
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrodrugs: an electrochemical prodrug activation strategy†
Daniel J.
Norman
 a,
Eva
González-Fernández
a,
Jessica
Clavadetscher
a,
Lulu
Tucker
a,
Matteo
Staderini
a,
Andrew R.
Mount
a,
Alan F.
Murray
b and
Mark
Bradley
*a
a,
Eva
González-Fernández
a,
Jessica
Clavadetscher
a,
Lulu
Tucker
a,
Matteo
Staderini
a,
Andrew R.
Mount
a,
Alan F.
Murray
b and
Mark
Bradley
*a
aEaStChem School of Chemistry, University of Edinburgh, David Brewster Rd, Edinburgh, EH9 3FJ, Scotland, UK. E-mail: m.bradley@ed.ac.uk
bSchool of Engineering, University of Edinburgh, Mayfield Road, Edinburgh, EH9 3JL, Scotland, UK
First published on 24th July 2018
Abstract
The term electroceutical has been used to describe implanted devices that deliver electrical stimuli to modify biological function. Herein, we describe a new concept in electroceuticals, demonstrating for the first time the electrochemical activation of metal-based prodrugs. This is illustrated by the controlled activation of Pt(IV) prodrugs into their active Pt(II) forms within a cellular context allowing selectivity and control of where, when and how much active drug is generated.
The emerging field of bio-electronic medicine or electroceuticals — a term coined by GSK in 20131 — encompasses the potential for diseases to be treated through the use of implantable devices capable of modulating biological functions using electrical signals.2 To date, the focus of electroceuticals has been the mimicking of natural neural signals present in living systems. Devices such as pacemakers3 and cochlear implants4 are commonplace but recent applications of electroceuticals are more ambitious and include so-called “closed-loop” systems — devices that can receive and interpret electrical nerve signals from the body and elicit a modulatory effect in response.1,2 This electroceutical approach directs biological function via electrical stimulation. Here, we present a new type of electroceutical and report for the first time an electrochemical prodrug activation strategy, leading to the site-selective generation of the anticancer agent, oxaliplatin.
Platinum-based cancer therapies have been studied extensively with the aim of improving their toxicity profiles while increasing selectivity.5,6 Pt(IV) compounds show reduced cytotoxicity compared to the “parent” Pt(II) complexes, where the axial ligands can function as protecting groups until they are released upon reduction of the Pt(IV) compound.7,8 In addition, the two additional axial ligands provide convenient handles for functionalising the metal centre that can be used to increase lipophilicity or introduce adjuvant bio-active ligands. Activation of these Pt(IV) prodrugs is generally accepted to be via reduction by biological reductants such as glutathione, ascorbic acid or cytochrome c, generating the active Pt(II) form.9 However, the widespread presence of such biological reductants in vivo means selectivity towards cancerous tissue will always be fraught with limited local control of the Pt(IV)–Pt(II) conversion.
For our proof-of-concept studies we used a prodrug of oxaliplatin, a drug currently approved for use in advanced colorectal cancer and in clinical trials for an array of other cancers.10,11
Certain ligand types attached to Pt(IV), such as carboxylates, can be effective in preventing inner-sphere reduction by biological reductants in comparison to halide and pseudo-halide axial ligands.12,13 To improve selectivity, targeting moieties have been incorporated as axial ligands, such as oestrogen, to exploit active uptake into cancer cells that overexpress the oestrogen receptor.14 More exotic drug delivery systems have been designed for similar purposes — this includes Pt(IV) complexes tethered to carbon nanotubes that enter cells via endocytosis,15 up-converting nanoparticles with DNA-bound Pt(IV) complexes shown to release the Pt(IV) complex upon illumination in the presence of ascorbic acid16 and Pt(IV)–NaYF4 nanoparticles decorated with folic acid have been used to enable folate receptor mediated targeting.17
Despite the elegancy of recent developments, they are all reliant on biological processes that are innately highly variant between tissue types. Pt(IV) carboxylate complexes typically exhibit irreversible reduction potentials in the range of −0.3 to −1.0 V (vs. Ag|AgCl|Cl−),18 which presents a unique opportunity to develop a prodrug activation system that is reliant, not upon biological stimuli, but upon an externally controlled electrochemical stimulus.
Herein, we report the first electrochemically-activated prodrug system, where an electrode was used to electrochemically activate a Pt(IV) prodrug allowing the generation of an active anticancer Pt(II) complex with spatial and temporal control. This was enabled by the use of a ruthenium-based redox mediator (identified through empirical screening of redox mediators with suitable redox potentials) coated onto an electrode surface that acts as an electron shuttle between the electrode surface and the Pt(IV) prodrug. This enabled its reduction to the active Pt(II) drug (at much lower applied potentials when compared to the potential required for its direct reduction at the electrode surface), leading to electrochemical-mediated controlled cell death. A Pt(IV) derivative 1 that contained two succinate axial ligands was synthesised which, when subjected to an electrochemical activation/reduction process utilising a redox mediator, efficiently generated oxaliplatin 2 (Scheme 1). The anionic nature of the succinate ligands at physiological pH is likely to contribute to the low cytotoxicity of the prodrug in relation to the Pt(II) species, by preventing passage through the cell membrane. This also afforded a “bio-inert” prodrug, as 1 was not efficiently reduced by biological reductants.19 In addition, prodrug 1 was kinetically inert towards biological reductants, in comparison to other Pt(IV) compounds with higher reduction potentials (Fig. S1, ESI†).
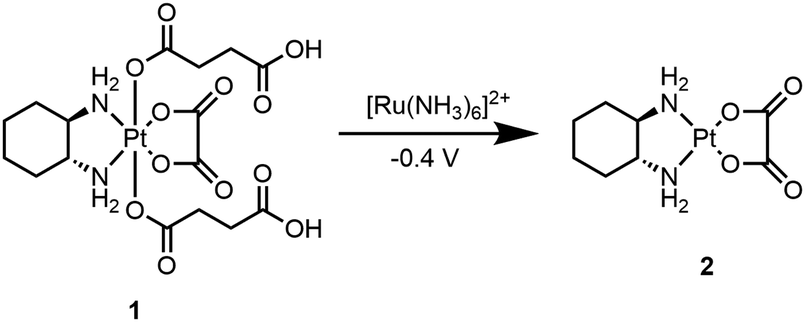 | ||
| Scheme 1 Prodrug 1 electrochemically activated to the active anti-cancer drug oxaliplatin, 2 mediated through electron transfer by [Ru(NH3)6]2+. | ||
Cyclic voltammetry (CV) of the Pt(IV) derivative 1 (2 mM in PBS at pH 7) showed an irreversible reduction with a peak potential of −0.65 V (vs. Ag | AgCl | KCl (3 M), Fig. S2, ESI†), corresponding to the reduction of Pt(IV) to Pt(II) with the loss of both axial ligands. This potential indicated that direct electrochemical reduction of prodrug 1 would require a relatively high reduction potential that in the context of a biological environment would be inefficient due to the interference of redox-active biomolecules, hindering specificity of the system towards the prodrug. However, CV analysis of a solution containing the redox mediator, hexaammineruthenium chloride [Ru(NH3)6]Cl3 (100 μM) in PBS showed reversible redox behaviour. With the addition of 1 (2 mM) to the same solution, the current associated with the reduction process of the mediator (IC) increases, as would be expected of an electro-catalytic system indicating generation of more oxidised mediator via reduction of 1 (Fig. 1a and b). Moreover, application of a reductive potential (−0.4 V) afforded good conversion of 1 (Fig. S3, ESI†).
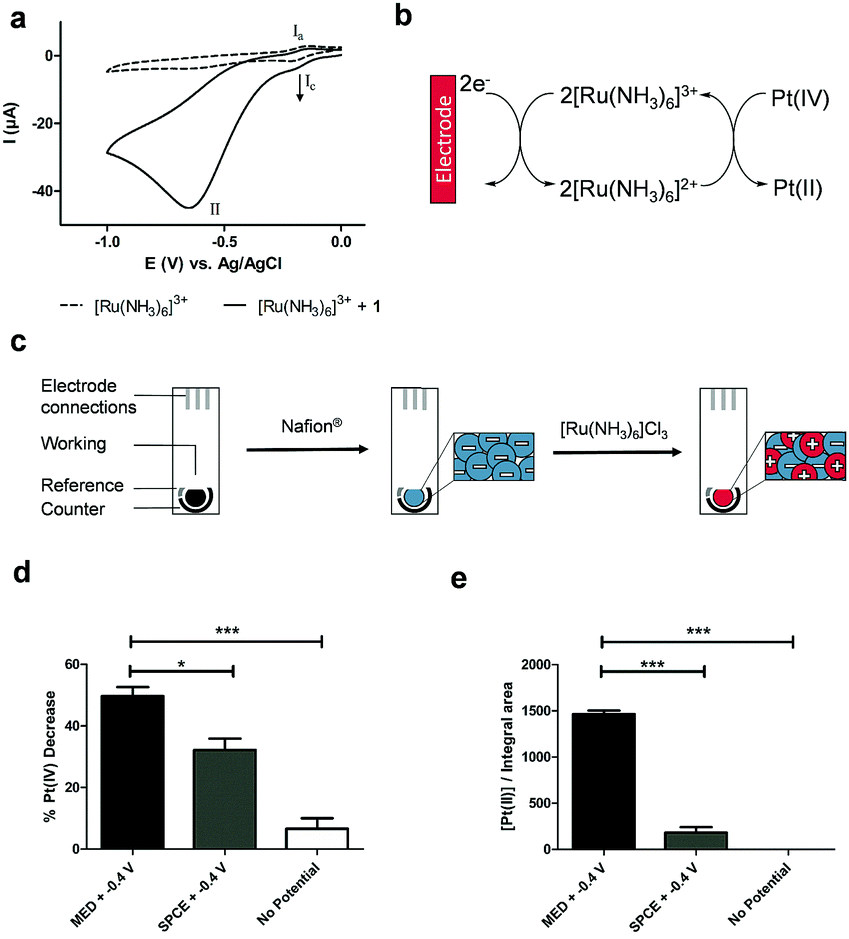 | ||
| Fig. 1 (a) Overlay of the cyclic voltammograms of [Ru(NH3)6]Cl3 (100 μM) in PBS (dashed line) alone, and following the addition of prodrug 1 (2 mM). A glassy carbon electrode was used as working electrode. Potentials are relative to the Ag/AgCl reference electrode employed. Processes (Ia) and (Ic) correspond to the reversible oxidation and reduction of the mediator, respectively, and (II) corresponds to the irreversible reduction of prodrug 1 (solid line); (b) scheme showing the electro-catalytic mechanism for the reduction of the Pt(IV) prodrug 1, where the ruthenium complex shuttles electrons from the electrode surface to the prodrug. The reduced ruthenium complex ([Ru(NH3)6]2+) is electrochemically regenerated through the continuous application of the reduction potential characteristic for the redox couple [Ru(NH3)6]3+/[Ru(NH3)6]2+; (c) the immobilisation of [Ru(NH3)6]3+ onto the carbon ink screen-printed electrodes (SPCE) was carried out by coating the working electrode with the sulfonated fluoropolymer Nafion®, followed by cation exchange entrapment of [Ru(NH3)6]3+. The successful entrapment of the redox mediator was confirmed by CV analysis in PBS with the immobilised [Ru(NH3)6]3+ showing the expected reversible redox behaviour (Fig. S4, ESI†); (d and e) electroactivation of prodrug 1 by the application of −0.4 V with modified electrode (MED) or un-modified electrode (SPCE) in PBS with HPLC monitoring of: (d) %Pt(IV) and (e) the Pt(II)–GMP bisadduct (n = 3). The data represent the mean ± S.D. ***P < 0.001 by one-way ANOVA with Tukey post-test. | ||
We thus devised a redox-mediated system to allow electrochemical activation of prodrug 1 at a lower reduction potential via [Ru(NH3)6]Cl3 electrostatically bound within a sulfonated fluoropolymer Nafion® film that was able to shuttle electrons from the electrode surface via electron hopping to reduce the Pt(IV) species in solution (Fig. 1b and c). Successful entrapment of the redox mediator was confirmed by CV analysis in PBS with the immobilised [Ru(NH3)6]3+ showing the expected reversible redox behaviour (Fig. S4, ESI†).
The oxidised form of the redox mediator, [Ru(NH3)6]3+, was reduced at the electrode surface by applying a potential of −0.4 V to afford [Ru(NH3)6]2+, which was able to reduce the Pt(IV) species, becoming re-oxidised in the process and closing the electro-catalytic cycle. A bulk electrolysis experiment consisting of 1 and [Ru(NH3)6]3+ (subjected to a constant potential of −0.4 V) was analysed by HPLC and showed good conversion of 1 to the active drug 2 in PBS (Fig. S3, ESI†).
The ability of the modified electrode to function in biological media was assessed in 10% foetal bovine serum (FBS) and in colorectal carcinoma HCT 116 cell lysate. CV analysis over 24 h showed that the modified electrode retained the reversible redox behaviour characteristic of the Ru(III)/Ru(II) couple (Fig. S5, ESI†), with only incubation in cell lysate showing a change in redox processes, attributed to protein binding affecting the redox mediator and its interactions within the Nafion® layer. Despite this, the electroactivation of 1 in cell lysate was not significantly affected. In addition, prodrug 1 was found to be relatively stable in 10% FBS at 37 °C (16% degradation after 24 h, n = 3, Fig. S6, ESI†) and towards reduction by glutathione and ascorbic acid with <10% decrease in Pt(IV) levels after 24 h at 37 °C (Fig. S1, ESI†). Leaching of the mediator from the electrode into solution was minimal in both solution and in cellular environments as shown by ICP-OES (Fig. S7 and Table S1, ESI†). The total content of the ruthenium complex electrostatically bound in the Nafion® layer was quantified by ICP-OES and the proportion that was redox-active was determined by chronoamperometry (Experimental details in Fig. S8 and S9, ESI†). The total content of mediator per electrode was 1.08 ± 0.02 μg, of which 0.84 μg or around 78% of the total content was found to be redox-active.
The conversion of Pt(IV) to Pt(II) was investigated using the modified electrode for the electroactivation of 1 by applying a reduction potential of −0.4 V for 24 h at 37 °C in PBS. Guanosine monophosphate (GMP) was included in the solutions to aid analysis as the generated active species oxaliplatin, 2, following aquation of the oxalate ligand is highly thermodynamically-driven to complex the N7 position of the guanosine monophosphate, allowing both Pt(IV) and Pt(II) species to be readily visualised by HPLC (Fig. S10, ESI†).
Electroactivation of 1 in PBS with the modified electrodes resulted in a clear increase of Pt(II) in solution, with 49% conversion of prodrug 1 after 24 h (Fig. 1d and e). The modified electrodes were also capable of electroactivating 1 in 10% FBS and HTC 116 cell lysate (Fig. S11, ESI†). The disparity between the conversion of Pt(IV) and the resultant yield of Pt(II) with the non-modified electrodes is attributed to undesired reactions occurring at the electrode surface. With the modified electrodes, where there is an electron shuttle between the electrode surface and 1, the conversion occurs consistently into oxaliplatin 2. These results illustrate the ability of the proposed system to electrochemically mediate the reduction of a Pt(IV) prodrug to its active Pt(II) form at a lower reduction potential compared to the non-mediated direct reduction. As the direct reduction of prodrug 1 has been established to have a pH-dependent reduction potential (Fig. S12, ESI†), we confirmed that no significant decrease in pH occurred during the electroactivation process (Table S2, ESI†).
Cell compatibility of the components of the modified electrode were assessed (see set-up in Fig. 2a) by immersing the modified electrode into a culture of HCT 116 cells, and applying a reduction potential of −0.4 V (Fig. 2b). This resulted in no cytotoxicity.
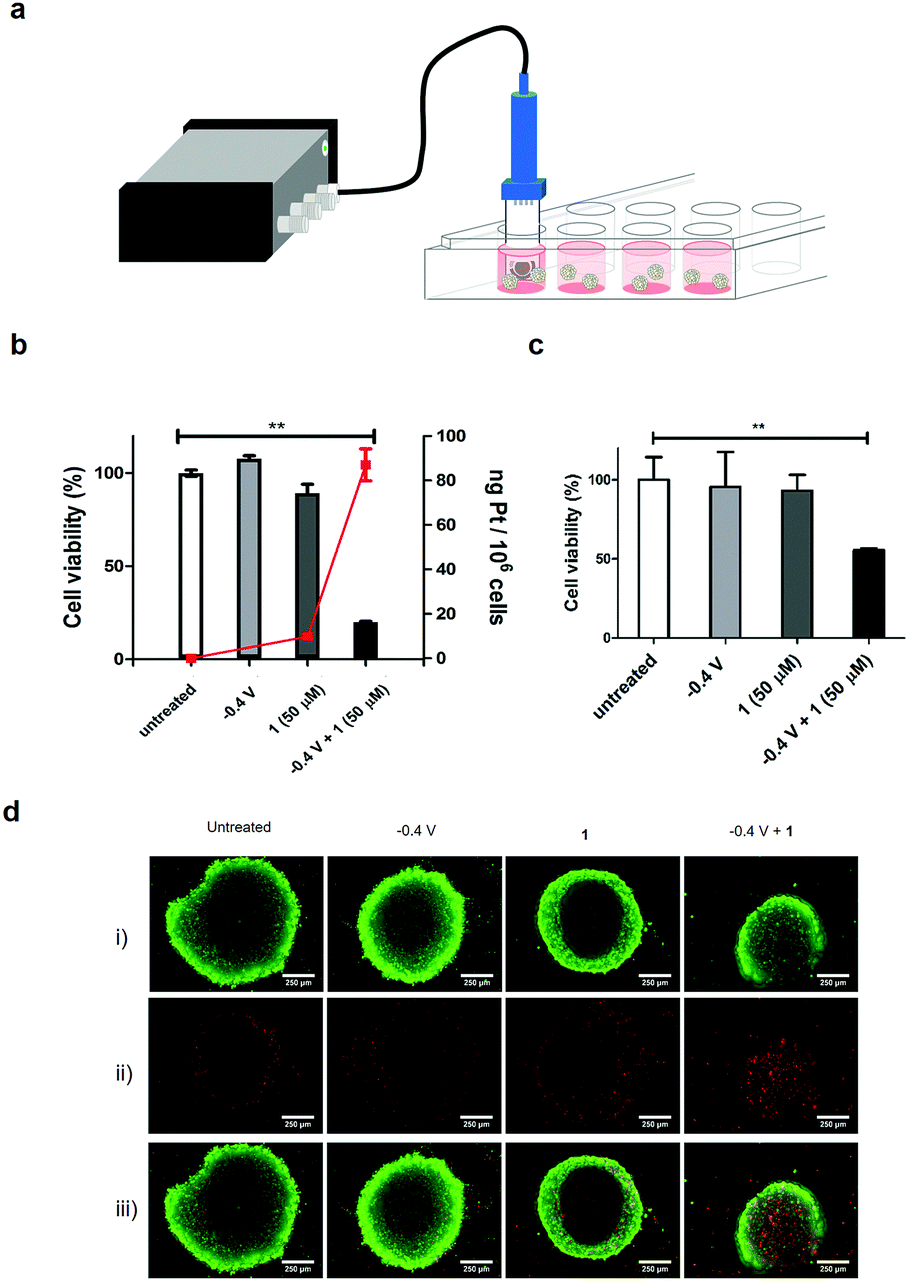 | ||
| Fig. 2 (a) Set-up used for the electroactivation of 1 in cell culture, showing the modified electrode immersed in cell culture and connected to a 4-channel portable potentiostat; (b) cell viability of HCT 116 cells treated with 1 (50 μM), with a reduction potential of −0.4 V applied for 1 h, and subsequently incubated for 3 days (MTT assay, n = 3). ***P < 0.001 by one-way ANOVA with Dunnett post-test, compared to the untreated control group; red line shows the quantification of Pt in the HCT 116 cells after treatment with 1 (50 μM) and −0.4 V for 1 h, and incubation for 4 h, after which the cells were harvested and the Pt content in the DNA isolated (Qiagen DNeasy blood and tissue kit) determined by ICP-MS; (c) cell viability of HCT 116 spheroids treated with 1 (50 μM) and −0.4 V applied for 1 h, and incubated for 3 days (CellTiter Glo 3D assay, n = 3). The data represent the mean ± S.D. ***P < 0.001 by one-way ANOVA with Dunnett post-test, compared with the untreated control group; (d) cells were incubated as above and stained with the green-fluorescent calcein-AM to indicate intracellular esterase activity and red-fluorescent ethidium homodimer-1 to show loss of plasma membrane integrity (LIVE/DEAD® assay) and imaged by fluorescence microscopy: (i) live cells (green), (ii) dead cells (red) and (iii) merged image. | ||
Importantly, 1 exhibited limited cytotoxicity up to 50 μM, thus displaying a good toxicity selectivity ratio between the Pt(IV) and Pt(II) states (the IC50 of 2 with HCT 116 cells was 0.19 μM, Fig. S13, ESI†). The electrochemical prodrug activation was validated in the same cell culture with the modified electrode immersed in the cell culture medium containing 1 (50 μM), and a reduction potential of −0.4 V was applied for 1 h with cell viability measured after 3 days. Cells treated with 1 in the absence of the reduction potential showed no decrease in viability, whereas 80% of cells treated with 1 and the application of the reduction potential died (Fig. 2b and Fig. S14, ESI†). These cells also showed an increased uptake of Pt (82 ng Pt/106 cells, n = 3) compared to cells treated only with 1 (10 ng Pt/106 cells, n = 3), as analysed by ICP-MS of the cell content (Fig. 2b), as well as an increase in platinated DNA (4 ng Pt per mg DNA vs. 0.75 ng Pt per mg DNA, Fig. S15, ESI†).
In order to mimic the 3D tumour environment multi-cellular tumour spheroids were subjected to the electroactivation process. Spheroids were generated from HCT 116 cells by the hanging drop method20,21 and treated with 1 followed by electroactivation at −0.4 V for 1 h on day 5. Cell viability was measured after 3 days which showed, as expected, no decrease in cell viability in spheroids treated only with 1; however, a 50% decrease in viability and increased number of apoptotic cells was observed when 1 was activated electrochemically (Fig. 2c and Fig. S16, ESI†). To further demonstrate the cytotoxic effect of the electroactivation of 1 on 3D tumour spheroids, live and dead cells were stained after electroactivation at −0.4 V for 1 h and incubating for 48 h (Fig. 2d), showing an increase in the number of dead cells.
In summary, an electrochemical prodrug activation system, that is stable in biological complex media, for platinum-based prodrugs has been devised and its efficacy in generating cytotoxic Pt(II) species examined in cell culture. The system retained its activating function within biological milieu and is amenable to miniaturisation for implantation directly into a tumour. Stereotactic placement of miniaturised electrochemical devices within a tumour, as part of the Implantable Microsystems for Personalised Anti-Cancer Therapy (IMPACT) project, could provide unequivocal control over prodrug activation in a site-selective, time-dependent and localised mediated manner. Further investigation will be focused on improving rate of reduction of Pt(IV) species by the nano-structuring of the electrode surface to increase active surface area. Expansion of the scope of this electrochemical prodrug activation technology is also being explored.
This work was supported by funding from University of Edinburgh, UK Engineering and Physical Sciences Research Council through the Implantable Microsystems for Personalised Anti-Cancer Therapy (IMPACT) programme grant (EP K034510/1) and the Rosetrees Trust (A865). Data used within this publication can be accessed at: http://dx.doi.org/10.7488/ds/2395.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Famm, B. Litt, K. J. Tracey, E. S. Boyden and M. Slaoui, Nature, 2013, 496, 159–161 CrossRef PubMed.
- P. S. Olofsson and K. J. Tracey, J. Intern. Med., 2017, 282, 3–4 CrossRef PubMed.
- S. Furman and J. B. Schwedel, N. Engl. J. Med., 1959, 261, 943–948 CrossRef PubMed.
- B. S. Wilson, C. C. Finley, D. T. Lawson, R. D. Wolford, D. K. Eddington and W. M. Rabinowitz, Nature, 1991, 352, 236–238 CrossRef PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef PubMed.
- X. Han, J. Sun, Y. Wang and Z. He, Med. Res. Rev., 2015, 35, 1268–1299 CrossRef PubMed.
- R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Eur. J. Inorg. Chem., 2017, 1596–1612 CrossRef.
- Y. Shi, S.-A. Liu, D. J. Kerwood, J. Goodisman and J. C. Dabrowiak, J. Inorg. Biochem., 2012, 107, 6–14 CrossRef PubMed.
- A. Lasorsa, O. Stuchlíková, V. Brabec, G. Natile and F. Arnesano, Mol. Pharmaceutics, 2016, 13, 3216–3223 CrossRef PubMed.
- E. Chu and V. T. DeVita Jr, Physician's Cancer Chemotherapy Drug Manual, Jones and Bartlett Publishers, Inc., 15th revised edn, 2015 Search PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- J. Z. Zhang, E. Wexselblatt, T. W. Hambley and D. Gibson, Chem. Commun., 2012, 48, 847–849 RSC.
- M. Sinisi, F. P. Intini and G. Natile, Inorg. Chem., 2012, 51, 9694–9704 CrossRef PubMed.
- K. R. Barnes, A. Kutikov and S. J. Lippard, Chem. Biol., 2004, 11, 557–564 CrossRef PubMed.
- R. P. Feazell, N. Nakayama-Ratchford, H. Dai and S. J. Lippard, J. Am. Chem. Soc., 2007, 129, 8438–8439 CrossRef PubMed.
- F. Ai, T. Sun, Z. Xu, Z. Wang, W. Kong, M. W. To, F. Wang and G. Zhu, Dalton Trans., 2016, 45, 13052–13060 RSC.
- Y. Dai, X. Kang, D. Yang, X. Li, X. Zhang, C. Li, Z. Hou, Z. Cheng, P. Ma and J. Lin, Adv. Healthcare Mater., 2013, 2, 514 CrossRef.
- P. Gramatica, E. Papa, M. Luini, E. Monti, M. B. Gariboldi, M. Ravera, E. Gabano, L. Gaviglio and D. Osella, JBIC, J. Biol. Inorg. Chem., 2010, 15, 1157–1169 CrossRef PubMed.
- G. Thiabaud, R. McCall, G. He, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Angew. Chem., Int. Ed., 2016, 55, 12626–12631 CrossRef PubMed.
- H. Li and A. B. Hummon, Anal. Chem., 2011, 83, 8794–8801 CrossRef PubMed.
- X. Liu, E. M. Weaver and A. B. Hummon, Anal. Chem., 2013, 85, 6295–6302 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc04151b |
| This journal is © The Royal Society of Chemistry 2018 |