
The impact of vinylene bridges and side chain alkyl groups on the solid state structures of tricyanovinyl-substituted thiophenes†
Phuong-Truc T.
Pham
a,
Victor G.
Young
Jr.
b and
Mamoun M.
Bader
 *c
*c
aDepartment of Chemistry, Penn State Worthington Scranton, PA 18512, USA
bDepartment of Chemistry, University of Minnesota, Minneapolis, MN 55455, USA
cDepartment of Chemistry, Alfaisal University, Riyadh 11533, Saudi Arabia. E-mail: mbader@alfaisal.edu
First published on 16th November 2017
Abstract
The goal of this work is to examine the solid state structures of compounds that have been designed for increased conjugation and solubility, as these factors are important if these compounds are to be used in the solid state. The impact of three commonly employed molecular design strategies on the solid state structures of three thiophene derivatives is reported herein. These strategies include: (i) introduction of a strong electron accepting group (2T–TCV, 1); (ii) increase in conjugation by introducing a vinylene bridge in the presence of a strong electron accepting group (2T–TCV with both a TCV group and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bridge, 2); and (iii) enhancing the solubility by introducing n-butyl side chain groups in the presence of both a strong electron accepting group and a CC bridge (2T–TCV containing a strong electron accepting group, a CC bridge and four n-butyl groups, 3). Compounds 1 and 2 crystallize with four molecules in the unit cell while the unit cell of compound 3 contains only two molecules. The torsion between the two thiophene rings increases from 4.39° to 5.50° to 5.75° for 1, 2, and 3, respectively. The short distances between adjacent molecules within the unit cell also increase from 2.84 Å in 2 to 3.47 Å in 3. We also note that while the sulfur atoms assume a syn conformation in both 1 and 2, they favor the anti-conformation in 3. DFT calculations show a small energy difference between the syn and anti-conformation for 1 and 2, i.e. 3.18 kJ mol−1 and 3.19 kJ mol−1, respectively; this energy difference is found to be greater for compound 3 with the anti-conformation being 17.47 kJ mol−1 more stable than the syn conformation.
C bridge, 2); and (iii) enhancing the solubility by introducing n-butyl side chain groups in the presence of both a strong electron accepting group and a CC bridge (2T–TCV containing a strong electron accepting group, a CC bridge and four n-butyl groups, 3). Compounds 1 and 2 crystallize with four molecules in the unit cell while the unit cell of compound 3 contains only two molecules. The torsion between the two thiophene rings increases from 4.39° to 5.50° to 5.75° for 1, 2, and 3, respectively. The short distances between adjacent molecules within the unit cell also increase from 2.84 Å in 2 to 3.47 Å in 3. We also note that while the sulfur atoms assume a syn conformation in both 1 and 2, they favor the anti-conformation in 3. DFT calculations show a small energy difference between the syn and anti-conformation for 1 and 2, i.e. 3.18 kJ mol−1 and 3.19 kJ mol−1, respectively; this energy difference is found to be greater for compound 3 with the anti-conformation being 17.47 kJ mol−1 more stable than the syn conformation.
Introduction
Thiophene-based materials such as oligothiophenes (Tn) and oligo-ethylenedioxythiophenes (EDOT)n and their derivatives have been widely investigated as they possess a wide range of interesting electrical and optical properties.1 Oligomers with well-defined structures are also viewed as model compounds to aid in the understanding of properties of corresponding polymers.2 In addition, the synthetic chemistry of thiophenes is well-developed, a feature that has allowed for facile preparation and subsequent investigation of numerous structures and studies of their properties. In particular, Suzuki and Stille coupling reactions have been widely utilized in these invesitigations.3 Synthetic flexibility has made systematic studies on series of structurally related thiophene molecules possible and has given them an advantage over other organic conjugated counterparts.4 Extensive studies on thiophenes over the years have led to a better understanding of molecular structure/property relationships and the way in which the structure affects the molecular assembly in the solid state.In this work, we set to investigate how the introduction of vinylene groups and side chain alkyl groups impacts the crystals structures of three closely related thiophene derivatives. These are two commonly employed strategies to enhance the conjugation and improve the solubility of oligomers and polymers alike. These molecules can also be viewed as model compounds for the corresponding polymers.
The structures of substituent-free oligothiophenes, some oligoethylene-dioxythiophenes and many substituted oligothiophenes are known.5 We have previously reported the electrochemical and structural features of some tricyanovinyl (TCV), dicyanovinyl (DCV)-, and bromo-substituted oligothiophenes.6 We have also reported the CN⋯S and Br⋯Br interactions in these materials7 and the structures of fused 7,7,8,8-tetracyanoquinodimethane (TCNQ) type organic acceptors.8 Our studies have demonstrated that the introduction of strong electron accepting groups provides a rather simple approach in modifying the electrical properties of oligothiophenes as evident from their electrochemical data, presumably by lowering the LUMO levels. We and others have also shown that electron acceptor groups also enforce molecular planarity, promote π-stack formation, and in some cases favor the syn conformation.6 Subsequently, TCV-substituted oligothiophenes were shown to have ambipolar transport properties.9
At the molecular level, two molecular design strategies have been widely employed by materials chemists to attain favorable intrinsic molecular properties. They are: (i) the introduction of conjugated CC bonds to increase the conjugation length10 and (ii) the introduction of alkyl side chains to enhance solubility.11 These strategies were widely employed in the 1990s especially in the design of organic and polymeric nonlinear optical materials to obtain processable low and high molecular weight conjugated organic materials.12 We herein examine how these strategies impact solid state structures.
This work is a continuation of our efforts to systematically study and address molecular features impacting the solid state structures of thiophenes. To address the impact of the above mentioned strategies on solid state structures, we report herein the results of our study on the single crystal structures of three structurally related molecules (Fig. 1): 2-([2,2′-bithiophen]-5-yl)-1,1,2-tricarbonitrile (1); (E)-2-(5-(2-(thiophen-2-yl)vinyl)-thiophen-2-yl)-ethene-1,1,2-tricarbonitrile (2); and (E)-2-(3,4-dibutyl-5-(2-(3,4-dibutylthiophen-2-yl)vinyl)thiophen-2-yl)ethene-1,1,2-tricarbonitrile (3). These molecules differ from one another by systematically introducing a TCV group, followed by a CC bridge, and then a TCV group, a CC bridge and four butyl groups (Fig. 1).
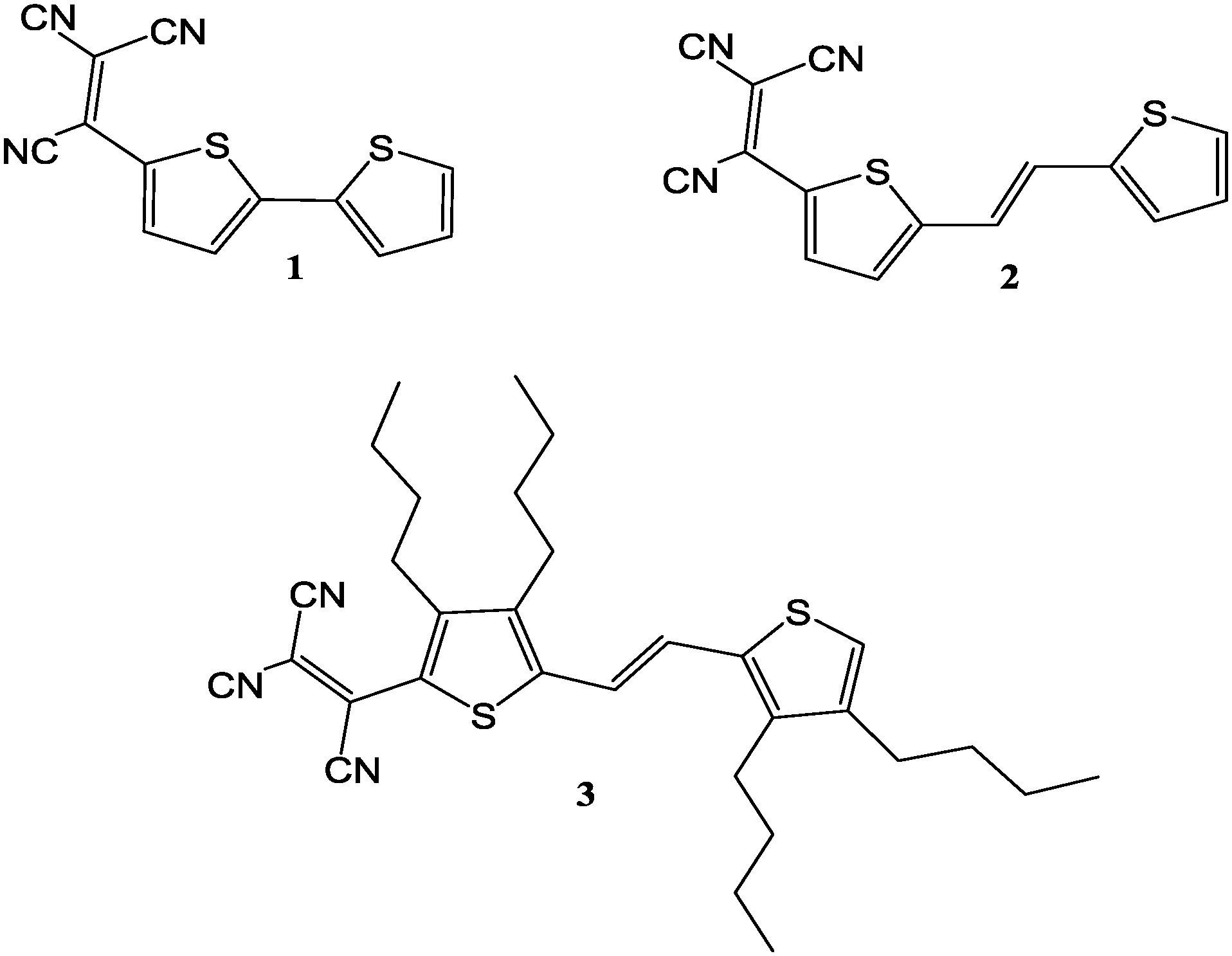 | ||
| Fig. 1 Structures of compounds 1–3. | ||
Results and discussion
Compounds 1–3 were prepared following published literature procedures.13 The TCV-free compound is reacted with tetracyanoethylene (TCNE) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at room temperature in dimethyl formamide (DMF) for 24 hours, followed by extraction using methylene chloride and purification using column chromatography. Crystals suitable for X-ray diffraction were grown by slow evaporation from acetonitrile or methylene chloride solution. We reported earlier the structure of compound 1 and we include it here only for comparison purposes.6a To the best of our knowledge, the structures of the TCV-free counterparts for compounds 2 and 3 are not known. We were unable to grow single crystals of these precursors. The crystallographic information along with selected structural features for compounds 1–3 is summarized in Table 1. Packing in the unit cells is shown in Fig. 2.
:1 ratio at room temperature in dimethyl formamide (DMF) for 24 hours, followed by extraction using methylene chloride and purification using column chromatography. Crystals suitable for X-ray diffraction were grown by slow evaporation from acetonitrile or methylene chloride solution. We reported earlier the structure of compound 1 and we include it here only for comparison purposes.6a To the best of our knowledge, the structures of the TCV-free counterparts for compounds 2 and 3 are not known. We were unable to grow single crystals of these precursors. The crystallographic information along with selected structural features for compounds 1–3 is summarized in Table 1. Packing in the unit cells is shown in Fig. 2.
| Compounds | 1 | 2 | 3 |
|---|---|---|---|
| 2T–TCV | T–CHCH–T–TCV |
Bu2–T–CHCH–T–(Bu2)–TCV |
|
| Empirical formula | C13H5N3S2 | C15H7N3S2 | C31H39N3S2 |
| Formula weight | 267.32 | 293.36 | 517.77 |
| Temperature K | 173(2) | 173(2) | 173(2) K |
| Crystal system | Orthorhombic | Monoclinic | 0.71073 |
| Space group | Pna21 | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a, Å | 7.8630(12) | 5.560(5) | 9.1187(16) |
| b, Å | 24.489(4) | 14.516(13) | 10.6243(18) |
| c, Å | 6.1193(9) | 16.611(15) | 16.361(3) |
| α ° | 90 | 90 | 73.912(3)° |
| β ° | 90 | 93.872(16) | 82.666(3)° |
| γ ° | 90 | 90 | 70.835(3)° |
| Volume, Å3 | 1178.3(3) | 1338(2) | 1437.4(4) |
| Z | 4 | 4 | 2 |
| Density (calc.), Mg m−3 | 1.507 | 1.457 | 1.196 |
| Crystal color, morphology | Red | Metallic purple, needle | Dark metallic green plate |
| Crystal size, mm | 0.39 × 0.36 × 0.07 | 0.44 × 0.14 × 0.06 | 0.28 × 0.19 × 0.06 |
| Reflections collected | 1.062 | 9166 | 10466 |
| Completeness to theta | R 1 = 0.0303, wR2 = 0.0691 | 99.70% | 97.9% |
| No. of parameters | 3.224 | 209 | 556 |
| Intramolecular CN–S distance | 3.224 | 3.391 | 3.323 |
| CCD number | 1584731 | 1502184 | 1502185 |
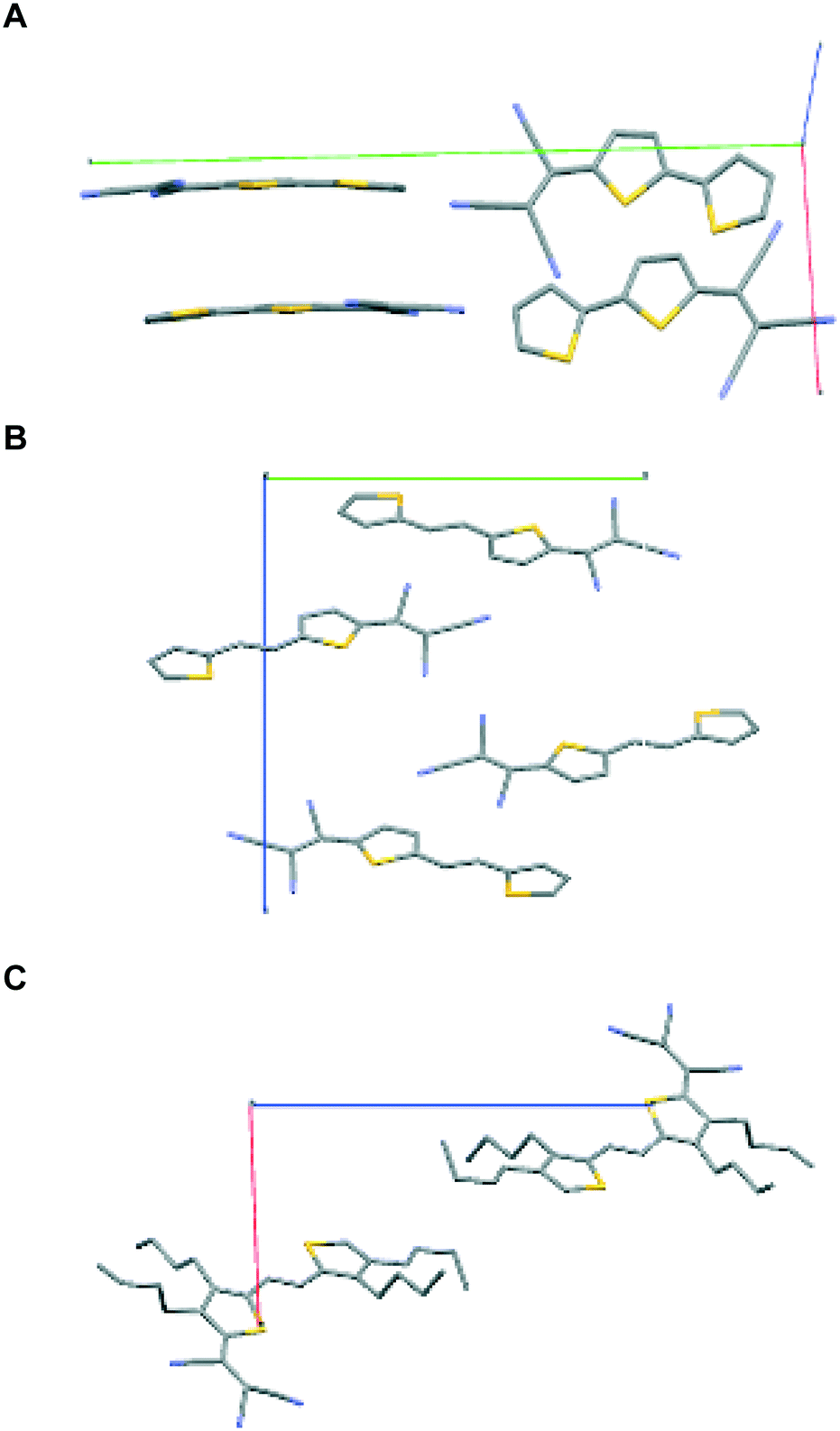 | ||
| Fig. 2 Unit cells for compounds 1–3, A–C, respectively. Note the syn conformation of the sulfur atoms in 1 and 2 and anti in 3. | ||
First, we note that compounds 1 and 2 crystallize with four molecules in the unit cell while the unit cell of compound 3 contains only two molecules. There are various reports on the structure of bithiophene.14 It crystallizes in the P21/c space group with 2 molecules in the unit cell and a density of 1.44 g mL−1. The calculated densities for 1–3 ranged from 1.2 to 1.5 g mL−1. The lower density for compound 3 (1.19 g mL−1) is probably due to the four bulky butyl groups which result in less compact packing due to the steric influence of the side chain groups. As one might anticipate, the enhancement of solubility comes at the expense of risking poorer charge transport. This has been reported earlier on other systems.15
The introduction of a TCV group in 2T (compound 1) results in the formation of stacks with the sulfur atoms assuming the syn conformation as opposed to the more common herringbone packing with the all anti conformation observed in bithiophene and the majority of known oligothiophene structures. The rare syn conformation was observed for example in 2,2′-bithiophene-5-carbaldehyde.16 Strong S⋯N interactions, both intramolecular and intermolecular, are evident with the latter being both within the stack and between adjacent stacks. Distances either close to or well below the sum of the van der Waals radii of the two atoms of 3.35 Å are observed. The molecules form stacks along the c-axis.
We now consider molecules 2 and 3: we note that in 2, the two sulfur atoms adopt the syn conformation whereas in 3 they adopt the anti-conformation. In 2, the plane of the thiophene portion relative to that of the tricyanovinyl portion is skewed by 4.05°, while the distance between layers is approximately 3.47 Å. Close intermolecular contacts are found between nitrogen atoms and various hydrogen atoms attached to carbon atoms ranging from 2.50 to 2.72 Å. On the other hand, molecule 3 forms stacks along the c-axis with torsion angles between TCV and the thiophene ring of 7.90° while the torsion angle of CC–T is 6.15° and 6.56° for the TCV bearing and remote thiophene rings, respectively (Fig. 3 and 4). The short distance between CN and C(CN)C(CN)2 is 3.217 Å, while that of S⋯S is 3.824 Å and inter-stack distances (3.43 and 3.46 Å) are found between S⋯N (Fig. 4). The layers further aggregate by dispersion interactions between the butyl chains to complete the 3D crystal architecture. We note that the butyl groups show an eclipsed conformation as opposed to the expected all anti conformation of long chain alkyl groups attached to thiophenes, a feature that has been observed usually in side chains with an even number of carbon atoms and thiophenes bearing strong electron-accepting groups. This is an interesting feature which may warrant further investigation.17
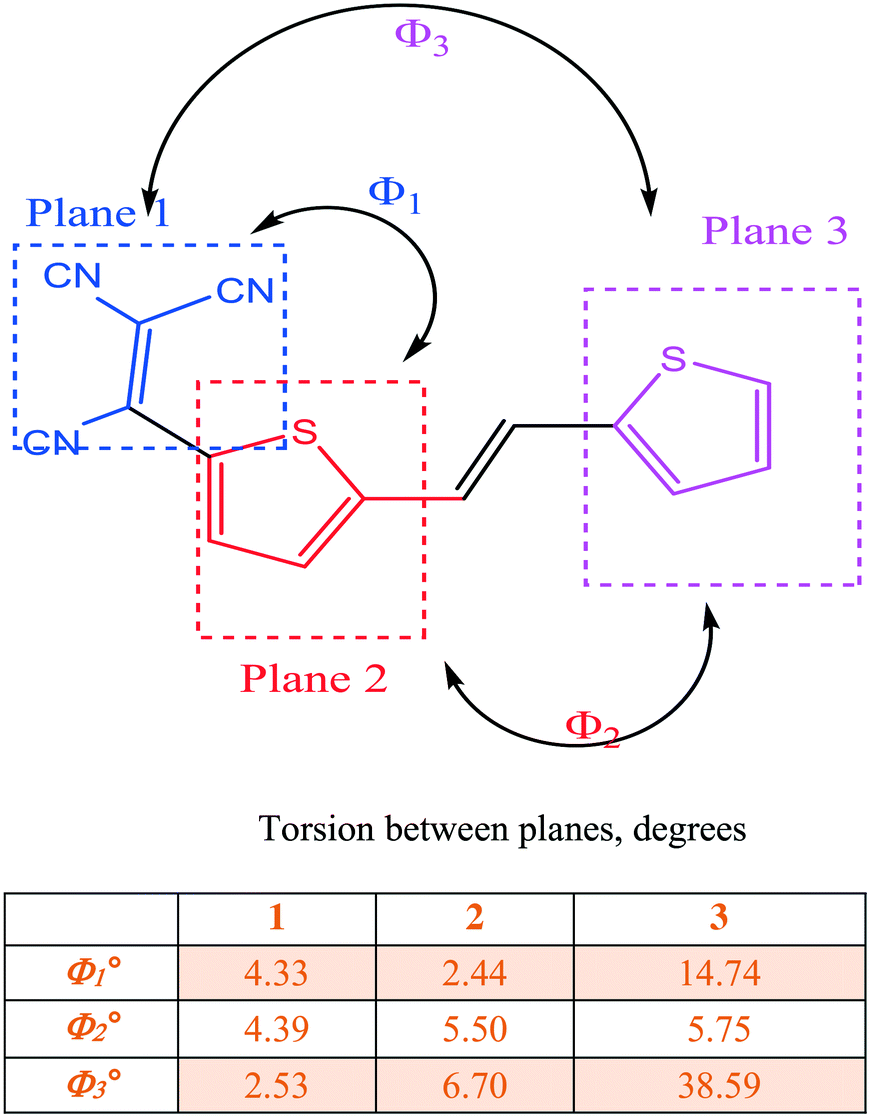 | ||
| Fig. 3 Planes and torsion angles in compounds 1–3. | ||
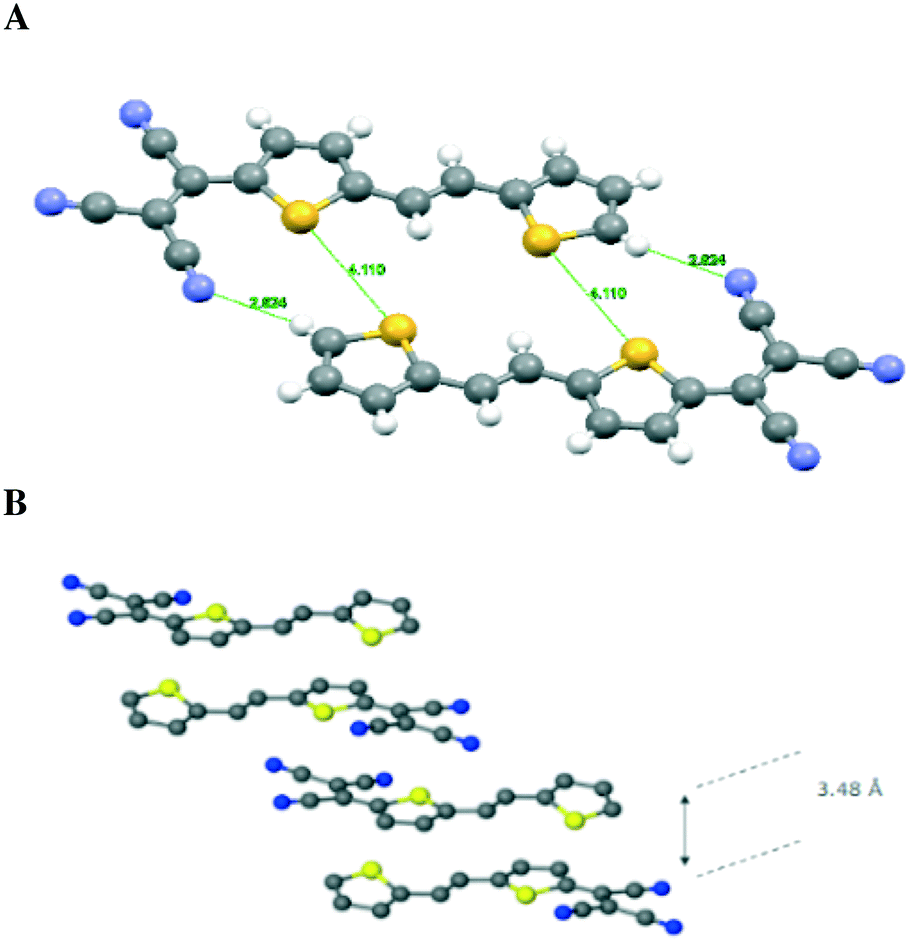 | ||
| Fig. 4 Stacks of 2 and 3 and some short contact distances. Structures and short distances in compounds 2 (A) and 3 (B); butyl groups are omitted for clarity. | ||
We also carried out DFT calculations for both the syn and anti-conformations of compounds 1–3.18 Results show that the anti-conformation for 1 is more stable by 3.39 kJ mol−1, whereas the opposite is seen for 2 where surprisingly the syn conformer is calculated to be more stable than the anti by 3.18 kJ mol−1. Yet both displayed a syn conformation in the crystalline structures. We note that these calculations were carried out on isolated gas phase molecules. The observation of the syn conformer in the solid state indicates that the crystal packing forces override the preference for the anti-conformer, consistent with previous theoretical studies that suggest comparable energies for the two forms.19 Although in the minority, the syn conformation has been observed in a few other oligothiophenes.20,21 Calculations for compound 3 showed the anti-conformation to be expectedly more stable by 17.47 kJ mol−1. We also note that the calculated band gaps did not change too much between the syn and anti-conformations (Table 2). We like to point out that during the preparation of this manuscript Briseno and co-workers reported a similar system with hexyl groups along with device performance. They reported that the hexyl-substituted molecules show marked differences in solid-state packing compared to the unsubstituted analogs. The alkylated monomer crystal structure exhibits terminal thiophenes in the syn conformation. In contrast, the unsubstituted monomer adopts the more common anti conformation. Gas phase conformations of oligomers rationalize the intrinsic conformational preferences. This renders confidence in our analysis and findings.21
| Compounds | ΔE = Eanti − Esyn (eV) | HOMO (eV) | LUMO (eV) | Band gap |
|---|---|---|---|---|
| (1) anti | −6.39 | −3.71 | 2.68 | |
| (1) syn | 3.39 | −6.4 | −3.7 | 2.7 |
| (2) anti | −6.07 | −3.65 | 2.42 | |
| (2) syn | −3.18 | −6.06 | −3.69 | 2.37 |
| (3) anti | −6.29 | −4.14 | 2.15 | |
| (3) syn | 17.47 | −6.36 | −4.04 | 2.32 |
Conclusions
In summary, we have shown that the introduction of a tricyanovinyl (TCV) group, a strong-electron accepting group, into thiophene molecules provides an efficient way to induce planarity and offers a new tool for molecular design and crystal engineering of these materials. The introduction of CC bridges reduces planarity as evidenced from the dihedral angles. The side chain solubilizing butyl groups push molecules further apart while at the same time inducing overall reduced planarity. This is seen in the evolution of the torsion angles in these molecules. DFT calculations confirm these results while not the syn and anti-conformations observed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge support from Alfaisal University internal research grant IRG-2014 project number 313021507131 (M. Bader); Penn State Worthington Scranton Research Development Grant (P.-T. Pham); and the MRSEC Program Funded by the National Science Foundation through the University of Minnesota under award number DMR-1420013. The authors also acknowledge Dr. Radu Custelcean, W. Brennessel and the X-Ray Crystallographic Laboratory, Department of Chemistry at the University of Minnesota and the Center for Drug Design, University of Minnesota (P. Pham). M. Bader acknowledges many useful discussions with professors Michael D. Ward and C. Daniel Frisbie.References
- (a) Handbook of Thiophene-Based Materials: Applications in Organic Electronics and Photonics, ed. I. F. Perepichka and D. F. Perepichka, Wiley, Weinheim, 2009 Search PubMed; (b) A. Mishra, C.-Q. Ma and P. Baeuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS PubMed; (c) L. Zhang, N. S. Colella, F. Liu, S. Trahan, J. K. Baral, H. H. Winter, S. C. B. Mannsfeld and A. L. Briseno, J. Am. Chem. Soc., 2013, 135, 844–854 CrossRef CAS PubMed; (d) A. Facchetti, M. H. Yoon, C. L. Stern, G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2004, 126, 13480–13501 CrossRef CAS PubMed; (e) L. S. M. William, W. Porter III and A. J. Matzger, Org. Lett., 2007, 9, 1005–1008 CrossRef PubMed; (f) J. T. Henssler and A. J. Matzger, J. Org. Chem., 2012, 77, 9298–9303 CrossRef CAS PubMed; (g) J. T. Henssler and A. J. Matzger, Org. Lett., 2009, 11, 3144–3147 CrossRef CAS PubMed; (h) K. Zong and J. R. Reynolds, J. Org. Chem., 2001, 66, 6873–6882 CrossRef CAS PubMed; (i) S. Das, P. K. Dutta, S. Panda and S. S. Zade, J. Org. Chem., 2010, 75, 4868–4871 CrossRef CAS PubMed; (j) M. T. P. Frère and J. Roncali, J. Org. Chem., 2003, 68, 5357–5360 CrossRef PubMed; (k) D. Demeter, P. Blanchard, M. Allain, I. Grosu and J. Roncali, J. Org. Chem., 2007, 72, 5285–5290 CrossRef; (l) C. H. Liu, H. Zhao and H. Yu, Org. Lett., 2011, 13, 4068–4071 CrossRef CAS PubMed; (m) T. Taerum, O. Lukoyanova, R. G. Wylie and D. F. Perepichka, Org. Lett., 2009, 11, 3230–3233 CrossRef CAS PubMed; (n) L. Fillaud, G. Trippé-Allard and J. C. Lacroix, Org. Lett., 2013, 15, 1028–1031 CrossRef CAS PubMed; (o) G. A. Sotzing, J. R. Reynolds and P. J. Steel, Chem. Mater., 1996, 8, 882–889 CrossRef CAS; (p) G. Zotti, B. Vercelli, A. Berlin, M. Pasini, T. L. Nelson, R. D. McCullough and T. Virgili, Chem. Mater., 2010, 22, 1521–1532 CrossRef CAS.
- Electronic Materials: The Oligomer Approach, ed. K. Mullen and G. Wegner, Wiley, Weinheim, 1998 Search PubMed.
- (a) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) O. L. Griffith, J. E. Anthony, A. G. Jones, Y. Shu and D. L. Lichtenberger, J. Am. Chem. Soc., 2012, 134, 14185–14194 CrossRef CAS PubMed; (b) O. L. Griffith, J. E. Anthony, A. G. Jones, Y. Shu and D. L. Lichtenberger, J. Am. Chem. Soc., 2010, 132, 580–586 CrossRef CAS PubMed; (c) B. Purushothaman, S. R. Parkin and J. E. Anthony, Org. Lett., 2010, 12, 2060–2063 CrossRef CAS PubMed.
- D. Fichou, J. Mater. Chem., 2000, 10, 571–588 RSC.
- (a) M. M. Bader, R. Custelcean and M. D. Ward, Chem. Mater., 2003, 15, 616–618 CrossRef CAS; (b) M. M. Bader, P. T. T. Pham and E. H. Elandaloussi, Cryst. Growth Des., 2010, 10, 5027–5030 CrossRef CAS.
- P.-T. T. Pham and M. M. Bader, Cryst. Growth Des., 2014, 14, 916–922 CAS.
- M. M. Bader, P. T. T. Pham, B. R. Nassar, H. Lin, Y. Xia and C. D. Frisbie, Cryst. Growth Des., 2009, 9, 4599–4601 CAS.
- X. Cai, M. W. Burand, C. R. Newman, D. A. da Silva Filho, T. M. Pappenfus, M. M. Bader, J.-L. Brédas, K. R. Mann and C. D. Frisbie, J. Phys. Chem. B, 2006, 110, 14590–14597 CrossRef CAS PubMed.
- For recent examples see: (a) W. Y. So, J. Hong, J. J. Kim, G. A. Sherwood, K. Chacon-Madrid, J. H. Werner, A. P. Shreve, L. A. Peteanu and J. Wildeman, J. Phys. Chem. B, 2012, 116, 10504–10513 CrossRef CAS PubMed; (b) W. Rodríguez-Córdoba, C. A. Sierra, C. O. Puentes, P. M. Lahti and J. Peon, J. Phys. Chem. B, 2012, 116, 3490–3503 CrossRef PubMed.
- See for example: (a) J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS; (b) T. Lei, J.-Y. Wang and J. Pei, Chem. Mater., 2014, 26, 594–603 CrossRef CAS.
- See for example: P. A. Sullivan, H. Rommel, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, K. A. Firestone, J.-W. Kang, J. Luo, J. A. Davies, D. H. Choi, B. E. Eichinger, P. J. Reid, A. Chen, A. K.-Y. Jen, B. H. Robinson and L. R. Dalton, J. Am. Chem. Soc., 2007, 129, 7523–7530 CrossRef CAS PubMed.
- (a) M. M. Bader, R. Custelcean and M. D. Ward, Chem. Mater., 2003, 15, 616–618 CrossRef CAS; (b) T. M. Pappenfus, M. W. Burand, D. E. Janzen and K. R. Mann, Org. Lett., 2003, 5, 1535–1538 CrossRef CAS PubMed; (c) T. M. Pappenfus, K. B. Schliep, A. Dissanayake, T. Ludden, B. Nieto-Ortega, J. T. L. Navarrete, M. C. R. Delgado and J. Casado, J. Chem. Educ., 2012, 89, 1461–1465 CrossRef CAS.
- (a) P. A. Chaloner, S. R. Gunatunga and P. B. Hitchcock, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1941–1942 CrossRef; (b) M. Pelletier and F. Brisse, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1942–1945 CrossRef.
- (a) T. W. Holcombe, J. E. Norton, J. Rivnay, C. H. Woo, L. Goris, C. Piliego, G. Griffini, A. Sellinger, J.-L. Brédas, A. Salleo and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 12106–12114 CrossRef CAS PubMed; (b) O. L. Griffith, J. E. Anthony, A. G. Jones, Y. Shu and D. L. Lichtenberger, J. Am. Chem. Soc., 2012, 134, 14185–14194 CrossRef CAS PubMed.
- P. A. Chaloner, P. B. Hitchcock and M. R. Simmons, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1945–1947 CrossRef.
- H. B. Akkerman, S. C. B. Mannsfeld, A. P. Kaushik, E. Verploegen, L. Burnier, A. P. Zoombelt, J. D. Saathoff, S. Hong, S. Atahan-Evrenk, X. Liu, A. Aspuru-Guzik, M. F. Toney, P. Clancy and Z. Bao, J. Am. Chem. Soc., 2013, 135, 11006–11014 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) Spartan 06, Wavefunction Inc., Irvine, CA, 2006 Search PubMed.
- G. A. Diaz-Quijada, N. Weinberg, S. Holdcroft and M. B. Pinto, J. Phys. Chem. A, 2002, 106, 1266 CrossRef CAS , and references therein.
- (a) D. B. Mitzi, Inorg. Chem., 2000, 39, 6107 CrossRef CAS PubMed; (b) H. Muguruma, K. Kobiro and S. Hotta, Chem. Mater., 1998, 10, 1459 CrossRef CAS; (c) P. A. Chaloner, S. R. Gunatunga and P. B. Hitchcock, J. Chem. Soc., Perkin Trans. 2, 1997, 1597 RSC; (d) D. B. Mitzi, K. Chondroudis and C. R. Kagan, Inorg. Chem., 1999, 38, 6246 CrossRef CAS PubMed; (e) S. P. Ames, P. A. Chaloner, P. B. Hitchcock and M. R. Simmons, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 1945 CrossRef.
- B. P. Cherniawski, S. A. Lopez, E. K. Burnett, I. Yavuz, L. Zhang, S. R. Parkin, K. N. Houk and A. L. Briseno, J. Mater. Chem. C, 2017, 5, 582–588 RSC.
Footnote |
| † CCDC CIF files for compounds 1–3. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ce01574g |
| This journal is © The Royal Society of Chemistry 2018 |