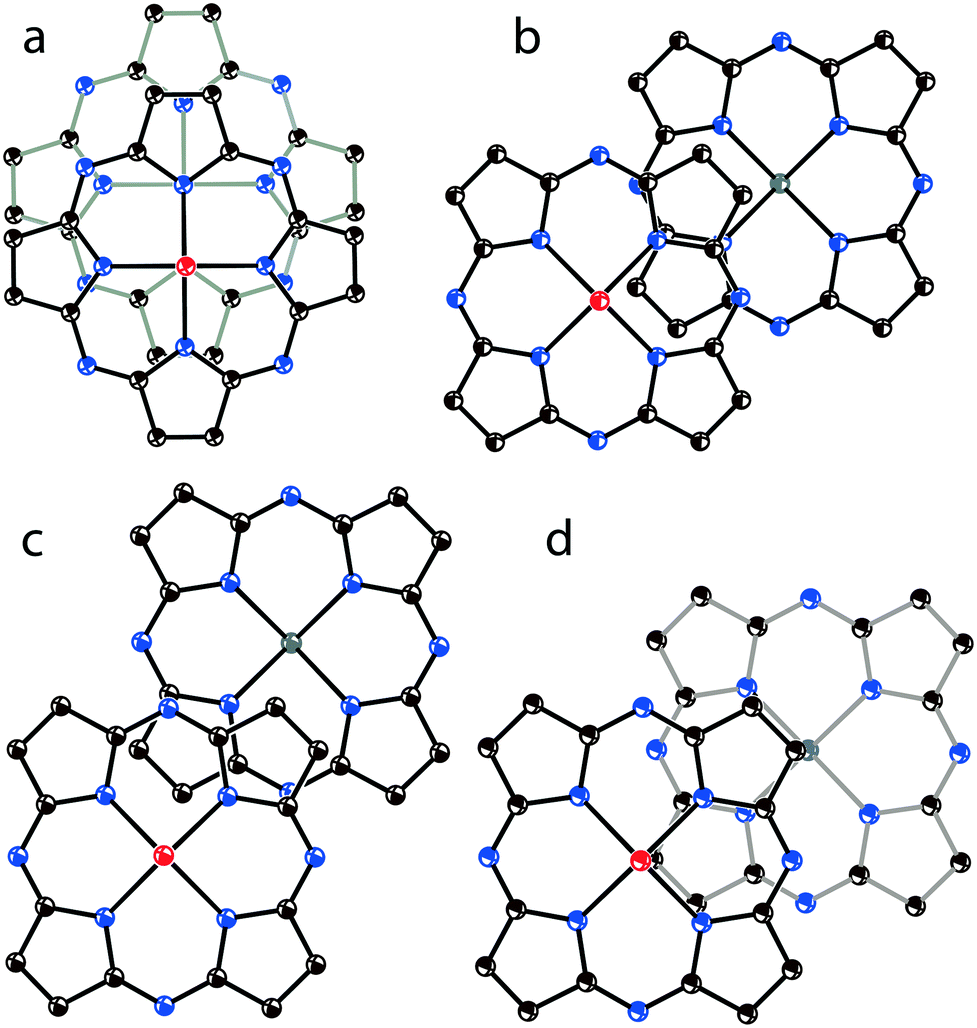 | ||
Fig. 12 View of the π-stacking [{TiIVO(Pc˙3−)}˙−]2 dimers along the M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds: (a) optimal overlapping observed in 1, 8 and 13; (b) partial overlapping observed in 2; (c) weak overlapping observed in 4 and 9; (d) intermediate overlapping in 12. Only 24-atom Pc planes are shown. O bonds: (a) optimal overlapping observed in 1, 8 and 13; (b) partial overlapping observed in 2; (c) weak overlapping observed in 4 and 9; (d) intermediate overlapping in 12. Only 24-atom Pc planes are shown. | ||
5. Jahn–Teller distorted molecular structure and effect of reduction on the optical properties of titanyl and vanadyl phthalocyanines
In this section we consider the effect of reduction on the geometry of titanyl and vanadyl phthalocyanines (Table 4) and their optical properties. The structure of pristine TiIVO(Pc2−) was studied by the Rietveld method42 but that of VIVO(Pc2−) was studied by single crystal X-ray diffraction43 (Table 4). There are two types of C–N bonds in the Pc macrocycles with pyrrole (pyr) and imine (im) nitrogen atoms. The C–Npyr bond lengths are 1.374(6) and 1.387(4) Å in TiIVO(Pc2−) and VIVO(Pc2−), respectively,42,43 which are longer than those of the C–Nim bonds. No alternation of the C-Npyr is manifested at the formation of {MIVO(Pc˙3−)}˙− but they are slightly elongated in the salts (Table 4). Shorter C–Nim bonds neither alternate in pristine phthalocyanines.42,43 However, the formation of the {MIVO(Pc˙3−)}˙− radical anions provides obvious alternation of these bonds since four bonds belonging to two oppositely located isoindole units become short but four other bonds belonging to two other oppositely located isoindole units become long (Table 4 and Fig. 13a). The difference between the short and long bonds of 0.022–0.038 (Table 4) essentially exceeds the error in the determination of the lengths of these bonds. Additionally, the alternation of the C–C bonds in the pyrrole rings (which are not involved in the phenylene substituents) is also observed, and among eight bonds four bonds are short and four other bonds are long as shown in Fig. 13a. The difference between the short and long bonds is in the 0.020–0.029 Å range (Table 4).| Compound | Average bond length in the macrocycle, Å (see Fig. 13a) | Average M–N(Pc) bond, Å | The MO bond, Å |
Displacement,a Å | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| C–Npyr | C–Nim | C–C bonds in the pyrrole ring | ||||||||
| Short | Long | Difference | Short | Long | Difference | |||||
| a Displacement of metal atoms from the 24-atom plane. b Made by the Rietveld analysis. | ||||||||||
| TiIVO(Pc2−)b (ref. 42b) | 1.375(5) | 1.329(5) | 0 | 1.442(4) | 0 | 2.071(5) | 1.643(3) | 0.742 | ||
| (MDABCO+) | 1.382(3) | 1.312(3) | 1.351(3) | 0.038 | 1.429(3) | 1.458(3) | 0.029 | 2.053(2) | 1.645(2) | 0.605 |
| {TiIVO(Pc˙3−)}˙− (8) | ||||||||||
| (MDABCO+)(TPC) {TiIVO(Pc˙3−)}˙− (12) | 1.388(3) | 1.320(3) | 1.343(3) | 0.023 | 1.438(3) | 1.458(3) | 0.020 | 2.055(2) | 1.649(2) | 0.683 |
| (MDABCO+)2 {TiIVO(Pc˙3−)}˙−(I−) (13) | 1.390(3) | 1.319(4) | 1.357(4) | 0.038 | 1.442(3) | 1.467(3) | 0.025 | 2.061(2) | 1.650(2) | 0.572 |
| (Pent4N+)1 {MIVO(Pc˙3−)}˙− (4) 2 | 1.386(3) | 1.313(4) | 1.351(3) | 0.038 | 1.439(3) | 1.463(3) | 0.024 | 2.067(2) | 1.647(2) | 0.754 |
| 1.386(3) | 1.315(4) | 1.352(4) | 0.038 | 1.434(3) | 1.458(3) | 0.024 | 2.064(2) | 1.649(2) | 0.768 | |
| VIVO(Pc2−) (ref. 43) | 1.387(4) | 1.334(4) | 0 | 1.442(4) | 0 | 2.044(4) | 1.595(4) | 0.696 | ||
| (MDABCO+)2 {VIVO(Pc˙3−)}˙−(I−) (14) | 1.392(2) | 1.318(2) | 1.355(2) | 0.037 | 1.439(2) | 1.467(2) | 0.028 | 2.031(2) | 1.613(1) | 0.522 |
| (Pent4N+) 1 {VIVO(Pc˙3−)}˙− (5) 2 | 1.393(3) | 1.315(3) | 1.352(3) | 0.037 | 1.440(3) | 1.466(3) | 0.026 | 2.037(2) | 1.605(2) | 0.690 |
| 1.388(3) | 1.318(3) | 1.354(3) | 0.036 | 1.439(3) | 1.462(3) | 0.023 | 2.038(2) | 1.610(2) | 0.699 | |
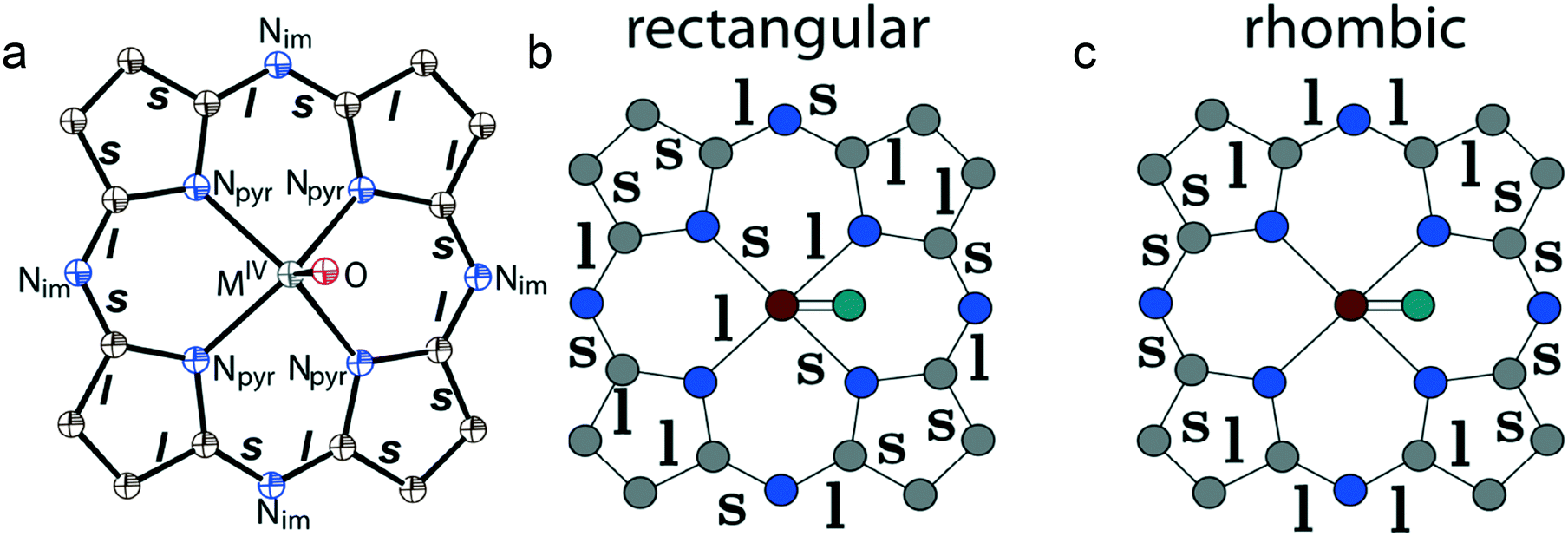 | ||
| Fig. 13 (a) Experimentally observed alternation of the C–N(imine) and C–C pyrrole bonds in the {MIVO(Pc˙3−)}˙− radical anions. Schematic diagrams of the theoretically predicted rectangular (a global minimum) (b) and rhombic (a saddle point). (c) The C2v JT deformations of the central part of the {MIVO(Pc˙3−)}˙− radical anions. The symbols l and s indicate C,N⋯C,N bonds which are elongated and shortened, respectively, at the reduction of Pc2− to Pc˙3−. | ||
The reason for the bond alternation can be clearly explained in terms of the Jahn–Teller (JT) theory. According to the theoretical works44–46 the calculated potential surface of phthalocyanine radical anions has four special points: two equivalent minima and two saddle points (two barriers between these minima). The potential minima correspond to rectangular deformations of the Pc macrocycles while transition states correspond to rhombic ones. All these deformations reduce the symmetry from C4v to C2v but they are characterized by different bond structures as shown in Fig. 13b and c. Recent studies show relatively small energy barriers between the JT deformations for free phthalocyanines in the gas phase, suggesting the dynamical JT phenomena. However, in the case of monoclinic crystals, the presence of a low-symmetry environment for the radical anions stabilizes the lower-symmetry JT configurations. It can be concluded from a comparative analysis of experimental data and the data of theoretical calculations that rectangular distortions are observed for the Pc˙3− macrocycles in all the salts with the alternation of the C–Nim and the C–C bonds in the pyrrole rings (Fig. 13a and b). However, no predicted alternation of the Npyr–MIV bonds (Fig. 13b) was found. Rhombic type distortions (Fig. 13c) were not found in the experimental structures of the {MIVO(Pc˙3−)}˙− radical anions.
Disruption of aromaticity can give an additional effect on the alternation of the bonds in the macrocycles. The Pc2− macrocycles in pristine phthalocyanines have a stable aromatic 18 π-electron system. The formation of a less stable 19 π-electron system in Pc˙3− should be accompanied by partial disruption of their aromaticity, which enhances the effect of alternation. From this point of view it is possible to explain the increase in the difference between short and long C–Nim bonds (0.060–0.071 Å) in the Pc4− macrocycles in the salts with substituted titanyl phthalocyanines and porphyrazines47 since in this case an even less stable 20 π-electron system is formed.
In addition to distortions, shortening of the M–Npyr bonds is found at the formation of {MIVO(Pc˙3−)}˙− (Table 4). Therefore, the appearance of an additional electron on the Pc˙3− macrocycles slightly enhances the bonding ability of a Pc ligand. In accordance with the shortening of the M–Npyr bonds the displacement of metal atoms from the 24-atom Pc plane decreases at the formation of salts (Table 4). It is also seen that the MO bonds are slightly elongated in the salts (Table 4) most probably due to stronger bonding of Pc˙3− to the metal atoms.
The spectra of the salts in the IR region are presented in Fig. S1–S9,† whereas those in the UV-visible-NIR range are presented in Fig. S10–S14.† The optical properties of salts 1–3, 10, 11, and 15 were published previously.12,27,35 Pristine TiIVO(Pc2−) shows the Soret band at 350 nm and the Q-band with a maximum at 713 nm (Fig. 14). Obviously, the Q-band is split into three bands but other bands are not well pronounced. There is no absorption in the spectrum of TiIVO(Pc2−) over 900 nm (Fig. 14). The spectra of the salts are similar and strongly different from those of pristine TiIVO(Pc2−) (Fig. 14). New bands are manifested in the spectra of the salts in the NIR range with maxima at 1000 nm (Fig. 14). These bands are associated with the population of the SOMO orbital of {TiIVO(Pc˙3−)}˙− from which a new electronic transition takes place to the above located orbitals. An essential blue shift of both Soret and Q-bands is seen at the formation of Pc˙3−. This indicates the increase of the energy of transition associated with the Soret and the Q-bands. The Soret band is positioned at 337–344 nm in the spectra of the salts but the Q-band is split into three bands observed at 592–599, 630–636 and 700–706 nm (Fig. 14). The intensity of relatively weak and broad charge transfer (CT) bands between {TiIVO(Pc˙3−)}˙− in the dimers is defined by the π-π interactions between Pc˙3−. Weak overlapping of Pc˙3− in 2, 4 and 6 does not allow the observation of CT bands. These bands are manifested in the spectra of salts 1 and 10 with noticeable overlap integrals. These bands are observed in the spectra of 1 (Fig. 14) and 10 at 1484 and 1467 nm, respectively.
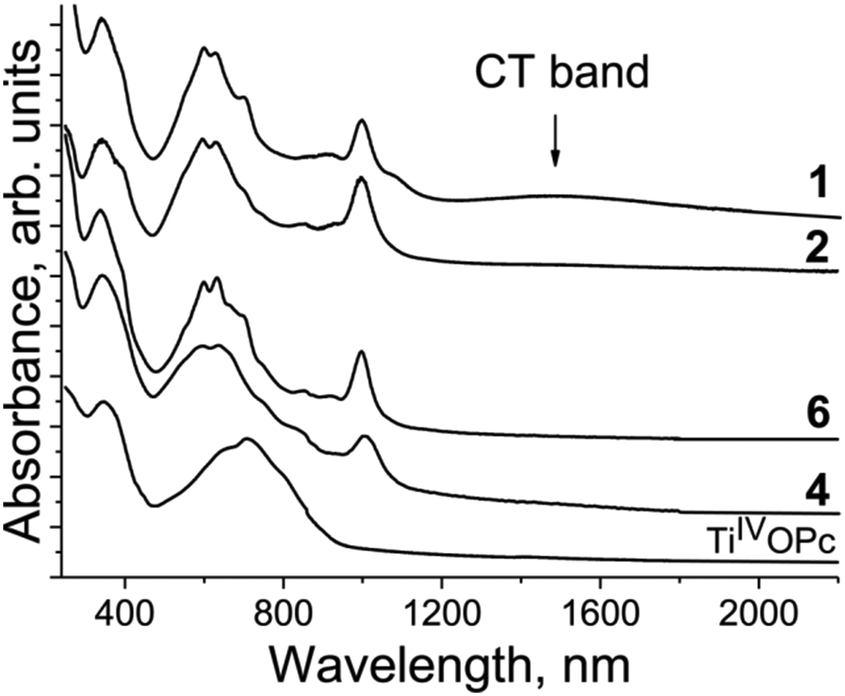 | ||
| Fig. 14 Spectra of pristine TiIVO(Pc2−) and selected salts 1, 2, 4 and 6 in the UV-visible-NIR ranges in KBr pellets. Pellets for the salts were prepared under anaerobic conditions. | ||
The spectrum of pristine VIVO(Pc2−) (Fig. 15) shows features similar to those of TiIVO(Pc2−) with the single Soret band at 351 nm and the split Q-band with a maximum at 723 nm. There is no absorption in the spectrum of VIVO(Pc2−) over 900 nm (Fig. 15). New bands appear in the spectra of the salts at 1000–1006 nm. Both Soret and Q-bands are noticeably blue shifted and appear in the spectra of the salts as a single band at 339 nm (the Soret band) and the split Q-band at 593–604 and 634–640, and the third band is well pronounced in the spectrum of 11 at 693 nm (Fig. 15). There is also a set of weak bands between 700 and 1000 nm (Fig. 15). Similarly to the {TiIVO(Pc˙3−)}˙− salts there are no CT bands in the spectra of 5 and 7 which have relatively weak Pc˙3− overlapping. At the same time an intense CT band with a maximum at 1447 nm is observed in the spectrum of 11 which has the largest π–π overlapping between Pc˙3− among the studied {VIVO(Pc˙3−)}˙− salts and shows effective magnetic coupling between spins. Thus, the presence of relatively intense CT bands in the spectra of the salts with phthalocyanine dimers can guarantee the observation of rather effective magnetic coupling between spins.
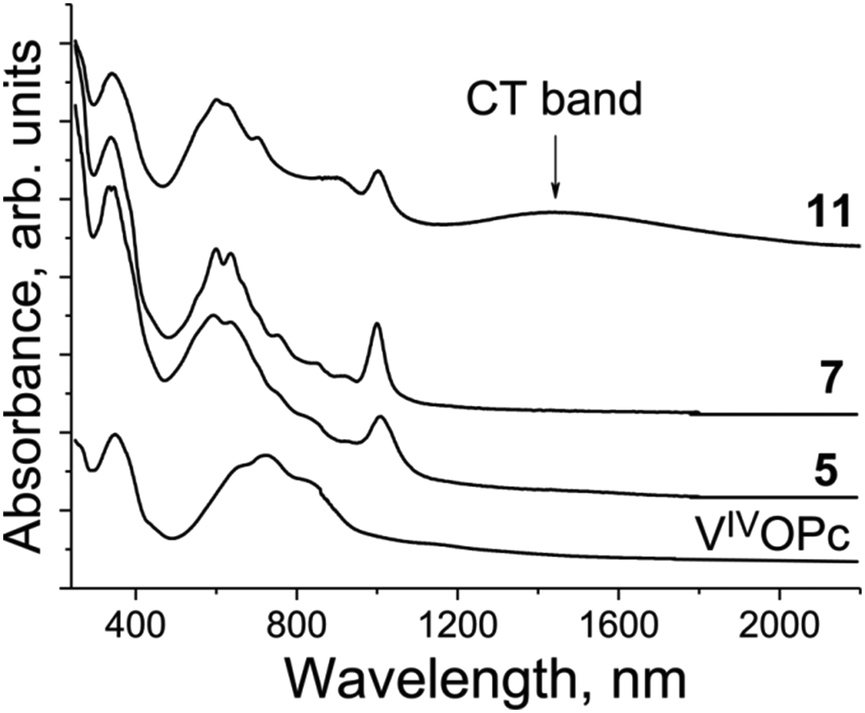 | ||
| Fig. 15 Spectra of pristine VIVO(Pc2−) and selected salts 5, 7 and 11 in the UV-visible-NIR ranges in KBr pellets. Pellets for the salts were prepared under anaerobic conditions. | ||
Conclusion
We developed several approaches for the synthesis of salts with titanyl and vanadyl phthalocyanine radical anions: the synthesis of 1:1 salts as solvent-free or solvent-containing phases with the cations of different sizes, the insertion of neutral components into the radical anion salts and finally the synthesis of the salts containing two cations and an additional counter anion per one phthalocyanine molecule. The reduction of these metallophthalocyanines centered on the macrocycles is accompanied by the formation of the Pc˙3− radical trianions. Titanyl and vanadyl phthalocyanines containing one axial oxygen ligand tend to form π-stacking [{MIVO(Pc˙3−)}˙−]2 dimers with effective π–π interactions between the Pc˙3− macrocycles. In most cases these dimers are packed in layers which alternate with the cationic layers, while the too large-sized tetrahexylammonium cation did not allow the dimer formation. The size of cationic layers defines the size of phthalocyanine layers composed of the dimers. As a result, the increase of the cation size provides the shift of the Pc˙3− macrocycles in the dimers. The decrease of the π–π interactions between the Pc˙3− macrocycles results in the decrease of magnetic coupling between spins and suppresses CT between Pc˙3− macrocycles. In this case it is possible to tune the magnetic and optical properties of the salts by a simple variation of cation size. Titanyl and vanadyl phthalocyanine radical anions have different magnetic states since TiIV is diamagnetic and the S = 1/2 spin is delocalized on Pc˙3− (π-radical), whereas both VIV and Pc˙3− have an S = 1/2 spin in {VIVO(Pc˙3−)}˙− (d- and π-electrons). It is shown that in the latter case spins are ferromagnetically coupled within the radical anion with an exchange interaction of +3.3 to +7.7 K. Intermolecular coupling between the macrocycles is antiferromagnetic and at effective π–π interaction between Pc˙3− is several times stronger than intramolecular ferromagnetic coupling. As a result, due to mutual suppression of ferro- and antiferromagnetic interactions, the resulting antiferromagnetic coupling is weaker for the {VIVO(Pc˙3−)}˙− salts in comparison with the {TiIVO(Pc˙3−)}˙− salts. Reduction provides alternation of the C–Nim bonds and the C–C bonds of the pyrrole rings in the Pc˙3− macrocycle, the appearance of new bands in the NIR range and the essential blue shift of both Soret and Q-bands. The salts obtained in some cases show effective magnetic coupling between spins especially when only π-electrons are present on the macrocycles. Further enhancement of this coupling at the extension of the π-systems of titanyl and vanadyl macroheterocycles is in progress.
Experimental
Materials
(Pent4N+)(Br−) (Aldrich, >99%), (Hex4N+)(Br−) (Aldrich, >99%), triptycene (TPC, Aldrich, 98%), 1,3-di-i-propylimidazolium chloride, ((i-Pr2Im+)(Cl−), Strem, >97%), and [VIVO(Pc2−)]0 (Acros 85%) were used as received. [TiIVO(Pc2−)]0 was obtained by 2 hour boiling of [TiIVCl2(Pc2−)]0 (95%, Aldrich) in wet pyridine (5% of H2O) as reported.48 Sodium fluorenone ketyl was obtained according to the literature.36 (MDABCO+)(I−) was obtained as previously described.49 Solvents were purified in an argon atmosphere and degassed. o-Dichlorobenzene (C6H4Cl2) was distilled over CaH2 under reduced pressure, benzonitrile was distilled over Na under reduced pressure, and n-hexane was distilled over Na/benzophenone. All manipulations for the syntheses of 4–9 and 12–14 were carried out in an MBraun 150B-G glove box with a controlled argon atmosphere and the content of H2O and O2 less than 1 ppm. The solvents and crystals were stored in the glove box. Polycrystalline samples of 4–7 were placed in quartz tubes of 2 mm diameter in anaerobic conditions (under argon) and sealed at ambient pressure or under a 10−5 torr vacuum for SQUID and EPR measurements. KBr pellets for IR- and UV-visible-NIR measurements were prepared in the glove box.Synthesis
Syntheses of crystals of 1–3,1210–11 (ref. 35) and 15 (ref. 27) were described previously. The crystals of other salts were obtained by diffusion technique. The reaction mixture was cooled to room temperature and filtered into a glass tube for diffusion, of 1.8 cm diameter and 50 mL volume with a ground glass plug, and then 30 mL of n-hexane was layered over the solution. Slow mixing of the solutions resulted in the precipitation of crystals over 1–2 months. The solvent was then decanted from the crystals, and they were washed with n-hexane. All crystals had a characteristic copper luster. The compositions of the obtained salts were determined from X-ray diffraction analysis on a single crystal. Several crystals from one synthesis were found to consist of a single crystalline phase. Due to the high air sensitivity of 4–9 and 12–14, elemental analysis could not be used to confirm the composition because these salts reacted with oxygen in the air before the quantitative oxidation procedure could be performed.The crystals of (Pent4N+){MIVO(Pc˙3−)}˙− (M = Ti (4), V (5)) and (Hex4N+){MIVO(Pc˙3−)}˙−·C6H4Cl2 (M = Ti (6), V (7)) were obtained by the following procedure. The reduction of titanyl or vanadyl phthalocyanines (24.4 mg, 0.042 mmol) in 16 ml of C6H4Cl2 with sodium fluorenone ketyl (14 mg, 0.069 mmol) in the presence of one equivalent of (Pent4N+)(Br−) (15.8 mg, 0.042 mmol for 4 and 5) or (Hex4N+)(Br−) (18.1 mg, 0.042 mmol for 6 and 7) for one day at 80 °C yielded a deep blue solution of the salts. Crystals were obtained as black prisms (4), blocks (5, 6) and black trapezoids (7) in 74%, 83%, 66% and 72% yields, respectively.
The crystals of (MDABCO+){TiIVO(Pc˙3−)}˙− (8) were obtained by the reduction of titanyl phthalocyanine (24.4 mg, 0.042 mmol) in 14 ml of C6H4Cl2 with sodium fluorenone ketyl (12 mg, 0.059 mmol) in the presence of exactly one equivalent of (MDABCO+)(I−) (10.6 mg, 0.042 mmol) for one day at 80 °C. The reduction yielded a deep blue solution of the salt. To increase the solubility of the salt benzonitrile was added (3 ml) to the solution. Salt 8 was obtained as black irregular crystals in 42% yield.
The crystals of (i-Pr2Im+){VIVO(Pc˙3−)}˙−·0.75C6H4Cl2 (9) were obtained by the reduction of vanadyl phthalocyanine (24.4 mg, 0.042 mmol) in 16 ml of C6H4Cl2 with sodium fluorenone ketyl (14 mg, 0.069 mmol) in the presence of a double excess of (i-Pr2Im+)(Cl−) (15.75 mg, 0.082 mmol) for one day at 100 °C. The reaction yielded a deep blue solution of the salt. Crystals of 9 were obtained as black blocks in 34% yield. The crystal structure of 9 contains six solvent o-C6H4Cl2 molecules per eight (i-Pr2Im+){VIVO(Pc˙3−)}˙− units. Therefore, the composition of 9 is (i-Pr2Im+){VIVO(Pc˙3−)}˙−·0.75C6H4Cl2.
The crystals of (MDABCO+)(TPC){TiIVO(Pc˙3−)}˙− (12) were obtained by the reduction of titanyl phthalocyanine (24.4 mg, 0.042 mmol) in 14 ml of C6H4Cl2 with sodium fluorenone ketyl (12 mg, 0.059 mmol) in the presence of exactly one equivalent of (MDABCO+)(I−) (10.6 mg, 0.042 mmol) for one day at 100 °C. The reaction yielded a deep blue solution of the salt. To increase its solubility benzonitrile was added (3 ml) to the solution. The solution was cooled to room temperature and 150 mg of TPC (0.590 mmol) was dissolved in the obtained solution at room temperature for 4 hours. The final solution was filtered into a tube for diffusion. Crystals of 12 were obtained as black blocks in 27% yield.
The crystals of (MDABCO+)2{MIVO(Pc˙3−)}˙−(I−) (M = Ti (13), V (14)) were obtained by the following procedure. The reduction of titanyl or vanadyl phthalocyanines (24.4 mg, 0.042 mmol) in 14 ml of C6H4Cl2 with sodium fluorenone ketyl (14 mg, 0.069 mmol) in the presence of the excess of (MDABCO+)(I−) (24 mg, 0.094 mmol) for one day at 100 °C yielded a deep blue solution of the salts. To increase the solubility of the salts benzonitrile was added (3 ml) to the solution. Crystals were obtained as dark black blocks (13) and plates (14) in 38% and 43% yields.
General
UV-visible-NIR spectra for 4–9 and 12–14 were measured in KBr pellets on a Perkin-Elmer Lambda 1050 spectrometer in the 250–2500 nm range. FT-IR spectra of 4–9 and 12–14 were obtained in KBr pellets with a Perkin-Elmer Spectrum 400 spectrometer (400–7800 cm−1). EPR spectra were recorded for sealed polycrystalline samples of 4–7, 13, and 14 from 4 up to 295 K with a JEOL JES-TE 200 X-band ESR spectrometer equipped with a JEOL ES-CT470 cryostat. A Quantum Design MPMS-XL SQUID magnetometer was used to measure the static magnetic susceptibility of 4–7 at 100 mT magnetic field under cooling and heating conditions in the 300–1.9 K range. A sample holder contribution and core temperature independent diamagnetic susceptibility (χd) were subtracted from the experimental values. The χd values were estimated by the extrapolation of the data in the high-temperature range by fitting the data with the expression χM = C/(T − Θ) + χd, where C is the Curie constant and Θ is the Weiss temperature. Effective magnetic moment (μeff) was calculated with the following formula: μeff = (8·χM·T)1/2.Crystal structure determination
X-ray diffraction data for 4–9 and 12–14 were collected on an Oxford diffraction “Gemini-R” CCD diffractometer with graphite monochromated MoKα radiation using an Oxford Instrument Cryojet system (Table 5). Raw data reduction to F2 was carried out using CrysAlisPro, Oxford Diffraction Ltd. The structures of 4–9 and 12–14 were solved by direct methods and refined by the full-matrix least-squares method against F2 using SHELX 2016/6, SHELX 2013 and Olex2 programs.50 Non-hydrogen atoms were refined in the anisotropic approximation. The positions of hydrogen were calculated geometrically. Views of the unit cells of salts of 4–9 and 12–14 are shown in Fig. S30–S38.† One pentyl substituent of one of two independent Pent4N+ cations is disordered in 4 between two positions with the 0.564(7)/0.436(7) occupancies. There are two independent Pent4N+ cations in 5. One of the pentyl substituents of the cation is disordered between two positions with the 0.839(4)/0.161(4) occupancies whereas two pentyl substituents of the second cation are disordered between two positions with the 0.681(4)/0.319(4) and 0.546(4)/0.454(4) occupancies. One of the hexyl substituents of Hex4N+ is disordered between two positions with the 0.588(5)/0.412(5) occupancies in 7. The MDABCO+ cation is disordered in 8 between three orientations with the 0.597(2)/0.246(3)/0.157(3) occupancies. The crystal structure of 9 contains six strongly disordered solvent o-C6H4Cl2 molecules per unit cell which were deleted by a standard SQEEZE procedure. The unit cell contains eight (i-Pr2Im+){VIVO(Pc˙3−)}˙− units. Therefore, the corrected composition of 9 should be (i-Pr2Im+){VIVO(Pc˙3−)}˙−·0.75C6H4Cl2. One of two i-Pr substituents of i-Pr2Im+ is disordered between two positions with the 0.522(11)/0.448(11) occupancies. Only major occupied orientations are shown for all disordered components in Fig. 2–5 and 8. To keep the anisotropic thermal parameters of the disordered atoms within reasonable limits the displacement components were restrained using ISOR, SIMU and DELU SHELXL instructions. This resulted in 699, 206, and 346 restraints used for the refinement of the crystal structures of 5, 7 and 8, respectively.| Compound | 4 | 5 | 6 | 7 | 8 | 9 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|
| Emp. formula | C104H120N18 O2Ti2 | C104H120N18O2V2 | C62H72Cl2N9OTi | C62H72Cl2N9OV | C39H31N10OTi | C41H36N10OV | C59H45N10OTi | C46H46IN12OTi | C46H46IN12OV |
| M r [g mol−1] | 1749.97 | 1756.05 | 1078.08 | 1081.12 | 703.64 | 732.71 | 957.95 | 957.75 | 960.79 |
| Color and shape | Black, prism | Black, block | Black, block | Black, trapezoid | Black, irregular | Black, block | Black, block | Black, block | Black, plate |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
Pc | Cc | C2/c | Pccn | P21/n | P21/c | P21/c |
| a, Å | 11.8378(3) | 11.8287(4) | 23.2181(5) | 22.9200(6) | 19.5955(11) | 11.8705(5) | 13.4984(3) | 12.5787(7) | 12.5922(3) |
| b, Å | 13.0103(3) | 13.0419(4) | 11.4301(2) | 11.7027(3) | 17.1171(10) | 31.689(2) | 24.7177(5) | 13.0731(5) | 13.0726(2) |
| c, Å | 16.5916(3) | 16.6492(5) | 21.4414(4) | 21.2051(6) | 19.1812(13) | 23.8325(9) | 13.8468(3) | 25.4452(17) | 25.4070(7) |
| α, ° | 67.7290(19) | 67.700(3) | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| β, ° | 71.634(2) | 71.527(3) | 96.059(2) | 95.783(3) | 99.444(6) | 90 | 98.086(2) | 91.733(5) | 91.730(2) |
| Γ, ° | 78.1997(19) | 78.147(2) | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| V, Å3 | 2233.71(9) | 2243.27(13) | 5658.44(19) | 5658.8(3) | 6346.5(7) | 8964.9(8) | 4574.04(17) | 4182.4(4) | 4180.41(17) |
| Z | 1 | 1 | 4 | 4 | 8 | 8 | 4 | 4 | 4 |
| ρ calc [g cm−3] | 1.301 | 1.300 | 1.266 | 1.269 | 1.473 | 1.086 | 1.391 | 1.521 | 1.527 |
| μ [mm−1] | 0.242 | 0.271 | 0.296 | 0.320 | 0.322 | 0.261 | 0.244 | 0.996 | 1.029 |
| F(000) | 930 | 932 | 2284 | 2288 | 2920 | 3040 | 1996 | 1956 | 1960 |
| T [K] | 117 | 117 | 100 | 110 | 150 | 293 | 150 | 130 | 130 |
| Max. 2Θ, ° | 61.016 | 61.016 | 58.552 | 62.054 | 58.222 | 52.75 | 58.108 | 59.248 | 61.746 |
| Reflns measured | 46042 |
44912 |
71322 |
22958 |
19120 |
46378 |
45629 |
50066 |
71075 |
| Unique reflns | 26916 |
26754 |
27307 |
12469 |
7370 | 8768 | 11107 |
10563 |
12334 |
| Parameter | 1163 | 1183 | 1360 | 692 | 585 | 496 | 640 | 552 | 552 |
| Restraints | 3 | 699 | 2 | 206 | 346 | 12 | 0 | 0 | 0 |
| Reflns [Fo > 2(Fo)] | 24196 |
22238 |
20292 |
10847 |
4546 | 5782 | 4654 | 8487 | 10408 |
| R 1 [Fo > 2σ(Fo)] | 0.0379 | 0.0428 | 0.0544 | 0.0406 | 0.0613 | 0.1261 | 0.0455 | 0.0485 | 0.0373 |
| wR2 (all data) | 0.0963 | 0.1019 | 0.1282 | 0.0898 | 0.1467 | 0.3212 | 0.0814 | 1.102 | 1.094 |
| G.O.F | 1.041 | 1.014 | 1.026 | 1.029 | 0.997 | 1.064 | 0.733 | 0.1006 | 0.0833 |
| CCDC number | 1581401 | 1581402 | 1583980 | 1581403 | 1581407 | 1581408 | 1581410 | 1581414 | 1581417 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the Russian Science Foundation (project N 17-13-01215), the JSPS KAKENHI Grant Number 26288035, and the JST (ACCEL) 27 (100150500010) project.Notes and references
- C. G. Claessens, W. J. Blau, M. Cook, M. Hanack, R. J. M. Nolte, T. Torres and D. Wöhrle, Monatsh. Chem., 2001, 132, 3 CrossRef CAS.
- D. Wöhrle, G. Schnurpfeil, S. G. Makarov, A. Kazarin and O. N. Suvorova, Macroheterocycles, 2012, 5, 191 CrossRef.
- J. L. Petersen, C. S. Schramm, D. R. Stojakovic, B. M. Hoffman and T. J. Marks, J. Am. Chem. Soc., 1977, 99, 286 CrossRef CAS.
- H. Hasegawa, T. Naito, T. Inabe, T. Akutagawa and T. Nakamura, J. Mater. Chem., 1998, 8, 1567 RSC.
- M. Matsuda, T. Naito, T. Inabe, N. Hanasaki, H. Tajima, T. Otsuka, K. Awaga, B. Narymbetov and H. Kobayashi, J. Mater. Chem., 2000, 10, 631 RSC.
- M. Matsuda, T. Naito, T. Inabe, N. Hanasaki and H. Tajima, J. Mater. Chem., 2001, 11, 2493 RSC.
- T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503 CrossRef CAS PubMed.
- J. S. Miller, C. Vazquez, J. C. Calabrese, R. S. McLean and A. J. Epstein, Adv. Mater., 1994, 6, 217 CrossRef CAS.
- D. K. Rittenberg, L. Baars-Hibbe, A. B. Böhm and J. S. Miller, J. Mater. Chem., 2000, 10, 241 RSC.
- E. Tosatti, M. Fabrizio, J. Tóbik and G. E. Santoro, Phys. Rev. Lett., 2004, 93, 117002 CrossRef PubMed.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, E. U. Hakimova and R. N. Lyubovskaya, New J. Chem., 2012, 36, 48 RSC.
- D. V. Konarev, A. V. Kuzmin, M. A. Faraonov, M. Ishikawa, Y. Nakano, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Eur. J., 2015, 21, 1014 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, Y. Nakano, M. A. Faraonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2016, 55, 1390 CrossRef CAS PubMed.
- (a) A. B. P. Lever, E. P. Milaeva and G. Speier, in Phthalocyanines, properties and applications, ed. C. C. Lezuoff and A. B. P. Lever, VCH: NewYork, 1993, vol. 3, pp. 1–70 Search PubMed; (b) R. H. Campbell, G. A. Heath, G. T. Hefter and R. C. S. McQueen, J. Chem. Soc., Chem. Commun., 1983, 1123 RSC.
- (a) D. W. Clack and J. R. Yandle, Inorg. Chem., 1972, 11, 1738 CrossRef CAS; (b) A. Louati, M. Elmeray, J. J. Andre, J. Simon, K. M. Kadish, M. Gross and A. Giraudeau, Inorg. Chem., 1985, 24, 1175 CrossRef CAS.
- M. Tahiri, P. Doppelt, J. Fischer and R. Weiss, Inorg. Chim. Acta, 1987, 127, L1 CrossRef CAS.
- H. Huckstadt and H. Homborg, Z. Anorg. Allg. Chem., 1998, 624, 715 CrossRef.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 9870 RSC.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2013, 52, 3851 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, S. V. Simonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2012, 41, 13841 RSC.
- D. V. Konarev, L. V. Zorina, M. Ishikawa, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Cryst. Growth Des., 2013, 13, 4930 CAS.
- D. V. Konarev, A. V. Kuzmin, M. Ishikawa, Y. Nakano, M. A. Faraonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2014, 3863 CrossRef CAS.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, A. L. Litvinov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 6810 RSC.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2017, 46, 3492 RSC.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2014, 43, 13061 RSC.
- D. V. Konarev, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Asian J., 2017, 12, 910 CrossRef CAS PubMed.
- D. V. Konarev, M. A. Faraonov, A. V. Kuzmin, S. S. Khasanov, Y. Nakano, M. S. Batov, S. I. Norko, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, New J. Chem., 2017, 41, 6866 RSC.
- D. V. Konarev, S. I. Troyanov, M. Ishikawa, M. Faraonov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, J. Porphyrins Phthalocyanines, 2014, 18, 1157 CrossRef CAS.
- E. W. Y. Wong and D. B. Leznoff, J. Porphyrins Phthalocyanines, 2012, 16, 154 CrossRef CAS.
- E. W. Y. Wong, C. J. Walsby, T. Storr and D. B. Leznoff, Inorg. Chem., 2010, 49, 3343 CrossRef CAS PubMed.
- J. A. Cissell, T. P. Vaid and A. L. Rheingold, Inorg. Chem., 2006, 45, 2367 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2016, 4099 CrossRef CAS.
- J. A. Cissell, T. P. Vaid, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2007, 46, 7713 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2016, 45, 10780 RSC.
- D. V. Konarev, S. S. Khasanov, A. V. Kuzmin, Y. Nakano, M. Ishikawa, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Cryst. Growth Des., 2017, 17, 753 CAS.
- D. V. Konarev, S. S. Khasanov, E. I. Yudanova and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2011, 816 CrossRef CAS.
- R. O. Loutfy, C. K. Hsiao, B. S. Ong and B. Keoshkerian, Can. J. Chem., 1984, 62, 1877 CrossRef CAS.
- M. E. Lines, J. Phys. Chem. Solids, 1970, 31, 101 CrossRef CAS.
- Y. Nakano, T. Yagyu, T. Hirayama, A. Ito and K. Tanaka, Polyhedron, 2005, 24, 2141 CrossRef CAS.
- J. S. Smart, in Magnetism III, ed. G. T. Rado and H. Suhl, Academic Press, NY, 1963, p. 63 Search PubMed.
- (a) D. V. Konarev, S. S. Khasanov, A. Otsuka, M. Maesato, G. Saito and R. N. Lyubovskaya, Angew. Chem., Int. Ed., 2010, 49, 4829 CrossRef CAS PubMed; (b) D. V. Konarev, S. S. Khasanov, A. Otsuka, M. Maesato, M. Uruichi, K. Yakushi, A. Shevchun, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Eur. J., 2014, 20, 7268 CrossRef CAS PubMed.
- (a) K. Oka, O. Okada and K. Nukada, Jpn. J. Appl. Phys., 1992, 31, 2181 CrossRef CAS; (b) O. Okada, K. Oka and M. Iijima, Jpn. J. Appl. Phys., 1993, 32, 3556 CrossRef CAS.
- A. J. Ramadan, L. A. Rochford, D. S. Keeble, P. Sullivan, M. P. Ryan, T. S. Jones and S. Heutz, J. Mater. Chem. C, 2015, 3, 461 RSC.
- J. Tóbik and E. Tosatti, J. Phys. Chem. A, 2007, 111, 12570 CrossRef PubMed.
- M. G. Cory, H. Hirose and M. C. Zerner, Inorg. Chem., 1995, 34, 2969 CrossRef CAS.
- L. Andjelković, S. Stepanović, F. Vlahović, M. Zlatar and M. Gruden, Phys. Chem. Chem. Phys., 2016, 18, 29122 RSC.
- M. A. Faraonov, D. V. Konarev, S. S. Khasanov, S. I. Troyanov and R. N. Lyubovskaya, Dalton Trans., 2017, 46, 3547 RSC.
- L. A. Tomachynsk, V. Y. Chernii and S. V. Volkov, Russ. J. Inorg. Chem., 2002, 47, 208 Search PubMed.
- D. V. Konarev, S. S. Khasanov, A. Otsuka, G. Saito and R. N. Lyubovskaya, Chem. – Eur. J., 2006, 12, 5225 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (b) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Data of magnetic measurements for salts 4–7. CCDC 1581401–1581403, 1583980, 1581407, 1581408, 1581410, 1581414 and 1581417. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ce01918a |
| This journal is © The Royal Society of Chemistry 2018 |