
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA catalyst-free, temperature controlled gelation system for in-mold fabrication of microgels†
Andreas J. D.
Krüger
a,
Jens
Köhler
 a,
Stefan
Cichosz
b,
Jonas C.
Rose
a,
David B.
Gehlen
a,
Tamás
Haraszti
a,
Martin
Möller
ac and
Laura
De Laporte
*a
a,
Stefan
Cichosz
b,
Jonas C.
Rose
a,
David B.
Gehlen
a,
Tamás
Haraszti
a,
Martin
Möller
ac and
Laura
De Laporte
*a
aDWI–Leibniz-Institute for Interactive Materials, 52074 Aachen, Germany. E-mail: delaporte@dwi.rwth-aachen.de
bTechnical University of Lodz (TUL), 90-924 Lodz, Poland
cInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, 52072 Aachen, Germany
First published on 1st June 2018
Abstract
Anisometric microgels are prepared via thermal crosslinking using an in-mold polymerization technique. Star-shaped poly(ethylene oxide-stat-propylene oxide) polymers, end-modified with amine and epoxy groups, form hydrogels, of which the mechanical properties and gelation rate can be adjusted by the temperature, duration of heating, and polymer concentration. Depending on the microgel stiffness, the rod-shaped microgels self-assemble into ordered or disordered structures.
Amine-epoxy chemistry is a long-established industrial technology, commonly applied in the fabrication of resins and adhesives. The reaction mechanism, mixing, and reaction kinetics have been extensively described before. These include the curing of epoxy resins with simple short amine linkers,1,2 kinetic experiments investigating the addition reaction of primary and secondary amines in relation to their stoichiometry,3,4 mixing behavior,5 and water uptake of crosslinked networks.6 To prepare hydrogels, the amine-epoxy click reaction was implemented for biocompatible poly(ethylene glycol) (PEG) or thermoresponsive poly(N-isopropylacrylamide) (pNIPAAm) based systems. In contrast to commonly applied (photo)chemical free radical acrylate polymerization or copper catalyzed azide–alkyne click chemistry, the amine-epoxy ring-opening addition works independently of an initiator or a catalyst. Macro- and microporous hydrogels were fabricated by crosslinking bifunctional PEG-epoxy with cystamine and diamino-poly(ε-caprolactone) using a salt as a sacrificial template.7 Alternatively, PEG hydrogels containing antibacterial quaternary ammoniums were developed by combining thiol-yne and amine-epoxy click chemistry.8 In the case of pNIPAAm, prepolymer backbones were functionalized with side-chain epoxy functions and coupled via degradable polyamidoamines to form an injectable hydrogel system.9 When epoxy functions in the pNIPAAm hydrogels were modified with proteins via their free amines, a positive effect on cell viability and proliferation was observed in vitro.10 As this nucleophilic addition reaction can be induced via a thermal trigger, it is a facile but highly effective crosslinking mechanism that can be applied in a variety of hydrogel fabrication methods.
One type of hydrogel that has recently gained increasing attention is anisometric microgels, as they can be applied as building blocks to induce anisotropy in macroscopic materials. They have been produced via in-mold polymerization techniques using perfluorpolyether (PFPE) molds with low surface energy and defined cavities.11,12 Commonly, (PEG)-acrylates are crosslinked into particles or microgels with varying size, shape, and geometry by photochemical curing using radical initiators in an inert atmosphere.13 Functional groups that enable post-process modification with, for example, bioactive domains, were implemented by mixing side-chain functionalized acrylate moieties pre-crosslinking.12,15 To fabricate soft rod-shaped microgels with polymer concentrations down to 10 wt/V%, sPEG-acrylates were dispersed in liquid inert PEG (200 g mol−1) as a non-volatile solvent.13Via Michael-type addition, these microgels can be functionalized with thiol-containing moieties.14
In this report, a simple and more versatile crosslinking alternative is presented to apply for in-mold polymerization. Star poly(ethylene oxide-stat-propylene oxide) (sPEG) polymers are functionalized with epoxy or amine groups to enable thermally induced catalyst and radical-free click chemistry as a versatile alternative to the established acrylate-based systems. The sPEG polymer backbone consists of 20% propylene oxide (PO) suppressing the characteristic crystallization of PEG at molecular weights exceeding 700 g mol−1 (Fig. 1a).16 Their fluid characteristics open up a broad range of potential applications, excelling those of commercially established PEG derivatives. Moreover, the multi-arm backbone of sPEGs enables fast crosslinking, while maintaining free endgroups to implement further modification. Amine-epoxy systems especially benefit from these properties as three different reactive groups can be provided: amine, epoxy, and hydroxyl moieties (Fig. 1). This allows for coupling a broad range of functions, e.g., acrylates, carboxylic acids, amines, epoxy compounds (e.g. fluorophors17), NHS-active esters, alcohols, or even anhydrides. As a result, amine-epoxy gels can be specifically modified according to the desired application.
 | ||
| Fig. 1 (a) The sPEG backbone contains a defined ratio of EO and PO moieties, inhibiting the crystallization at elevated molecular weight; (b) synthesis procedure for the production of sPEG-NH2 and sPEG-epoxy (detailed procedures are provided in the ESI†); (c) gelation mechanism: a nucleophilic addition of amine to epoxy function. | ||
To obtain sPEGs with epoxy and amine endgroups, synthesis procedures are established to functionalize the liquid sPEG (Fig. 1b and ESI†). Briefly, sPEG-epoxy (1) is produced by the activation of sPEG with a strong inorganic base, followed by stirring with epichlorohydrin overnight. The procedure is adapted from previous reports.18 sPEG-amine (4) is synthesised via a three-step reaction procedure, adapted from the literature.19 The sPEG substrate is converted with triethylamine and mesylchloride leading to sPEG-mesylate (2). After reflux overnight with sodiumazide, compound 3 is obtained. Alternatively to an elaborate homogeneous catalysed Staudinger reduction, a heterogeneous catalysis with palladium on carbon in an 85 bar hydrogen atmosphere is performed in a high pressure autoclave reactor to reduce 3 to sPEG-amine.
The sPEG, modified with amine and epoxy endgroups, provides a system where three different parameters can be altered to control the mechanical properties, swelling behaviour, and gelation time of the hydrogels in an aqueous environment: polymer concentration, heating time, and the reaction temperature. The gelation time and mechanical properties are quantified by rheology. Prepolymer solutions with an equimolar ratio of amine to epoxy are applied at different polymer concentrations. The duration of heating and temperature are varied, while a solvent trap is installed around the sample to minimize water evaporation. First, the influence of the prepolymer concentration is investigated at 60 °C (Fig. 2a). This reveals that a minimum sPEG content of 7.5 wt/V% is necessary to achieve crosslinked hydrogels, which have a maximum G′ of 1.8 kPa. By increasing the polymer concentration, a range of different G′ is achieved up to 92 kPa at 20 wt/V%. In addition to the increase in storage modulus, the initiation period for gelation decreases from above 2000 s to less than 600 s. Secondly, different heating times are compared to initiate and stop the gelation reaction and adjust the stiffness of the hydrogels (Fig. 2b). 10 or 20 wt/V% sPEG solutions are applied to the rheometer's heating plate at 25 °C and consequently heated to 60 °C for specific durations. In the case of 20 wt/V%, a range of gels with G′ maximum of several kPa up to 62 kPa is realized as the heating time increases from 600 to 3500 s, respectively. When reducing the polymer concentration by half, the G′ maximum ranges from approximately 100 Pa to 10 kPa, respectively. Thirdly, the effect of the dispersing agent on the storage modulus and gelation rate is investigated as water will be replaced by a non-volatile PEG filler for the microgel fabrication (Fig. 2c and chapter 6, ESI†). When comparing different heating temperatures, similar G′ values are achieved independent of the dispersing agent or temperature. On the other hand, the gelation point is strongly related to the heat input and differs depending on the dispersing agent. In aqueous solution, a minimum gelation time of 261 s is detected at 80 °C, while for decreasing temperature, the gelation time prolongs up to 1236 s at 50 °C. In contrast, in the non-volatile PEG filler, gelation occurs four to eight times slower and the maximum G′ is still not reached after 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s in the case of 50 °C.
000 s in the case of 50 °C.
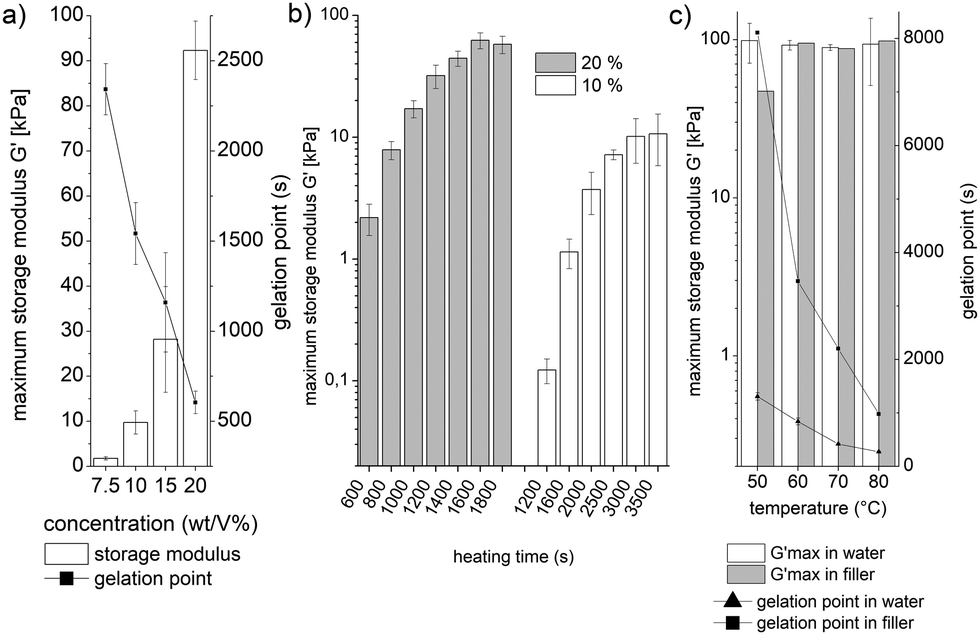 | ||
| Fig. 2 Hydrogel characterization (maximum G′ and gelation point) (procedures and detailed conditions are listed in the ESI†). (a) Variation of sPEG concentration for heating at 60 °C, (b) the effect of different heating times at 60 °C for both 10 and 20 wt/V%, (c) gelation at different temperatures for both water and non-volatile PEG filler as a dispersing agent (20 wt/V% reactive sPEG); measurements are reproduced three times except for 2c with the filler due to long gelation times. | ||
To compare hydrogels prepared with sPEG-acrylate versus sPEG-amine-epoxy, hydrogel discs are prepared in polydimethylsiloxane (PDMS) molds. In the case of the sPEG-amine-epoxy hydrogels, the solution is crosslinked at 60 °C for 45 minutes inside an oven. When using non-volatile PEG as the dispersing agent, the filler is successfully washed out post-gelation (Fig. S10b in the ESI†). As demonstrated before, hydrogel discs prepared with an sPEG-acrylate/linear PEG solution are, in contrast to water-based gelation, white and non-transparent (Fig. 3a).13 This change in optical properties indicates a reaction induced phase separation,20 leading to hydrogels with a predominantly mesoporous character. In contrast, hydrogel discs fabricated with sPEG-epoxy and sPEG-amine are comparably transparent, regardless of whether the reactive polymer is dispersed in water or the inert PEG filler (Fig. 3a). This distinction in internal network structure may be explained by the different crosslinking mechanisms (Fig. 3b). Acrylate groups crosslink with the formation of linear polymer chains and distributed side functions, which connect the linear chains together to form a crosslinked network. Amine-epoxy click reactions (Fig. 1c) on the other hand result in a step-wise polymerization process, leading to a more ordered network.21 The similar appearance of crosslinked hydrogels from sPEG-amine and sPEG-epoxy, when either water or the filler is used to disperse the polymer, is reflected in a similar swelling behaviour of the hydrogel discs at the same polymer concentration (Fig. S10a in the ESI†). Post-modification of the hydrogels is demonstrated by the coupling of rhodamine B-acrylate, indicating the presence of free amines after thermal crosslinking (Fig. S11 in the ESI†).
 | ||
| Fig. 3 Transition from crosslinking in bulk to in-mold polymerization for the production of microgel rods. (a) Comparison of bulk-hydrogels: discs are produced in PDMS molds in either water or linear PEG (200 g mol−1) as an inert filler, polymer concentration: 20 wt/V%, scale bar: 12 mm; (b) schematic of network formation using sPEG-acrylate or sPEG-amine-epoxy gelation systems: the acrylate produces heterogeneous networks, while the amine-epoxy networks are more ordered with more defined porosity. (c) STED-microscopy of microgels, fabricated with 20 wt/V% of the different pre-polymers and rhodamine B-acrylate, scale bar: 20 μm; below: magnification of porous microgel structure, scale bar: 1 μm. | ||
In addition to hydrogel discs, rod-shaped microgels (5 × 5 × 50 μm) are produced via in-mold polymerization using the PEG filler (details in chapter 9, ESI†).13 In the case of sPEG-amine and sPEG-epoxy, the filled mold is placed in an oven at 60 °C for 45 minutes. When compared to the sPEG-acrylate microgels, the thermally crosslinked microgels differ significantly in internal structure as imaged with stimulated emission depletion (STED) microscopy (Fig. 3c). To visualize the structure, rhodamine B-acrylate is coupled to the microgels by mixing the dye into the prepolymer solution before crosslinking. These results demonstrate that amine-epoxy microgels contain a more homogenous porosity with a mesh size in the range of 1–100 nm, while the pores for the sPEG-acrylate microgels are more heterogeneous and in the micrometer range. Similar to the hydrogels, the stiffness of the microgel rods is altered by the polymer concentration and measured using atomic force microscopy (AFM) (Fig. 4b, procedure details in the ESI†). Solutions with sPEG-amine and sPEG-epoxy at 10 wt/V% result in softer rods with an elastic modulus of 17 ± 2.6 kPa, whereas rigid and stiff microgels with moduli of 40 ± 6.2 kPa and 114 ± 12 kPa, respectively, are obtained with 50 and 100 wt/V% functional sPEG.
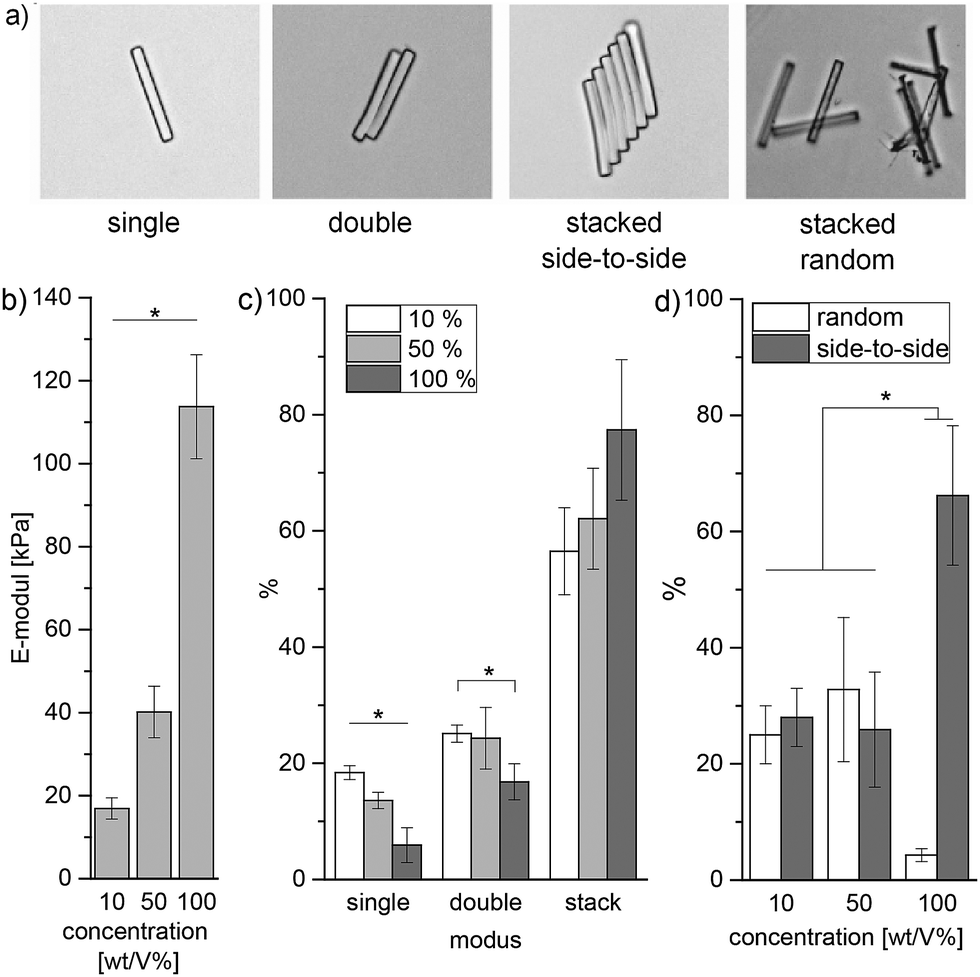 | ||
| Fig. 4 Analysis of microgel self-assembly: (a) stacking modes, the microgels are imaged using bright field microscopy; (b) individual microgel stiffness depending on the reactive polymer concentration, determined using AFM (procedure in the ESI†); (c) self-assembly is correlated with the reactive pre-polymer concentration used to prepared the microgels. The number of rods is counted and categorized according to the groups defined in (a); (d) comparison of random versus side-by-side microgel aggregation with respect to the polymer concentration. All measurements are reproduced three times. | ||
Interestingly, different self-assembly behaviours in water of the purified microgels are observed depending on the prepolymer concentration, and therefore on the stiffness of the individual microgels. Concentrated microgel solutions are compared for three prepolymer concentrations, 10, 50, and 100 wt/V%. The rods are counted and categorized by their degree of aggregation into four modes: single, double, stacked random, and stacked side-to-side. Stacks contain 3 or more microgels. A direct correlation between individual microgel stiffness and aggregation behaviour is observed when comparing the stacking modes and the rods’ prepolymer content (Fig. 4b). With increasing microgel stiffness, the tendency to build aggregates rises and the amount of isolated microgels drops significantly. In the case of 10 and 50 wt/V% microgels, the stacks show similar amounts of random and ordered stacked gels, whereas for the stiffer 100 wt/V% rods, 66 ± 12% of the microgels are part of side-to-side stacks with a minimal tendency for random aggregation for only 4.3 ± 1.1% of the microgels (Fig. S10 in the ESI†). As microgels with an increased polymer concentration are more hydrophobic and behave more like solid particles, elevated attraction forces may be induced.22 The interfacial free energy inside the more hydrophilic liquid is minimized via self-assembly of the microgels into ordered structures. Here, the high aspect ratio provided by the anisometric shape of the microgels further promotes this assembly behavior.22
In summary, a thermally induced gelation system is established, based on liquid sPEGs that crosslink via catalyst free amine-epoxy click chemistry. The system excels by its versatility, as the stiffness and gelation rate can be adjusted by three different parameters: polymer concentration, gelation temperature, and heating time. In addition, a broad range of possibilities are available to implement post-modifications. The thermal crosslinking mechanism facilitates many hydrogel fabrication methods, such as in-mold polymerization to fabricate soft microgels. In contrast to the UV polymerized microgels using this technique, the microgels formed via the click-reaction possess a more homogeneous nanometer mesh size. Depending on the individual microgel stiffness, self-assembly behaviour in an aqueous solution is altered. Therefore, the presented gelation system opens new avenues to prepare a platform of hydrogels and microgels with variable properties depending on the application.
We acknowledge funding from the Deutsche Forschungs-gemeinschaft (DFG) as part of the SFB 985 -functional microgels and microgel systems, and from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (Anisogel, grant No 637853; Jellyclock, No 695716). This work was performed in part at the Center for Chemical Polymer Technology (CPT), which was supported by the EU and the Federal State of North Rhine-Westphalia (grant EFRE 30 00 883 02).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. M. Laza, C. A. Julian, E. Larrauri, M. Rodriguez and L. M. Leon, Polymer, 1999, 40, 35–45 CrossRef.
- F. González Garcia, B. G. Soares, M. E. Leyva and A. Z. Simões, J. Appl. Polym. Sci., 2010, 117, 2762–2770 CrossRef.
- J. P. Bell, J. Polym. Sci., Part A-2, 1970, 8, 417–436 CrossRef.
- D. H. Kim and S. C. Kim, Polym. Bull., 1987, 18, 533–539 CrossRef.
- M. R. Vanlandingham, R. F. Eduljee and J. W. Gillespie Jr, J. Appl. Polym. Sci., 1999, 71, 699–712 CrossRef.
- P. O. S. B. Cursaru and M. Teodorescu, U.P.B. Sci. Bull., Series B, 2010, 72, 99–114 Search PubMed.
- Z. A. A. Hamid, A. Blencowe, B. Ozcelik, J. A. Palmer, G. W. Stevens, K. M. Abberton, W. A. Morrison, A. J. Penington and G. G. Qiao, Biomaterials, 2010, 31, 6454–6467 CrossRef PubMed.
- C. Zhou, V. X. Truong, Y. Qu, T. Lithgow, G. Fu and J. S. Forsythe, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 656–667 CrossRef.
- A. K. Ekenseair, K. W. M. Boere, S. N. Tzouanas, T. N. Vo, F. K. Kasper and A. G. Mikos, Biomacromolecules, 2012, 13, 1908–1915 CrossRef PubMed.
- M. O. Pehlivaner Kara and A. K. Ekenseair, Biomacromolecules, 2017, 18, 1473–1481 CrossRef PubMed.
- J. P. Rolland, B. W. Maynor, L. E. Euliss, A. E. Exner, G. M. Denison and J. M. DeSimone, J. Am. Chem. Soc., 2005, 127, 10096–10100 CrossRef PubMed.
- M. P. Kai, A. W. Keeler, J. L. Perry, K. G. Reuter, J. C. Luft, S. K. O'Neal, W. C. Zamboni and J. M. DeSimone, J. Controlled Release, 2015, 204, 70–77 CrossRef PubMed.
- J. C. Rose, M. Cámara-Torres, K. Rahimi, J. Köhler, M. Möller and L. De Laporte, Nano Lett., 2017, 17, 3782–3791 CrossRef PubMed.
- J. C. Rose, D. B. Gehlen, T. Haraszti, J. Köhler, C. J. Licht and L. De Laporte, Biomaterials, 2018, 163, 128–141 CrossRef PubMed.
- K. P. Herlihy, J. Nunes and J. M. DeSimone, Langmuir, 2008, 24, 8421–8426 CrossRef PubMed.
- J. Groll, J. Fiedler, E. Engelhard, T. Ameringer, S. Tugulu, H.-A. Klok, R. E. Brenner and M. Moeller, J. Biomed. Mater. Res., Part A, 2005, 74A, 607–617 CrossRef PubMed.
- J. Yu, Z. Su, H. Xu, X. Ma, J. Yin and X. Jiang, Polym. Chem., 2015, 6, 6946–6954 RSC.
- R. M. Laine, S. G. Kim, J. Rush, R. Tamaki, E. Wong, M. Mollan, H. J. Sun and M. Lodaya, Macromolecules, 2004, 37, 4525–4532 CrossRef.
- R. Mahou and C. Wandrey, Polymers, 2012, 4, 561 CrossRef.
- E. Girard-Reydet, H. Sautereau, J. P. Pascault, P. Keates, P. Navard, G. Thollet and G. Vigier, Polymer, 1998, 39, 2269–2279 CrossRef.
- V. M. Litvinov and A. A. Dias, Macromolecules, 2001, 34, 4051–4060 CrossRef.
- Y. Du, E. Lo, S. Ali and A. Khademhosseini, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9522 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Prepolymer functionalization and analysis, rheology and microscopy procedures. See DOI: 10.1039/c8cc02478b |
| This journal is © The Royal Society of Chemistry 2018 |