
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward universal protein post-translational modification detection in high throughput format†
Harri
Härmä
 *a,
Natalia
Tong-Ochoa
a,
Arjan J.
van Adrichem‡
c,
Ilian
Jelesarov
b,
Krister
Wennerberg
c and
Kari
Kopra
a
*a,
Natalia
Tong-Ochoa
a,
Arjan J.
van Adrichem‡
c,
Ilian
Jelesarov
b,
Krister
Wennerberg
c and
Kari
Kopra
a
aMaterials Chemistry and Chemical Analysis, University of Turku, Turku, Finland. E-mail: harri.harma@utu.fi
bDepartment of Biochemistry, University of Zurich, Zurich, Switzerland
cInstitute for Molecular Medicine Finland, FIMM, Helsinki, Finland
First published on 2nd March 2018
Abstract
Post-translational modification (PTM) of proteins plays essential regulatory roles in a variety of pathological conditions. Reliable and practical assays are required to accelerate the discovery of inhibitors and activators for PTM related diseases. Today, methodologies are based on specific or group-specific PTM recognition of e.g. phosphate for kinase activity without extending to other type of PTMs. Here we have established a universal time-resolved luminescence assay on a peptide-break platform for the direct detection of wide variety of PTMs. The developed assay is based on the leucine zipper concept wherein a europium-chelate labeled detection peptide and a non-labeled peptide substrate form a highly luminescent dimer. As an active PTM enzyme at sub or low nanomolar concentration modifies the substrate peptide, the luminescent signal of the detached detection peptide is quenched in the presence of soluble quenchers. The functionality of this universal assay technique has been demonstrated for the monitoring of phosphorylation, dephosphorylation, deacetylation, and citrullination with high applicability also to other PTMs in a high throughput format.
Post-translational modifications (PTMs) play pivotal roles in generating functional protein diversity for cellular function regulation. PTMs can define new recognition patterns, enzyme activities, localization and protein turnover controlling biological processes and, therefore, also disease conditions.1 One of the main challenges in studying protein PTMs on a large scale for drug discovery applications is the development of universal detection methods to investigate multiple different PTMs with a single platform. A variety of sensitive luminescence-based systems are commercially available for specific PTMs. Traditional approaches rely on the use of specific antibodies using detection techniques such as time-resolved luminescence resonance energy transfer (TR-LRET).2–4 Universal assays for enzyme activity have been developed using nucleotide detection methods for a variety of enzymes depending on the product being formed such as AMP for ligases, ADP for kinases, and UDP for glycosyltransferases.5,6 However, these non-direct approaches are limited to nucleotide-dependent enzymes and secondary products are detected. Other group-specific assays for kinases and phosphatases have been successfully developed but these methods have been limited to a single type of post-translational modification.7–9 Universal single-platform methods enable cost-effective assays and rapid turn-over with minimal method steps using fixed reagents when seeking novel drug candidates from molecular libraries.
Herein, we report a universal peptide-break technology for enzyme activity monitoring based on the leucine zipper peptides. Leucine zippers are coiled-coil dimerization domains of the basic-region leucine zipper (bZIP) transcription factor proteins (Fig. 1a). Leucine zippers are typically formed from approx. 30 amino acids in heptad repeats denoted as (abcdefg)n with leucines in d positions contributing strongly to the peptide-pair interaction having KD values ranging from pM to high μM.10,11 The universal peptide-break detection platform for PTMs was constructed on our quenching resonance energy transfer (QRET) detection technology.12–16 The QRET technique distinguishes between the leucine zipper complex and the dissociated post-translationally modified peptides in the presence of a soluble quencher molecule (Fig. 1b). High binding peptide motifs consisting of labeled peptide and enzyme substrate peptide interact when the substrate peptide carry no PTMs. At peptide pairing a high luminescence signal is measured as the europium-chelate label is protected from the soluble quenchers. Using a non-modified peptide, an active enzyme transfers a given modification to the peptide substrate, the peptides dissociate and a decreased luminescence signal is monitored. The peptide pair re-associates when the PTM is cleaved from the target site providing a method to investigate PTMs to both directions of modification. The functionality of the concept was demonstrated with four different PTM enzyme groups: kinase, phosphatase, deacetylase and amidinotransferase.
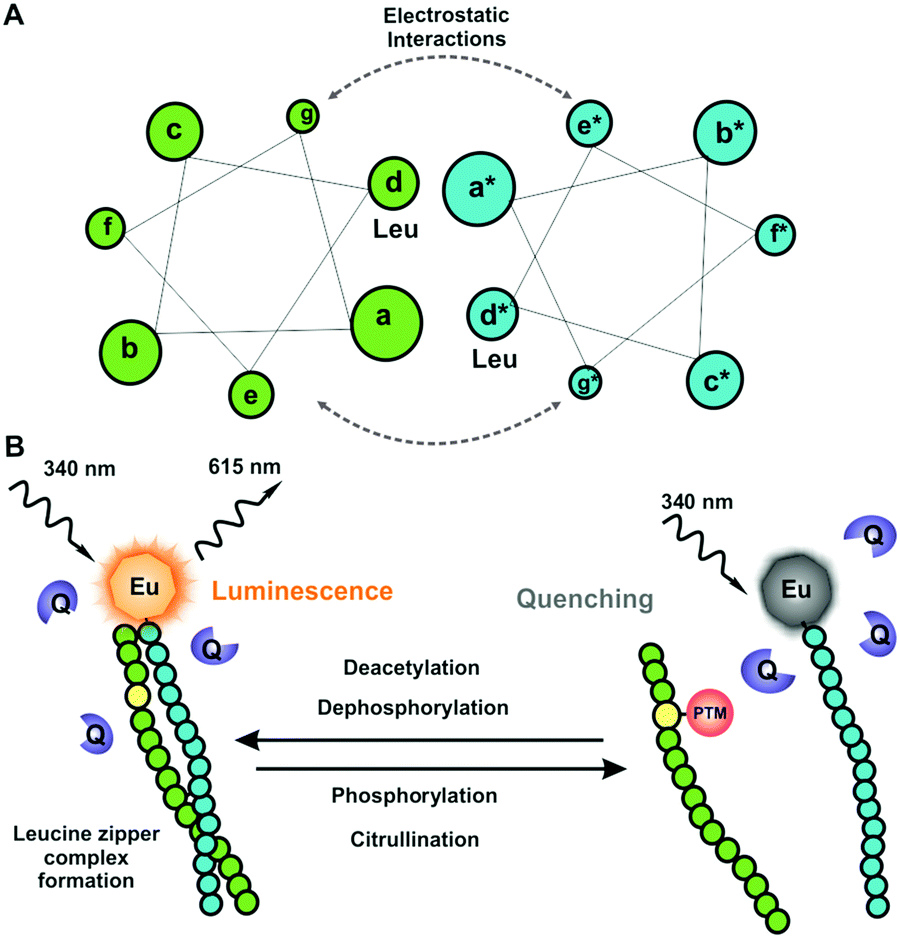 | ||
| Fig. 1 Principle of the universal peptide-break technology and leucine zipper interaction. (A) In the heptad-repeat leucine zipper complex structure d-positions are occupied with leucines and typically a-positions with hydrophobic amino acids. Positions e and g contain typically charged residues further contributing to the binding. (B) In the peptide-break technology, high europium (Eu) luminescence is detected upon leucine zipper complex formation and dimer detachment promotes luminescence quenching. | ||
The initial work was based on the peptide design using highly characterized leucine zipper from the Fos and Jun proteins which form a heterodimeric complex referred to as transcription factor AP-1.11 Jun and Fos sequences of 36 amino acid length from the leucine zipper domain were selected to serve as peptide substrate and detection peptide respectively. A tyrosine phosphorylated Jun (p-Jun), non-phosphorylated Jun and non-modified Fos were commercially synthesized. We first investigated the helicity of the peptides using circular dichroism (CD) at 30 μM peptide concentration. The CD spectrum showed that the non-phosphorylated Jun/Fos pair formed a typical helical structure while the helical structure was lost using the phosphorylated p-Jun peptide (Fig. 2a). This corroborated that the principle of the method was valid. A critical point in the development was the redesign of the leucine zipper sequences to achieve optimal peptide-pair binding with sufficient affinity and dissociation properties. Peptides with higher affinity were in demand to develop the concept to measure PTMs with higher sensitivity by shifting the peptide break-down point to nanomolar concentration. As an initial target we chose tyrosine kinase EGFR, which has shown not to possess high sequence specificity in vitro. Phosphorylated/non-phosphorylated tyrosine peptides were synthesized for these initial experiments and this provided a basis to investigate the concept without large sequence and structural limitations for the binding tests and subsequent enzymatic assay optimization. CD measurements for phosphotyrosine (TyroP-LZ) or non-phosphotyrosine peptide (Tyro-LZ) and EuLZ (non-labeled peptide for CD measurements) peptide pairs indicated that both TyroP-LZ and Tyro-LZ form a pair with the detection peptide at 30 μM concentration. The (222 nm/208 nm) ratios for Tyro-LZ/TyroP-LZ with non-labeled EuLZ were 0.75 and 0.77 respectively. The corresponding values for Jun/p-Jun and Fos were 0.89 and 0.56, respectively. This indicates substantial α-helicity for the modified Tyro-peptide pairs and Jun/Fos pair (Fig. 2a and b). Thereafter, the low affinity Jun/p-Jun and high affinity TyroP-LZ/Tyro-LZ peptides were investigated using the labeled detection peptides (Fos and EuLZ) in a homogeneous QRET binding assay. The substrate peptides were titrated in the assay and tested against the corresponding europium-labeled peptides at a fixed concentration (Fig. 2c and d). The binding of Tyro-LZ/EuLZ shifted from low nanomolar to high nanomolar binding of TyroP-LZ/EuLZ providing an EC50 ratio of approximately 100 between the phosphorylated vs. non-phosphorylated peptides (Fig. 2d). From this data one can conclude that the binding of the phosphorylated peptide observed at micromolar range also explains the peptide pairing at 30 μM peptide concentration in the CD studies. The binding affinity of the Tyro-LZ/EuLZ pair was improved approximately 1000-fold compared to the Jun/Fos peptides in the QRET assay (Fig. 2c), providing the basis to measure PTMs at nanomolar level in a high throughput screening format.
 | ||
| Fig. 2 Structural and binding studies of phosphorylated and non-phosphorylated LZ peptides. CD spectra of peptides with (A) Jun (black)/p-Jun (red) with Fos and (B) Tyro-LZ (red)/TyroP-LZ (black) with non-labeled EuLZ show no helicity for p-Jun/Fos pair compared to the remaining pairs. The homogeneous QRET binding tests (C) for Jun (black)/p-Jun (red) with Eu-Fos and (D) for Tyro-LZ (black)/TyroP-LZ (red) with EuLZ resulted in higher nanomolar affinity for Tyro-LZ/EuLZ peptides compared to Jun/Eu-Fos pairs. | ||
To demonstrate the functionality of the method we run enzymatic phosphorylation assay for EGFR. The assay confirmed the result obtained with the commercially phosphorylated TyroP-peptide (Fig. 3a). Next, the universality of the method was demonstrated using a variety of substrate sequences suitable for different enzymes (Table 1). Peptides were selected to contain the consensus sequence and thus required specificity to each enzymatic activity. With the designed peptide substrates we ran dose–response measurements for selective and non-selective inhibitors and an activator against PKA and EGFR kinases, PTP1B phosphatase, Sirtuin1 and HDAC3 deacetylases and PAD4 amidinotransferase for citrullination. The dose–response curves were measured using 0.5–3 nM of enzyme and typically 10 nM of substrate and detection peptides in a total volume of 50 μl (Table S1, ESI†). The obtained IC50 values were comparable to the literature values and the assay signal-to-background (S/B) ratios range between 4–97 (Table S2, ESI†). The data provided suggests that the peptide-break detection platform can be extrapolated to other PTMs regardless of the product being formed using the single-label approach.
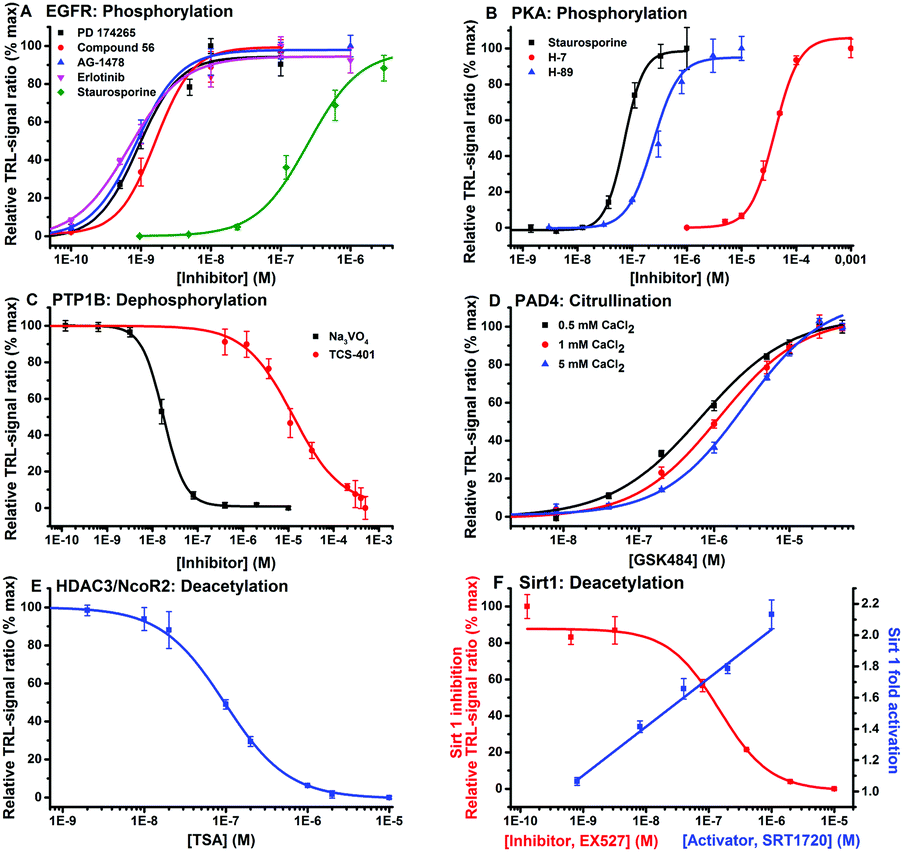 | ||
| Fig. 3 Dose–response measurements for the target enzymes at 0.5–3.0 nM concentration level: (A) EGFR and (B) PKA kinases, (C) PTP1B phosphatase, (D) PAD4 amidinotransferase at varying CaCl2 concentrations, (E) HDAC3 and (F) SIRT1 deacetylases using specific and universal inhibitors and an activator. Calculated IC50 values were comparable to the references (Table S2, ESI†). | ||
| Peptide | Enzyme target | PTM | Consensus sequence | EC50 ratio (modified/non-modified peptide) |
|---|---|---|---|---|
| Tyro-LZ | EGFR | Phosphorylation | –Y– | 100 |
| Ser-LZ | PKA | Phosphorylation | –RRxS– | n.d. |
| TyroP-LZ | PTP1B | Dephosphorylation | –pY– | 100 |
| LysAc-LZ | Sirt 1 | Deacetylation | –(Ac)KGA– | 50 |
| LysAc-LZ | HDAC3 | Deacetylation | –(Ac)KGA– | 50 |
| Arg-LZ | PAD4 | Citrullination | –R– | n.d. |
Typically, we measured the luminescence signal in an endpoint fashion but without reaction stopping agent. To enable real-time kinetic monitoring of the enzyme activity the protocol was modified so that ATP and kinase (EGFR and PKA) were added to the wells at the end to initiate the reaction. The data suggests that the method is highly applicable to continuous signal reading to measure reaction kinetics (Fig. S1, ESI†). This is in line with our previous QRET assay development where kinetic measurements have been demonstrated.13,14
Assay performance parameters were further studied with phosphorylation monitoring prior to a compound library screening. Compound library stocks are generally stored in DMSO and assays with high DMSO tolerance are prerequisite. Thus a varying concentration of DMSO (0–10%) was tested in the PKA assay. Despite the fact that luminescence signal levels increased approximately 4-fold at the highest DMSO concentration, compared to 0% DMSO concentration, the S/B remained unchanged providing an equal assay performance at all tested DMSO concentrations (Fig. S2, ESI†). The final assay validation was performed by running a small-scale HTS screen with the Published Kinase Inhibitor Set (PKIS) library. Prior to this, the PKA and EGFR kinase assays were miniaturized to 10 μl for the screening of 356 compounds at 1 μM concentration. We found 10/13 known PKA inhibitors in the screen having IC50 values lower than 1 μM considering 3SD threshold (Fig. 4); whereas in the EGFR assay screen, 42/48 known inhibitors were found considering 6SD threshold (Fig. S3, ESI†). Higher threshold value was chosen to the EGFR assay due to higher number of active compounds in the library. Assay robustness was investigated during the PKA screen and an average Z′ value of 0.67 was obtained using 96 negative and positive controls (0 and 1 μM staurosporine) measured from three plates each having Z′ value of 0.68, 0.68 and 0.65. Low assay variation shows not only high reliability and HTS compatibility but reduces also the need for high reagent concentrations resulting in high sensitivity (Fig. 4 and Fig. S3, ESI†).
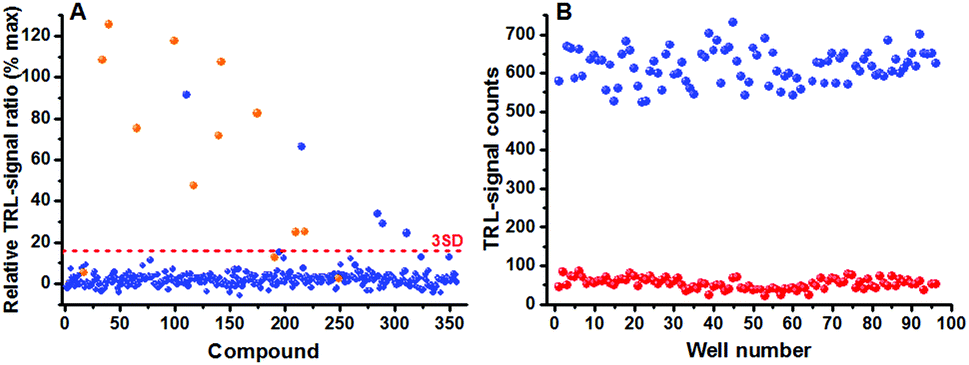 | ||
| Fig. 4 Validation of the universal peptide-break technology using PKIS library using PKA. (A) Screening was performed at 1 μM compound concentration with 3SD threshold for PKA. The number of detected compounds are comparable with the Nanosyn Caliper screening information in the literature.17 (B) Average Z′ value of 0.67 was calculated for the PKA kinase assay and the S/B ratio was 12. | ||
We have successfully developed a detection method potentially suitable for universal PTM detection. The key benefit of the method utilizing the peptide-break technology is its versatility. The method is based on the peptide pairing and dissociation as the substrate peptide is modified with an active PTM enzyme. For the end-user, the assay development is simple as the substrate peptide is solely modified to contain target-specific sequence for enzyme while the detection peptide is kept the same for all peptide pairs. The detection platform is potentially applicable to different PTMs irrespective of the product being formed during the PTM enzymatic assay. Moreover, the HTS compatible technique has high sensitivity and low cost due to low enzyme concentration requirements and the single label concept. Our data suggests that the methodology may provide an inexpensive luminescence-based detection concept to high throughput screening for multiple PTM targets using a single platform.
We are grateful for the support by the Academy of Finland (grant no. 270010, 296093, and 296225). Jani Turunen, Tiina Kuusisto, Laura Hietala, Samuli Tyvelä, Mari Laine and Mervi Salminen are acknowledged for their valuable laboratory work.
Conflicts of interest
Kari Kopra and Harri Härmä are involved in the QRET Technologies Ltd. company.References
- M. Mann and O. N. Jensen, Nat. Biotechnol., 2003, 21, 255 CrossRef CAS PubMed.
- J. Karvinen, P. Hurskainen, S. Gopalakrishnan, D. Burns, U. Warrior and I. Hemmilä, J. Biomol. Screening, 2002, 7, 223 CrossRef CAS PubMed.
- F. Degorce, A. Card, S. Soh, E. Trinquet, G. P. Knapik and B. Xie, Curr. Chem. Genomics, 2009, 3, 22 CrossRef CAS PubMed.
- R. A. Horton and K. W. Vogel, J. Biomol. Screening, 2010, 15, 1008 CrossRef CAS PubMed.
- S. Mondal, K. Hsiao and S. A. Goueli, Anal. Biochem., 2016, 510, 41 CrossRef CAS PubMed.
- T. Zielinski, M. Reichman, P. S. Donover and R. G. Lowery, Assay Drug Dev. Technol., 2016, 14, 240 CrossRef CAS PubMed.
- M. D. Shults and B. Imperiali, J. Am. Chem. Soc., 2003, 125, 14248 CrossRef CAS PubMed.
- M. D. Shults, D. Carrico-Moniz and B. Imperiali, Anal. Biochem., 2006, 352, 198 CrossRef CAS PubMed.
- Z. L. Wu, PLoS One, 2011, 6, e23172 CAS.
- D. L. Daugherty and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 4325 CrossRef CAS.
- J. B. Kaplan, A. W. Reinke and A. E. Keating, Protein Sci., 2014, 23, 940 CrossRef CAS PubMed.
- H. Härmä, A. Rozwandowicz-Jansen, E. Martikkala, H. Frang, I. Hemmilä, N. Sahlberg, V. Fey, M. Perälä and P. Hänninen, J. Biomol. Screening, 2009, 14, 936 CrossRef PubMed.
- K. Kopra, A. Ligabue, Q. Wang, M. Syrjänpää, O. Blaževitš, S. Veltel, A. J. van Adrichem, P. Hänninen, D. Abankwa and H. Härmä, Anal. Bioanal. Chem., 2014, 406, 4147 CrossRef CAS PubMed.
- K. Kopra, M. Syrjänpää, P. Hänninen and H. Härmä, Analyst, 2014, 139(8), 2016 RSC.
- K. Kopra and H. Härmä, Nat. Biotechnol., 2015, 32, 575 CAS.
- N. Tong-Ochoa, K. Kopra, M. Syrjänpää, N. Legrand and H. Härmä, Anal. Chim. Acta, 2015, 897, 96 CrossRef CAS PubMed.
- J. M. Elkins, V. Fedele, M. Szklarz, K. R. Abdul Azeez, E. Salah, J. Mikolajczyk, S. Romanov, N. Sepetov, X. P. Huang, B. L. Roth, A. Al Haj Zen, D. Fourches, E. Muratov, A. Tropsha, J. Morris, B. A. Teicher, M. Kunkel, E. Polley, K. E. Lackey, F. L. Atkinson, J. P. Overington, P. Bamborough, S. Muller, D. J. Price, T. M. Willson, D. H. Drewry, S. Knapp and W. J. Zuercher, Nat. Biotechnol., 2016, 34, 95 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1, Fig. S1–S3, materials and methods. See DOI: 10.1039/c7cc09575a |
| ‡ Present address: Clinical Chemistry and Hematology Department, Zuyderland Medical Centre, Sittard, Netherlands. |
| This journal is © The Royal Society of Chemistry 2018 |