
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-surface synthesis of polyethylenedioxythiophene†
Iolanda
Di Bernardo
 ab,
Peter
Hines
c,
Maryam
Abyazisani
b,
Nunzio
Motta
bc,
Jennifer
MacLeod
*bc and
Josh
Lipton-Duffin
*bc
ab,
Peter
Hines
c,
Maryam
Abyazisani
b,
Nunzio
Motta
bc,
Jennifer
MacLeod
*bc and
Josh
Lipton-Duffin
*bc
aDepartment of Physics, University of Rome “La Sapienza”, Rome, Italy
bSchool of Chemistry, Physics, and Mechanical Engineering, Queensland University of Technology (QUT), Brisbane, QLD, 4000, Australia. E-mail: jennifer.macleod@qut.edu.au; josh.liptonduffin@qut.edu.au
cInstitute for Future Environments, Queensland University of Technology (QUT), Brisbane, QLD, 4000, Australia
First published on 26th March 2018
Abstract
On-surface synthesis of conjugated polymers is made challenging by the need to promote the desired reaction while preventing or minimizing unwanted ancillary reactions that compromise the product integrity. We perform a comprehensive study of the reactions of 2,5-dichloro-3,4-ethylenedioxythiophene on coinage metal surfaces, and demonstrate that only on Ag(111) can we obtain a planar polymer product, polyethylenedioxythiophene (PEDOT).
The surface-confined synthesis of covalently bonded polymers is a rapidly emerging field whose growth is fuelled by the possibility for growing novel polymer structures with bespoke characteristics.1 One of the main goals of this approach is the tailored synthesis of graphene-like materials, which can be bandgap-engineered according to the choice of monomer and the overall topology of the resulting polymer structure.2 However, controlling the long-range crystallinity of 2D polymers remains a challenge, owing to the wide variety of defects that can be kinetically trapped into the sheets.3 In this respect the growth of 1D polymers can offer an intrinsic advantage, since the symmetry reduction associated with a bidentate monomer concomitantly reduces the range of possible defect structures. 1D polymers therefore represent an opportunity to examine and refine on-surface reaction mechanisms while forming a well-defined product.
Recent investigations have explored a variety of coupling mechanisms for 1D polymers, including aliphatic C–H activation,4 [3+2] cycloaddition,5 imidization,6 and Bergman cyclization followed by radical polymerization.7 However, 1D polymerization is most frequently approached via dehalogenation reactions, which have been used to produce oligo-8 and polyphenylenes,9,10 poly-porphyrins11 and, in conjunction with cyclodehydrogenation, to synthesize graphene nanoribbons.12 As this range of products demonstrates, surface-synthesized polymers can be designed a priori with specific functionalities targeted through rational building block design. However, on catalytic surfaces it is critical to consider not only the desired polymerization reaction, but also the possibility for unintended reactions. In particular, sulphur abstraction can occur in molecules containing thiophene moieties, critically changing the molecular properties while at the same time poisoning the catalytic surface through sulphur adsorption.13
Here, we report on the surface-confined reactions of a halogenated, thiophene-containing molecule: 2,5-dichloro-3,4-ethylenedioxythiophene (DCEDOT, Fig. 1). The polymer product of EDOT is poly-3,4-ethylenedioxythiophene (PEDOT, Fig. 1), a 1D polymer that is traditionally synthesized in the solution phase and is used in a range of industrial and research applications.14 Although the brominated monomer (dibromo-EDOT, DBEDOT) can undergo solid-state polymerization from thin films using thermal activation,15 DCEDOT has not been reported to polymerize under similar conditions. The surface-confined polymerization of EDOT could, given a judicious choice of substrate symmetry, provide a route to the formation of ordered, oriented PEDOT films, allowing the directional transport along the 1D polymer backbone to be fully exploited, and demonstrating the relevance of on-surface polymerization approach to the synthesis of complex, industrially-relevant polymers.
 | ||
| Fig. 1 Structure of DCEDOT, metal-coordinated EDOT, and PEDOT. The carbon atoms in each structure are color-coded. All structures contain β-carbon (gray) and ethylene–carbon (red). In the monomer, the α-carbon (pink) is bonded to Cl, whereas in the metal-coordinated EDOT it is coordinated to a metal center (yellow), and in PEDOT it is bonded to a neighbouring EDOT monomer (purple open circles). | ||
A previous study of on-surface polymerization of diiodo-EDOT (DIEDOT) suggested that short polymers with the monomer backbones oriented nearly perpendicular to the surface may be formed.16 In this configuration, one of the key competitive advantages of surface-confined polymerization, planarity, is lost, and hence the challenge of synthesizing planar, extended PEDOT remains open.
To provide a comprehensive picture of the surface-confined chemistry of DCEDOT we have deposited this precursor on Cu(111), Au(111) and Ag(111), which span a range of intrinsic reactivities from high (Cu) to low (Ag).17 The choice of halogen has been recently demonstrated to drive the morphology of resultant structures on the surface, insomuch as the adsorbed halogen and polymer must be simultaneously accommodated.18 We also note that the use of a lighter halogen facilitates dosing onto the surface, and thus the selection of DCEDOT is largely pragmatic. The substrate is of paramount importance in surface-confined chemistry, since it dictates both reactivity and molecular diffusion,19 and through lack of specificity can produce unwanted reactions in the monomer.20 We have characterized the molecular reactions using high-resolution synchrotron-radiation photoemission spectroscopy (SRPES), near-edge X-ray absorption for fine structure spectroscopy (NEXAFS), scanning tunnelling microscopy (STM) and density functional theory (DFT) calculations.21
SRPES: core level SRPES spectra for DCEDOT on Cu(111), Au(111) and Ag(111) were collected at C 1s, S 2p, Cl 2p and O 1s for various annealing temperatures. Qualitatively, the C 1s core level was used as an indicator for molecular adsorption and for the calibration of coverage: at room temperature DCEDOT adsorbs readily onto Cu(111), adsorbs weakly on Au(111), and does not adsorb on Ag(111) at all. Uptake on Ag(111) was successfully achieved only when heating the substrate at 150 °C, and even then only at doses substantially larger than 200 L. All data presented are for surfaces with near-monolayer coverage, which was estimated from the abrupt reduction in the rate of accumulation of C 1s signal intensity with time, indicating saturation of the metal surface and the onset of a lower sticking probability for second-layer molecules.
Fig. 2 shows high resolution scans of the C 1s core level of DCEDOT on Cu(111), Au(111) and Ag(111), stacked as a function of substrate annealing temperature. A number of color-coded synthetic peaks have been fitted to the C 1s data, corresponding to the chemical environments of the originating atoms: yellow for organometallic-associated α-carbons, pink for chlorine-associated α-carbons, grey for β-carbons and red for ethylene carbons. The DCEDOT undergoes bond cleavage in the thiophene ring starting at room temperature on Cu(111), as evidenced by the sp3-like state at approximately 284.5 eV (synthetic component coloured in green), and the substantial surface-bound state of sulphur seen in the RT spectrum (see ESI†). The components associated with ethylene–carbon and β-carbon have approximately equal intensity at room temperature, as expected for the intact molecule. Although the entire chemistry of the DCEDOT is reflected in the C 1s core level, our deconvolution of the C 1s signal is informed by the data collected at the core levels associated with the O, S and Cl heteroatoms. When the molecule adsorbs on the surface intact the α-carbons will bear the signature of bonding to chlorine, which is expected to result in a high-BE component.9 On Cu(111) there is spectral weight at high BE (pink), which we correlate with C–Cl bonds, evidence of which is also present in the Cl 2p spectra (see ESI†). Hence on Cu(111), annealing the DCEDOT molecule causes it to rupture at the thiophene ring without significant aryl coupling. This fragmentation of the molecule precludes PEDOT formation.
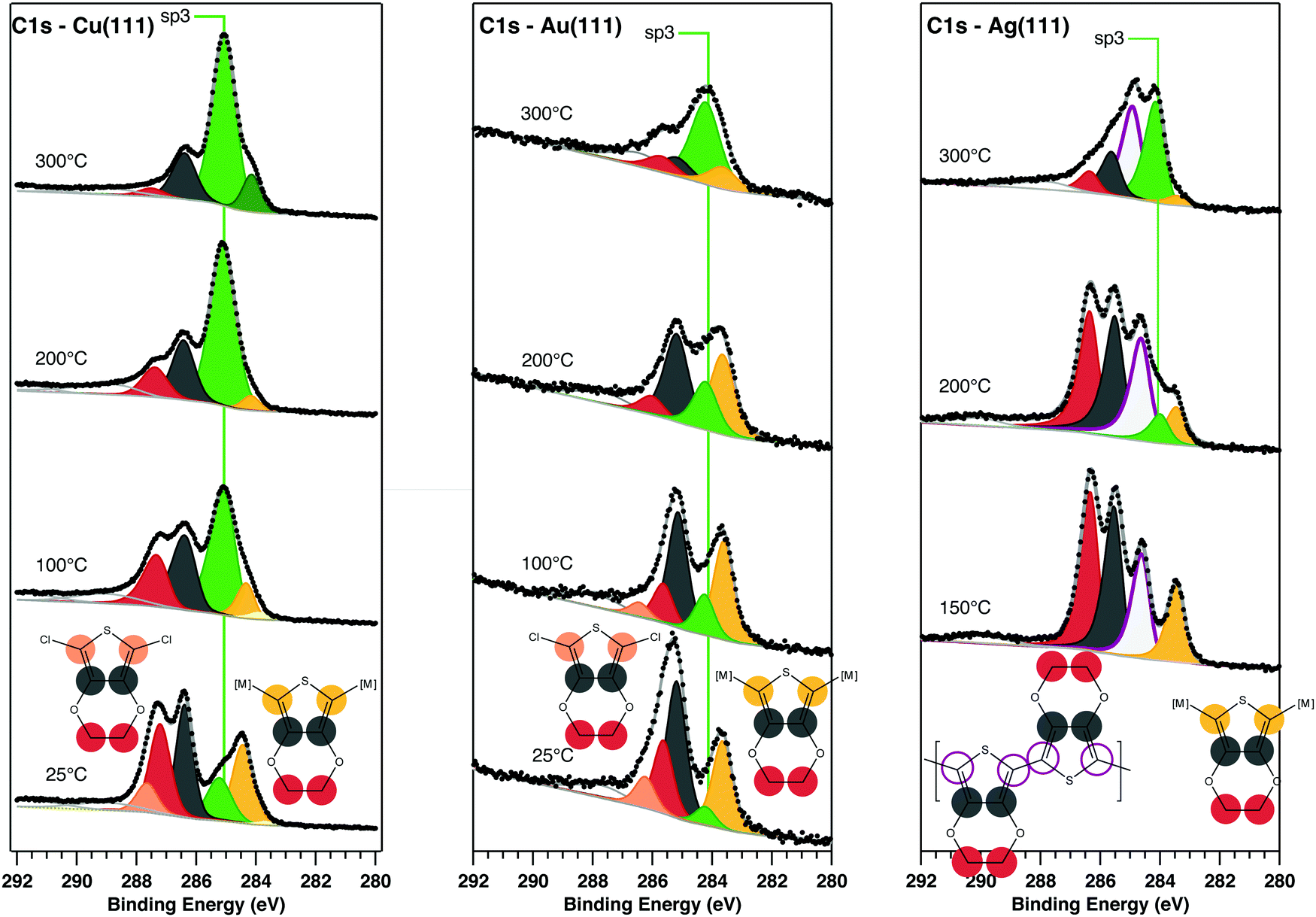 | ||
| Fig. 2 C 1s core level spectra of DCEDOT on Cu(111), Au(111), and Ag(111), stacked as a function of annealing temperature. Synthetic components correspond to features of the intact molecule are indicated by the color-coded molecular models shown at the bottom; the green sp3-like component is labelled in the figure. | ||
The C 1s spectrum on the RT Au(111) sample has a substantially reduced ethylene–carbon![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β-carbon ratio, which we attribute to fragmenting of the molecule under the intense synchrotron beam (see ESI†), an effect that has been previously observed in hydrogen-terminated EDOT species.22 After annealing at 200 °C both the α- and the ethylene–carbons are significantly altered. This temperature causes the rupture of the thiophene ring and the loss of the ethyl group, and no contributions related to the formation of new C–C bonds can be identified. The green sp3 component at 285.16 eV is assigned to molecular fragments adsorbed on the metal surface. Its area increases with subsequent annealing, indicating that on Au(111), as on Cu(111), fragmentation of the molecule prevents the formation of a PEDOT product.
:β-carbon ratio, which we attribute to fragmenting of the molecule under the intense synchrotron beam (see ESI†), an effect that has been previously observed in hydrogen-terminated EDOT species.22 After annealing at 200 °C both the α- and the ethylene–carbons are significantly altered. This temperature causes the rupture of the thiophene ring and the loss of the ethyl group, and no contributions related to the formation of new C–C bonds can be identified. The green sp3 component at 285.16 eV is assigned to molecular fragments adsorbed on the metal surface. Its area increases with subsequent annealing, indicating that on Au(111), as on Cu(111), fragmentation of the molecule prevents the formation of a PEDOT product.
Deposition of DCEDOT on Ag(111) at RT did not yield a measureable C 1s signal, indicating that the molecule was not adsorbed. However, following deposition onto a surface held at 150 °C, molecular features were observed in the high resolution scans of the C 1s core level. A new C 1s component can be identified at 284.6 eV; we ascribe this new feature to α-carbons in PEDOT. Following annealing to 200 °C, the C 1s spectra show a shifting of spectral weight from the α-C to a new peak near 285 eV, consistent with the sp3-like state of broken molecules. Further annealing increases the weight in this fragment signal. We propose that polymerization of DCEDOT occurs during the adsorption at 150 °C, and that the reaction may constrain the thiophene moiety into a configuration that protects against desulphurisation. This interpretation is supported by analysis of the S 2p, Cl 2p, and O 1s core levels (see ESI†).
NEXAFS: prior work on the EDOT monomer and films polymerized ex situ indicate that the three carbon types give rise to three distinct resonances in the NEXAFS spectrum, albeit with overlap in the spectral features.22,23 Angle-dependent NEXAFS collected from Ag(111) following deposition of DCEDOT onto the hot surface, shown in Fig. 3, are analysed to determine the adsorption geometry of the molecule.24 The spectra are fit with a series of peaks to produce a synthetic fit to the dataset.25 This decomposition is quite complex, as expected by the number of features in XPS core levels. Nevertheless, fitting the dichroism for the first three π* resonances24 leads to tilt angles of 6° ± 5°, 9° ± 8° and 50° ± 5° with respect to the surface normal. This is consistent with the growth of planar PEDOT, with the ethylene tail of each monomer twisted out of the surface (see ESI†).
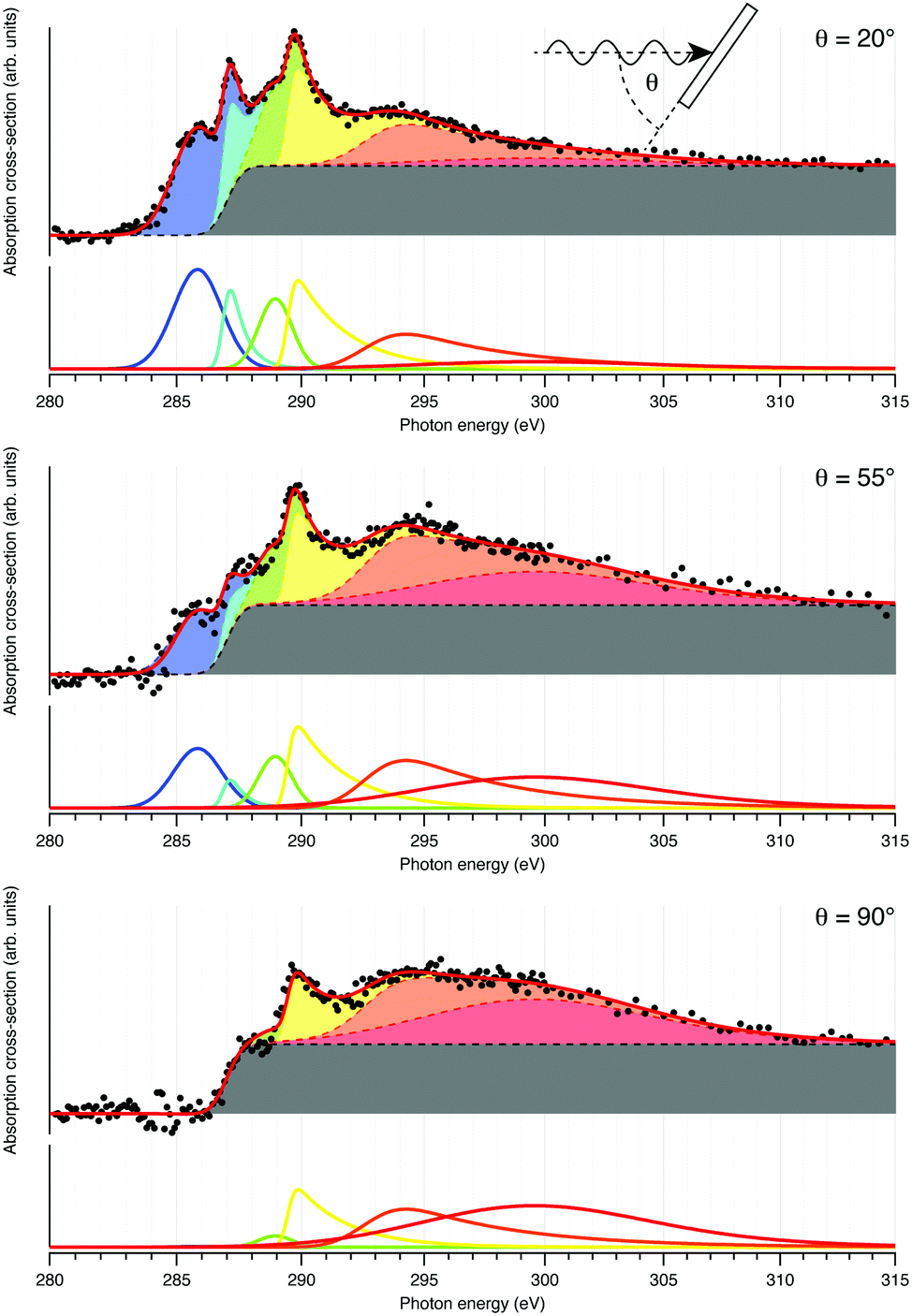 | ||
| Fig. 3 Angle-dependence of NEXAFS from DCEDOT on Ag(111). The spectra (black points) are fit with a linear combination of peaks and a constant absorption edge. | ||
STM and DFT: to confirm the polymerization suggested by the Ag(111) SRPES and NEXAFS data, we used scanning tunnelling microscopy to visualize the PEDOT product in real space. Typical images are show in Fig. 4: large-scale images show linear features extending along three directions, consistent with the symmetry of the underlying Ag(111). Higher resolution images reveal a zig-zag appearance to the lines, and confirm the spacing between individual units of 0.78 ± 0.04 nm, which agrees with the expected gas-phase periodicity of 0.797 nm.
 | ||
| Fig. 4 STM images of the annealed DCEDOT/Ag(111) surface (50 nm × 50 nm Vb = −1.2 V, It = 500 pA). The white arrows indicate the inferred high-symmetry directions of the Ag(111) substrate. Zoomed inset shows details of the rows (7.1 nm × 7.1 nm). | ||
Obtaining an accurate model of the adsorption is challenging because the 1 × 1 substrate lattice could not be imaged, but a simple assumption of commensurability allows a best-fit orientation to be extracted by matching the gas-phase spacing of the polymer to the substrate. This is achieved along the (1,−3) lattice direction, corresponding to the 〈2,1,−3〉 azimuth of the Ag crystal, and this orientation was used to obtain an optimized structure through DFT calculation, shown in Fig. 5, as well as to infer substrate directions in the micrographs of Fig. 4. Simulated STM images26 generated from the optimized structure are shown in Fig. 5, and exhibit qualitative agreement.
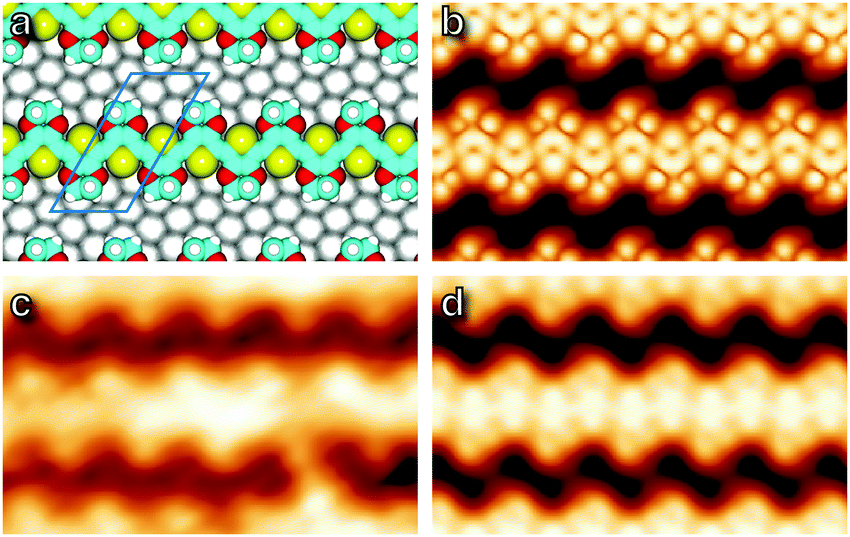 | ||
| Fig. 5 (a) Model structure optimised by DFT. The unit cell is indicated with a blue outline. (b) Simulated STM image corresponding to the geometry in (a). (c) STM image (Vb = −1.2 V, It = 500 pA), and (d) simulated STM image with a Gaussian blur applied to simulate the finite radius of the STM tip. All images are 4.2 nm × 2.6 nm field of view. | ||
This work demonstrates the fine balance that must be achieved to successfully implement the on-surface polymerization approach for complex molecules: the reactivity of the surface must be tuned to target only the polymerization reaction without incurring collateral damage to the molecular structure.
The authors thank the SXR beamline staff, Drs, E. Gann, A. Tadich, L. Thomsen, and B. Cowie, for their assistance during experiments. The authors acknowledge financial support from the Australian Synchrotron and from Queensland University of Technology (QUT) through the Science and Engineering Faculty's (SEF) School of Chemistry, Physics and Engineering (CPME), and through the Institute for Future Environments (IFE). Some of the data in this paper were obtained at the Central Analytical Research Facility (CARF) operated by the IFE. Computing resources and support were made available through QUT's High Performance Computing (HPC) center. JMM acknowledges funding from the ARC through DE170101170. J. L.-D. acknowledges funding from the ARC through DP160103116.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
C. Nacci, S. Hecht and L. Grill, in On-Surface Synthesis, Springer, 2016, pp. 1 CrossRef CAS
; M. El Garah, J. M. MacLeod and F. Rosei, Surf. Sci., 2013, 613, 6 CrossRef CAS
- A. Du, Z. Zhu and S. C. Smith, J. Am. Chem. Soc., 2010, 132, 2876 CrossRef CAS PubMed
- N. A. Zwaneveld, R. Pawlak, M. Abel, D. Catalin, D. Gigmes, D. Bertin and L. Porte, J. Am. Chem. Soc., 2008, 130, 6678 CrossRef CAS PubMed
- D. Zhong, J.-H. Franke, S. K. Podiyanachari, T. Blömker, H. Zhang, G. Kehr, G. Erker, H. Fuchs and L. Chi, Science, 2011, 334, 213 CrossRef CAS PubMed
- O. Diaz Arado, H. Mönig, H. Wagner, J. r.-H. Franke, G. Langewisch, P. A. Held, A. Studer and H. Fuchs, ACS Nano, 2013, 7, 8509 CrossRef CAS PubMed
- M. Treier, N. V. Richardson and R. Fasel, J. Am. Chem. Soc., 2008, 130, 14054 CrossRef CAS PubMed
- Q. Sun, C. Zhang, Z. Li, H. Kong, Q. Tan, A. Hu and W. Xu, J. Am. Chem. Soc., 2013, 135, 8448 CrossRef CAS PubMed
- A. R. Lahrood, J. Björk, W. M. Heckl and M. Lackinger, Chem. Commun., 2015, 51, 13301 RSC
- M. Di Giovannantonio, M. El Garah, J. Lipton-Duffin, V. Meunier, L. Cardenas, Y. Fagot Revurat, A. Cossaro, A. Verdini, D. F. Perepichka and F. Rosei, ACS Nano, 2013, 7, 8190 CrossRef CAS PubMed
- J. Lipton-Duffin, O. Ivasenko, D. Perepichka and F. Rosei, Small, 2009, 5, 592 CrossRef CAS PubMed
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed
- R. Gutzler, L. Cardenas, J. Lipton-Duffin, M. El Garah, L. E. Dinca, C. E. Szakacs, C. Fu, M. Gallagher, M. Vondracek, M. Rybachuk, D. F. Perepichka and F. Rosei, Nanoscale, 2014, 6, 2660 RSC
- Y. H. Kim, C. Sachse, M. L. Machala, C. May, L. Müller-Meskamp and K. Leo, Adv. Funct. Mater., 2011, 21, 1076 CrossRef CAS
- H. Meng, D. F. Perepichka, M. Bendikov, F. Wudl, G. Z. Pan, W. Yu, W. Dong and S. Brown, J. Am. Chem. Soc., 2003, 125, 15151 CrossRef CAS PubMed
- J. Lipton-Duffin, J. Miwa, M. Kondratenko, F. Cicoira, B. Sumpter, V. Meunier, D. Perepichka and F. Rosei, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11200 CrossRef CAS PubMed
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71 CAS
- G. Galeotti, M. Di Giovannantonio, J. Lipton-Duffin, M. Ebrahimi, S. Tebi, A. Verdini, L. Floreano, Y. Fagot-Revurat, D. F. Perepichka, F. Rosei and G. Contini, Faraday Discuss., 2017, 204, 453 RSC
- M. Bieri, M.-T. Nguyen, O. Gröning, J. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669 CrossRef CAS PubMed
- L. Jiang, A. C. Papageorgiou, S. C. Oh, O. z. Sağlam, J. Reichert, D. A. Duncan, Y.-Q. Zhang, F. Klappenberger, Y. Guo, F. Allegretti, S. More, R. Bhosale, A. Mateo-Alonso and J. V. Barth, ACS Nano, 2015, 10, 1033 CrossRef PubMed
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef CAS PubMed
- L. Pasquali, F. Terzi, M. Montecchi, B. Doyle, J. Lukkari, B. Zanfrognini, R. Seeber and S. Nannarone, J. Electron Spectrosc. Relat. Phenom., 2009, 172, 114 CrossRef CAS
- L. Pasquali, F. Terzi, B. P. Doyle and R. Seeber, J. Phys. Chem. C, 2012, 116, 15010 CAS
-
J. Stöhr, NEXAFS Spectroscopy, U.S. Government Printing Office, 1992 Search PubMed
- E. Gann, C. R. McNeill, A. Tadich, B. C. Cowie and L. Thomsen, J. Synchrotron Radiat., 2016, 23, 374 CAS
-
J. Tersoff and D. Hamann, Scanning Tunneling Microscopy, Springer, 1985, p. 59 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methodology, additional XPS spectra, additional NEXAFS spectra, calculated model geometry. See DOI: 10.1039/c8cc01465e |
| This journal is © The Royal Society of Chemistry 2018 |