
The near-UV absorber OSSO and its isomers†

Abstract
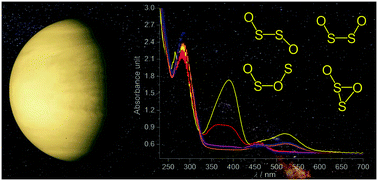
Disulfur dioxide, OSSO, has been proposed as the enigmatic “near-UV absorber” in the yellowish atmosphere of Venus. However, the fundamentally important spectroscopic properties and photochemistry of OSSO are scarcely documented. By either condensing gaseous SO or 266 laser photolysis of an S2⋯O2 complex in Ar or N2 at 15 K, syn-OSSO, anti-OSSO, and cyclic OS(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)S were identified by IR and UV/Vis spectroscopy for the first time. The observed absorptions (λmax) for OSSO at 517 and 390 nm coincide with the near-UV absorption (320–400 nm) found in the Venus clouds by photometric measurements with the Pioneer Venus orbiter. Subsequent UV light irradiation (365 nm) depletes syn-OSSO and anti-OSSO and yields a fourth isomer, syn-OSOS, with concomitant dissociation into SO2 and elemental sulfur.
O)S were identified by IR and UV/Vis spectroscopy for the first time. The observed absorptions (λmax) for OSSO at 517 and 390 nm coincide with the near-UV absorption (320–400 nm) found in the Venus clouds by photometric measurements with the Pioneer Venus orbiter. Subsequent UV light irradiation (365 nm) depletes syn-OSSO and anti-OSSO and yields a fourth isomer, syn-OSOS, with concomitant dissociation into SO2 and elemental sulfur.
- This article is part of the themed collection: SU 120: Celebrating 120 Years of Soochow University
 Please wait while we load your content...
Please wait while we load your content...

