
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convergent synthesis of trifunctional molecules by three sequential azido-type-selective cycloadditions†
Suguru
Yoshida
 a,
Kimiyuki
Kanno
b,
Isao
Kii‡
c,
Yoshihiro
Misawa
a,
Masatoshi
Hagiwara
c and
Takamitsu
Hosoya
*a
a,
Kimiyuki
Kanno
b,
Isao
Kii‡
c,
Yoshihiro
Misawa
a,
Masatoshi
Hagiwara
c and
Takamitsu
Hosoya
*a
aLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: thosoya.cb@tmd.ac.jp
bDepartment of Biological Information, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan
cDepartment of Anatomy and Developmental Biology, Graduate School of Medicine, Kyoto University, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan
First published on 5th March 2018
Abstract
A facile strategy for the synthesis of trifunctional molecules involving three sequential selective triazole-forming reactions is proposed. This method exploits three kinds of mechanistically different azido-type-selective cycloadditions. Three different azidophiles could be efficiently connected to a triazido platform molecule with three types of azido groups in a consecutive manner, which rendered a practical trifunctional molecule readily available.
In recent years, multifunctional molecules that are capable of playing multiple roles have received great attention in a broad range of disciplines.1 However, these well-designed molecules are usually prepared by time-consuming, linear, and multi-step synthetic routes that include many cumbersome protection/deprotection procedures and functional group transformations (Fig. 1A). This incurs low overall yields for the desired molecules and makes it difficult to prepare libraries of related candidates for certain purposes, being a bottleneck in the development of optimal molecules. Therefore, novel strategies that make multifunctional molecules more easily accessible are increasingly sought-after.
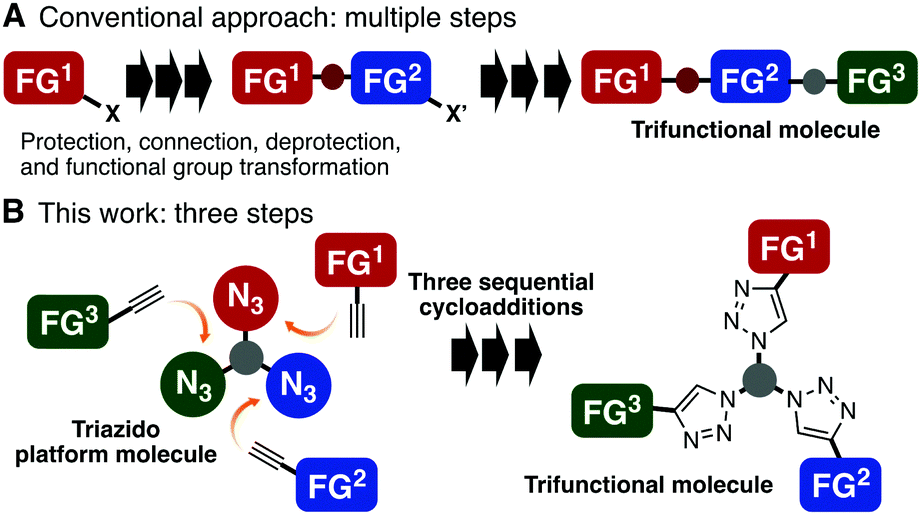 | ||
| Fig. 1 Synthetic approaches to trifunctional molecules. (A) General scheme of a conventional synthesis by a linear multi-step route. (B) The proposed convergent method based on three sequential triazole formations using a triazido platform molecule. FG = functional group. | ||
In principle, the easiest way to prepare a multifunctional molecule is to assemble multiple monofunctional components into a single molecule. Over the last decade, click reactions,2 such as copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC)3 and the copper-free variant, strain-promoted azide–alkyne cycloaddition (SPAAC),4,5 have become some of the most reliable methods for connecting two molecules. Although several efficient click-like reactions other than azide–alkyne combination have been developed by many other groups and have been successfully used for conjugating two molecules,6 synthesis of multifunctional molecules with three or more functional moieties in short steps is not easy using these methods, and only limited examples have been reported.7 To address this issue, we conceived the idea of performing a sequential triple-click reaction for the synthesis of trifunctional molecules by advancing our double-click strategies mediated by diynes8 (Fig. 1B).
Based on azido chemistry,9 including our previous findings on two-step photoaffinity labeling using a diazido probe and the dual reactivity of 2,6-disubstituted phenyl azides,10 we inferred that using a suitably designed triazido platform compound, three sequential selective cycloadditions would be possible if we could exploit three different types of cycloadditions that are orthogonal to each other. This convergent triple-click assembly approach shows significant advantages over the conventional linear conjugation approaches because it proceeds via reliable cycloaddition reactions of azides with a broad substrate scope. Furthermore, a variety of functional azidophiles, the reaction partners of azides, are readily available. However, due to the high reactivity of azides, discrimination between three or more azido groups is a challenging issue. Herein, we demonstrate three good combinations of azido groups and azidophiles applicable to the synthesis of trifunctional molecules by three sequential azido-type-selective cycloadditions.
We chose three simple azides, 2,6-diisopropylphenyl azide (1a), phenyl azide (1b), and benzyl azide (1c), as the representatives for the three types of azides, i.e., doubly sterically-hindered aromatic azides, standard aromatic azides, and aliphatic azides, respectively, to explore their orthogonal reactivity under different conditions. Using an equimolar mixture of azides 1a–c, we performed competitive experiments for various triazole-forming reactions to find a reaction that consumed one of the three azides selectively. After extensive screening, we identified three kinds of mechanistically different cycloadditions that preferred a specific type of azide (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Azidophile | Conditions | Product | Yielda (%) | ||
| a Yields were determined by 1H NMR analysis. b Benzyl azide (1c) was completely consumed. | ||||||
| 1 |
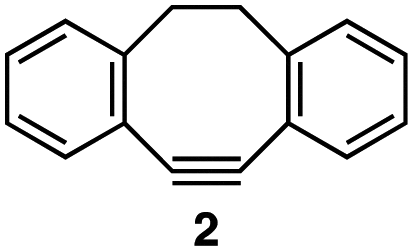
|
1a–c (1.2 equiv. each) |
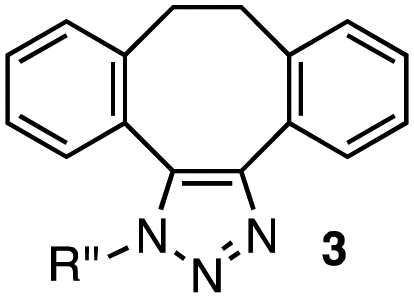
|
3a | 3b | 3c |
| 2 (1.0 equiv.) | ||||||
| MeOH, r.t., 1 h | 85 | <1 | 14 | |||
| 2 |
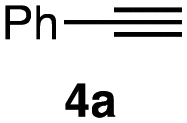
|
1a–c (1.2 equiv. each) |
|
5a | 5b | 5c |
| 4a (1.0 equiv.) | ||||||
| (MeCN)4CuBF4 (5 mol%) | 26 | 13 | 57 | |||
| TBTA (5 mol%) | ||||||
| DMSO, r.t., 24 h | ||||||
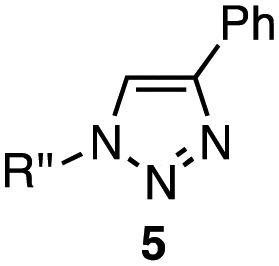
| 3 | 4a | 1a–c (1.2 equiv. each) | 5 | 5a | 5b | 5c |
| 4a (1.0 equiv.) | ||||||
| IMesCuBr (5 mol%) | 19 | 14 | 65 | |||
| t-BuOH–H2O, r.t., 24 h | ||||||
| 4 | 4a | 1a–c (1.0 equiv. each) |
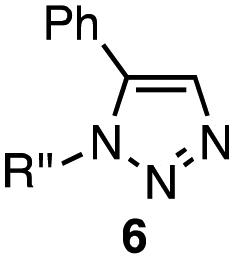
|
6a | 6b | 6c |
| 4a (4.0 equiv.) | ||||||
| Cp*Ru(PPh3)2Cl (8 mol%) | 0 | 17 | 83 | |||
| Benzene, r.t., 24 h | ||||||
| 5 | 4a | 1a–c (1.2 equiv. each) | 6 | 6a | 6b | 6c |
| 4a (1.0 equiv.) | ||||||
| Me4NOH (10 mol%) | 7 | 72 | 0 | |||
| DMSO, r.t., 48 h | ||||||
| 6 |
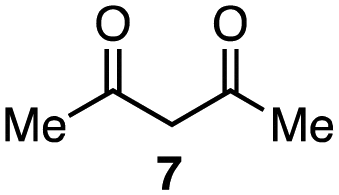
|
1a–c (1.0 equiv. each) |
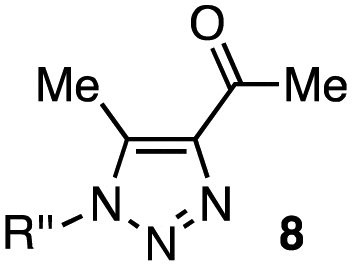
|
8a | 8b | 8c |
| 7 (1.0 equiv.) | ||||||
| K2CO3 (18 mol%) | 7 | 84 | 0 | |||
| DMF, r.t., 24 h | ||||||
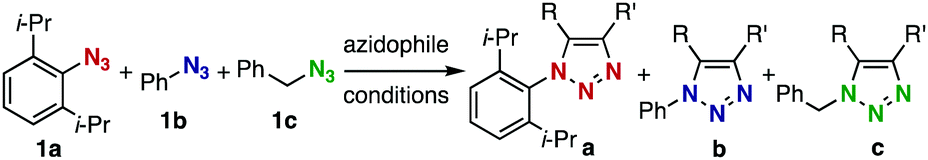
In the reaction with cyclooctyne 2, which proceeds in a concerted manner, sterically-hindered aromatic azide 1a was preferentially converted to the corresponding triazole 3a (entry 1). While a small amount of the benzyl azide-derived product 3c was also obtained, only a trace amount of triazole 3b was formed from the phenyl azide, demonstrating the high azido-type selectivity. This result is in good agreement with our previous observations, where the order of clickability for azides in reactions with a strained alkyne was strongly dominated by the distortability of the azido groups.10f In transition-metal-catalyzed cycloadditions with terminal alkyne 4a, benzyl azide (1c) showed the highest reactivity (entries 2–4). Although the selectivity was moderate under the copper-catalyzed conditions11 (entries 2 and 3), higher selectivity was achieved using a ruthenium catalyst,12 affording 1,5-substituted 1,2,3-triazoles 6 (entry 4). These results indicate that the selectivity depends on the steric environment of the azido groups, with the most sterically unhindered azide 1c reacting more favorably than the others. Furthermore, base-catalyzed cycloadditions of azides with anionic azidophiles showed high selectivity toward aromatic azides, particularly toward the more unhindered 1b (entries 5 and 6). The anionic intermediates of these reactions, which are generated by the nucleophilic attack of the anions on the azides, are likely to be more stabilized by the aryl group, resulting in high selectivity. While the cycloaddition with the terminal alkyne 4a under ammonium hydroxide-catalyzed conditions13 proceeded preferentially with aromatic azide 1b in high selectivity, complete decomposition of benzyl azide (1c) was also observed (entry 5), which was unsuitable for our purpose. To our delight, cycloaddition with 1,3-diketone 7,14 employing a weaker base such as potassium carbonate, proceeded smoothly with high 1b-selectivity, leaving 1c intact (entry 6).
We designed and synthesized triazide 11 bearing three types of azido groups, and examined the feasibility of this molecule as a platform molecule for three sequential selective cycloadditions (Fig. 2). Triazide 11 was easily prepared in a convergent manner by the Suzuki–Miyaura cross-coupling reaction between diazide 9 bearing an iodo group10g and readily synthesized arylboronic acid pinacol ester 10 (Fig. 2A). The three sequential cycloadditions of triazide 11 with three kinds of azidophiles proceeded smoothly to afford the desired tristriazole compound in high yield (Fig. 2B). For example, cycloaddition of triazide 11 with 1,3-diketone 7 catalyzed by potassium carbonate proceeded predominantly at the aromatic azido group to provide the monotriazole product in high yield. Subsequent ruthenium-catalyzed cycloaddition of the monotriazole bearing two unreacted azido groups with terminal alkyne 4b proceeded selectively at the aliphatic azido moiety to afford the bistriazole in excellent yield. Finally, the remaining sterically-hindered azido group reacted efficiently with strained alkyne 2, affording tristriazole 12a. In this scheme, the three sequential cycloadditions were achieved in 76% overall yield. Furthermore, the order of the cycloadditions was exchangeable; tristriazole 12b was prepared in 69% overall yield from triazide 11 by the three sequential reactions, i.e., the strain-promoted click reaction with 2, base-catalyzed cycloaddition with 7, and copper-catalyzed cycloaddition with 4a.
 | ||
| Fig. 2 Three sequential azido-type-selective cycloadditions. (A) Synthesis of triazido platform molecule 11. (B) Examples of the three-step synthesis of tristriazoles in different orders. See the ESI† for details. | ||
Three sequential azido-type-selective cycloadditions of triazide 11 with three functional modules enabled to develop a practical trifunctional probe in a short period of time. We demonstrated the utility of the strategy by preparing HaloTag ligands bearing a fluorescent BODIPY moiety and a biotinyl group (Fig. 3).15 Initially, we prepared simple azidophilic modules that contained the respective functional groups with different kinds of linkers. These included the two β-ketoamide-type HaloTag ligands 13a and 13b, terminal alkyne-type BODIPY derivative 14, and three kinds of biotinylated cyclooctyne derivatives 15a–c (Fig. 3A). Using these monofunctional modules, we synthesized four kinds of trifunctional tristriazoles 16a–d from triazide 11 by (1) base-catalyzed cycloaddition with 13, (2) ruthenium-catalyzed cycloaddition with 14, and (3) strain-promoted click reaction with 15 (Fig. 3B). In all of these cases, the three sequential cycloadditions proceeded efficiently without affecting the functional groups to afford the trifunctional tristriazoles 16a–d in 71–75% overall yields. The performances of the synthesized probe candidates 16a–d were evaluated. Each of them was added to a cell lysate that contained a GST-fused HaloTag protein (59 kDa) to ligate the probe candidates, followed by SDS-PAGE analysis. The gels were analyzed by fluorescence detection, and then analyzed by western blot with horseradish peroxidase (HRP)-conjugated streptavidin or Coomassie brilliant blue (CBB)-staining (Fig. 3C; ESI,† Fig. S1 and S2). The intensity of the fluorescent bands reflected the efficiency of the covalent binding of the probe candidates to the HaloTag protein. The weak fluorescence signal observed when using 16a without a linker in its HaloTag ligand moiety indicates that the covalent bond formation between 16a and the HaloTag protein was prevented, possibly due to the bulkiness of the platform core structure (Fig. 3C, lane 2). This is supported by the remarkable enhancement of the fluorescence intensity observed when 16b–d bearing a triethyleneoxy linker were used (Fig. 3C, lanes 3–5). Western blot analysis using HRP-streptavidin also indicates that a linker between the biotinyl group and the platform core with a sufficient length is critical for efficient recognition of the biotinylated protein by streptavidin (Fig. 3C, lanes 3–5). Resultantly, among the four probe candidates examined, 16d showed the best performance for dual modification of the HaloTag protein with BODIPY and biotin (Fig. 3D).16
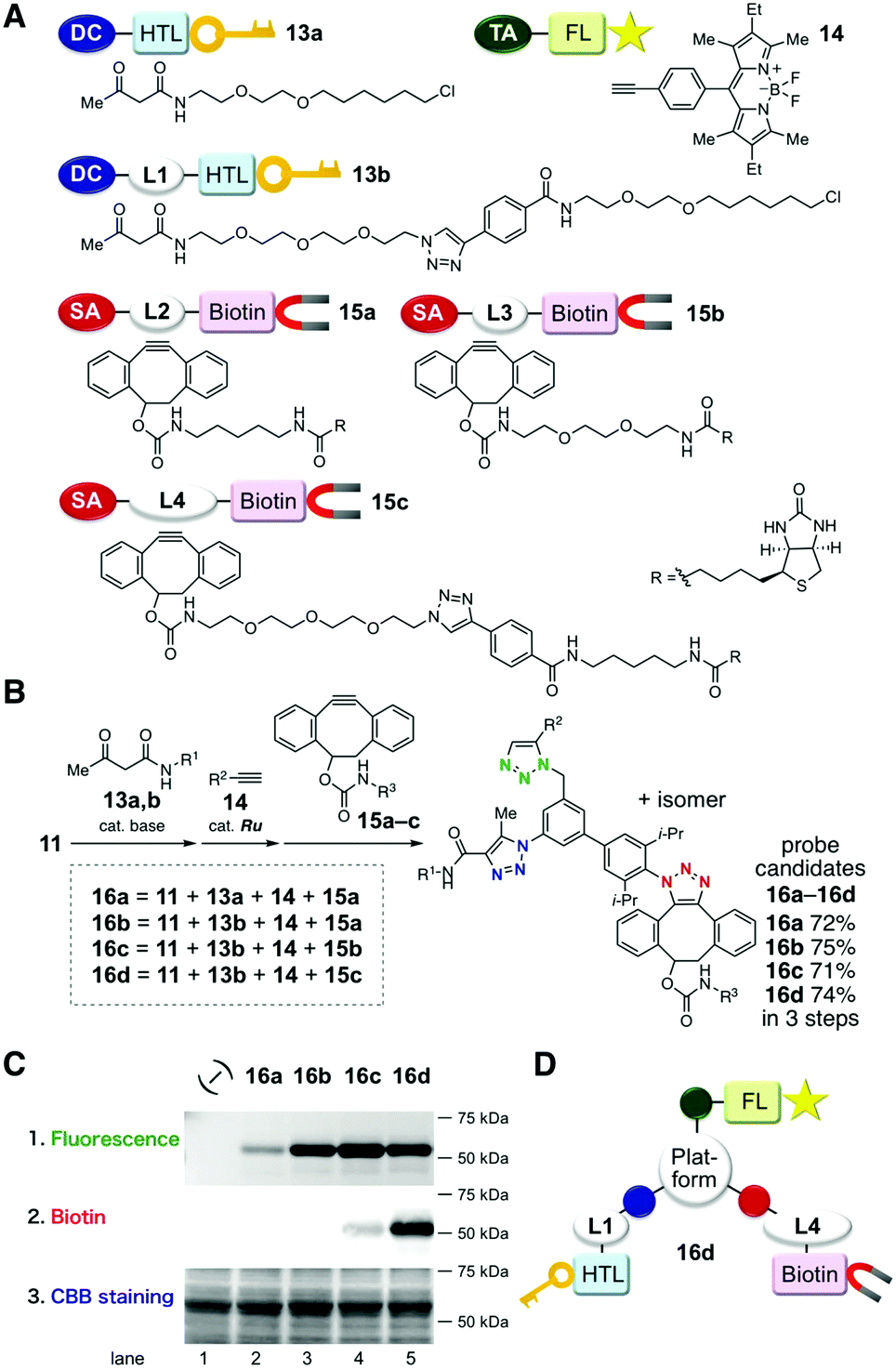 | ||
| Fig. 3 Synthesis of trifunctional probe candidates and functional evaluations. (A) Structures and schematic diagrams of azidophilic modules. (B) Synthesis of probe candidates 16a–d. (C) SDS-PAGE analysis of the GST-HaloTag proteins labeled with trifunctional probe candidates 16a–d. The gels were (1) scanned with a fluorescence image analyzer, and then (2) analyzed by western blot, or (3) stained with CBB. (D) The schematic diagram of trifunctional probe 16d. DC, HTL, TA, FL, SA, and L1–L4 indicate 1,3-dicarbonyl, HaloTag ligand, terminal alkyne, fluorescent, strained alkyne, and linker moieties, respectively. | ||
In summary, we have demonstrated that a triazido platform molecule bearing three types of sterically and electronically different azido groups is useful for the synthesis of trifunctional molecules by three sequential azido-type-selective cycloadditions. Although ruthenium- or base-catalyzed cycloadditions do not precisely meet the criteria of click chemistry, the broad scope of the cycloaddition reactions would allow for the facile synthesis of diverse trifunctional molecules. Further studies to enhance the selectivity of each triazole-forming reaction and applications to develop a variety of practical multifunctional molecules are currently underway in our laboratory.
This work was supported by JSPS KAKENHI Grant Numbers 15H03118 (B; T. H.), 16H01133 (Middle Molecular Strategy; T. H.), 17H06414 (Organelle Zone; T. H.), and 26350971 (C; S. Y.); Japan Agency for Medical Research and Development (AMED) under Grant Numbers JP17am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research) and JP17am0301024 (the Basic Science and Platform Technology Program for Innovative Biological Medicine); the Cooperative Research Project of Research Center for Biomedical Engineering; and Naito Foundation (S. Y.).
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- (a) A. Louie, Chem. Rev., 2010, 110, 3146–3195 CrossRef CAS PubMed; (b) M. Rudin and R. Weissleder, Nat. Rev. Drug Discovery, 2003, 2, 123–131 CrossRef CAS PubMed; (c) R. H. Kimura, Z. Miao, Z. Cheng, S. S. Gambhir and J. R. Cochran, Bioconjugate Chem., 2010, 21, 436–444 CrossRef CAS PubMed; (d) G. Bottari, Od. Trukhina, M. Ince and T. Torres, Coord. Chem. Rev., 2012, 256, 2453–2477 CrossRef CAS.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed; (c) J. Lahann, Click Chemistry for Biotechnology and Materials Science, John Wiley & Sons, West Sussex, 2009 Search PubMed.
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (c) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed; (b) M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168–1184 CrossRef CAS PubMed; (c) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC; (d) J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- (a) G. Wittig and A. Krebs, Chem. Ber., 1961, 94, 3260–3275 CrossRef CAS; (b) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed; (c) S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS PubMed; (d) J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS PubMed; (e) X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS PubMed; (f) A. A. Poloukhtine, N. E. Mbua, M. A. Wolfert, G.-J. Boons and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 15769–15776 CrossRef CAS PubMed; (g) J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688–3690 CrossRef CAS PubMed; (h) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425 CrossRef CAS PubMed; (i) J. Dommerholt, O. van Rooijen, A. Borrmann, C. F. Guerra, F. M. Bickelhaupt and F. L. van Delft, Nat. Commun., 2014, 5, 5378 CrossRef CAS PubMed.
- (a) A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC; (b) C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed; (c) M. R. J. Vallée, L. M. Artner, J. Dernedde and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2013, 52, 9504–9508 CrossRef PubMed; (d) H. Liu, D. Audisio, L. Plougastel, E. Decuypere, D.-A. Buisson, O. Koniev, S. Kolodych, A. Wagner, M. Elhabiri, A. Krzyczmonik, S. Forsback, O. Solin, V. Gouverneur and F. Taran, Angew. Chem., Int. Ed., 2016, 55, 12073–12077 CrossRef CAS PubMed.
- (a) D. M. Beal and L. H. Jones, Angew. Chem., Int. Ed., 2012, 51, 6320–6326 CrossRef CAS PubMed; (b) L. I. Willems, N. Li, B. I. Florea, M. Ruben, G. A. van der Marel and H. S. Overkleeft, Angew. Chem., Int. Ed., 2012, 51, 4431–4434 CrossRef CAS PubMed; (c) D. M. Beal, V. E. Albrow, G. Burslem, L. Hitchen, C. Fernandes, C. Lapthorn, L. R. Roberts, M. D. Selby and L. H. Jones, Org. Biomol. Chem., 2012, 10, 548–554 RSC; (d) Y. Sun, X. Ma, K. Cheng, B. Wu, J. Duan, H. Chen, L. Bu, R. Zhang, X. Hu, Z. Deng, L. Xing, X. Hong and Z. Cheng, Angew. Chem., Int. Ed., 2015, 54, 5981–59842 CrossRef CAS PubMed; (e) V. Vaněk, J. Pícha, B. Fabre, M. Buděšínský, M. Lepšík and J. Jiráček, Eur. J. Org. Chem., 2015, 3689–3701 CrossRef; (f) A.-C. Knall, M. Hollauf, R. Saf and C. Slugovc, Org. Biomol. Chem., 2016, 14, 10576–10580 RSC.
- (a) I. Kii, A. Shiraishi, T. Hiramatsu, T. Matsushita, H. Uekusa, S. Yoshida, M. Yamamoto, A. Kudo, M. Hagiwara and T. Hosoya, Org. Biomol. Chem., 2010, 8, 4051–4055 RSC; (b) S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590–13593 CrossRef CAS PubMed.
- (a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed; (b) S. Bräse and K. Banert, Organic Azides: Syntheses and Applications, John Wiley & Sons, West Sussex, 2010 Search PubMed.
- (a) T. Hosoya, T. Hiramatsu, T. Ikemoto, M. Nakanishi, H. Aoyama, A. Hosoya, T. Iwata, K. Maruyama, M. Endo and M. Suzuki, Org. Biomol. Chem., 2004, 2, 637–641 RSC; (b) T. Hosoya, T. Hiramatsu, T. Ikemoto, H. Aoyama, T. Ohmae, M. Endo and M. Suzuki, Bioorg. Med. Chem. Lett., 2005, 15, 1289–1294 CrossRef CAS PubMed; (c) T. Ikemoto, T. Hosoya, K. Takata, H. Aoyama, T. Hiramatsu, H. Onoe, M. Suzuki and M. Endo, Diabetes, 2009, 58, 2802–2812 CrossRef CAS PubMed; (d) T. Hosoya, A. Inoue, T. Hiramatsu, H. Aoyama, T. Ikemoto and M. Suzuki, Bioorg. Med. Chem., 2009, 17, 2490–2496 CrossRef CAS PubMed; (e) R. Kohta, Y. Kotake, T. Hosoya, T. Hiramatsu, Y. Otsubo, H. Koyama, Y. Hirokane, Y. Yokoyama, H. Ikeshoji, K. Oofusa, M. Suzuki and S. Ohta, J. Neurochem., 2010, 114, 1291–1301 CAS; (f) S. Yoshida, A. Shiraishi, K. Kanno, T. Matsushita, K. Johmoto, H. Uekusa and T. Hosoya, Sci. Rep., 2011, 1, 82 CrossRef PubMed; (g) S. Yoshida, Y. Misawa and T. Hosoya, Eur. J. Org. Chem., 2014, 3991–3995 CrossRef CAS.
- (a) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed; (b) S. Díez-González, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 7595–7606 RSC.
- (a) L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998–15999 CrossRef CAS PubMed; (b) B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 8923–8930 CrossRef CAS PubMed.
- S. W. Kwok, J. R. Fotsing, R. J. Fraser, V. O. Rodionov and V. V. Fokin, Org. Lett., 2010, 12, 4217–4219 CrossRef CAS PubMed.
- E. P. J. Ng, Y.-F. Wang, B. W.-Q. Hui, G. Lapointe and S. Chiba, Tetrahedron, 2011, 67, 7728–7737 CrossRef CAS.
- C. G. England, H. Luo and W. Cai, Bioconjugate Chem., 2015, 26, 975–986 CrossRef CAS PubMed.
- Similarly, we prepared three bifunctional tristriazoles, in which each functional group of 16d is replaced with a functionless dummy group, clearly demonstrating that each functional group of 16d worked practically. See the ESI† for the details.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization for new compounds including NMR spectra. See DOI: 10.1039/c8cc01195h |
| ‡ Present address: Common Facilities Unit, Compass to Healthy Life Research Complex Program, RIKEN Cluster for Science and Technology Hub, 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan. |
| This journal is © The Royal Society of Chemistry 2018 |