
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mild, calcium catalysed Beckmann rearrangements†
H. J.
Kiely-Collins
a,
I.
Sechi
a,
P. E.
Brennan
 *ab and
M. G.
McLaughlin
*ac
*ab and
M. G.
McLaughlin
*ac
aStructural Genomics Consortium & Target Discovery Institute, University of Oxford, NDM Research Building, Roosevelt Drive, Oxford, OX3 7FZ, UK. E-mail: P.brennan@sgc.ox.ac.uk
bARUK Oxford Drug Discovery Institute, University of Oxford Oxford, OX3 7FZ, UK
cFaculty of Science & Engineering, Division of Chemistry & Environmental Science, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, UK. E-mail: M.mclaughlin@mmu.ac.uk
First published on 21st December 2017
Abstract
A mild calcium catalysed Beckmann rearrangement has been realised, which forgoes the more traditional harsh reactions conditions associated with the transformation. The catalyst system is shown to be tolerant towards a wide variety of functional groups relevant to natural product synthesis and medicinal chemistry and the synthetic utility of the reaction has also been investigated. A preliminary mechanistic investigation was performed to understand the nature of the incoming nucleophile and a possible reaction pathway is described.
The Beckmann rearrangement of aldoximes and ketoximes to the corresponding amides under acidic conditions is an inherently elegant transformation, and has been used to great success in the synthesis of natural products and pharmaceuticals alike.1 Although the reaction has clear utility in synthetic organic chemistry, the need for harsh reaction conditions limits its usefulness to carefully chosen substrates.
In response to this problem, many groups have reported modifications of the reaction that allows the transformation to proceed under milder conditions. For example, Giacomelli reported that cyanuric chloride afforded the desired amides in good yield,2 and Yamamoto and co-workers realized the catalytic variant using an acid co-catalyst.3 Other catalytic variants have been reported such as the use of TPAC,4 gold,5 chloral,6 mercury7 and iodine.8 Recently Mhaske and co-workers reported a facile radical Beckmann rearrangement,9 while Yadav and colleagues harnessed visible light to power the reaction.10
Although all these methodologies provide the amides in good yields, they suffer from intolerance to various functional groups. Additionally, many of the reagents and catalysts used could conceivably take part in side reactions, thus limiting their uptake in industry (Scheme 1). During the course of a medicinal chemistry project targeting epigenetic enzymes, we required a mild and functional group tolerant method to synthesize a library of amides11 for structure activity relationship studies.
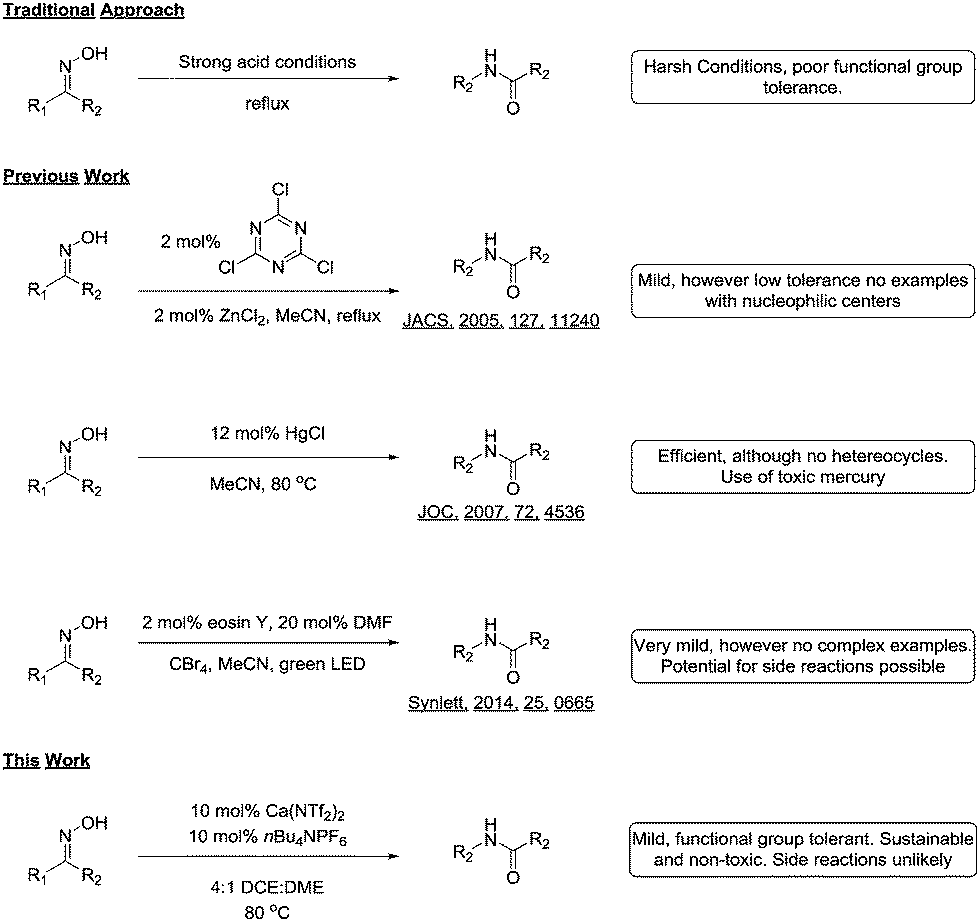 | ||
| Scheme 1 Comparison with previously reported methodology. | ||
Among the group 2 metals, calcium has been largely ignored by the synthetic community, which is surprising as calcium is a readily available and non-toxic metal.12 One major drawback associated with using calcium as a reagent in synthesis is its inherent lack of solubility in traditional organic solvents. However over the past decade, pioneering work by the groups of Niggemann,13 Kobayashi,14 France,15 and others16 have shown that calcium can be a highly powerful, yet mild catalyst for a range of transformations.
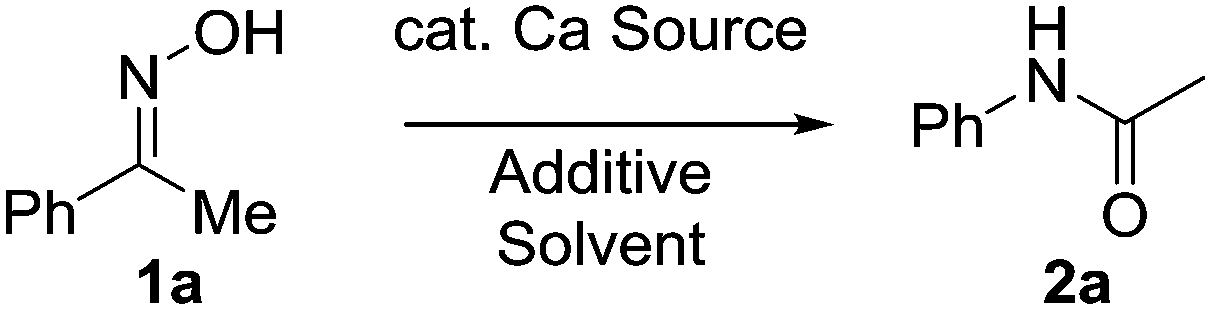
Our group has a burgeoning interest in using alkaline earth metals such as calcium as redox neutral catalysts to power organic transformations, and we reasoned that through judicious choice of catalyst system we could perform a mild Beckmann rearrangement to afford a wide variety of amides. We anticipated that the Lewis acidic nature of the calcium catalyst would be sufficient in generating the nitrillium ion which subsequently provides the requisite amide.
With this in mind we conducted an initial feasibility study to determine if the reaction could proceed (Table 1). Pleasingly, 10 mol% Ca(NTf2)2/nBu4NPF6 at room temperature provided the amide in 10% isolated yield. We were initially concerned that the reaction was proceeding stoichiometrically; however upon heating the mixture at 40 °C, the product was obtained in 49% yield. Increasing the temperature further to 80 °C increased both the rate of reaction and isolated yield, affording the product in 93% yield in 2 hours. We next explored the possibility of decreasing the catalyst loading, but we observed a steady decrease in isolated yield with decreasing catalyst loading; however under prolonged reaction times the yield can be recovered. Additionally, the reaction was set-up without either the calcium or ammonium salt, and as shown the reaction did not proceed. Furthermore, we also attempted the reaction in the presence of HNTf2, however only trace amounts of product were obtained in all cases.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ca source | Additive | Temp. (°C) | Time (h) | Yield (%) |
| 1 | Ca(NTf2)2 | nBu4NPF6 | r.t. | 12 | 10 |
| 2 | Ca(NTf2)2 | nBu4NPF6 | 40 | 12 | 49 |
| 3 | Ca(NTf2)2 | nBu4NPF6 | 60 | 12 | 73 |
| 4 | Ca(NTf 2 ) 2 | nBu 4 NPF 6 | 80 | 2 | 93 |
| 5 | Ca(NTf2)2 | nBu4NPF6 | 80 | 12 | 93 |
| 6 | Ca(NTf2)2 | None | 80 | 12 | 0 |
| 7 | None | nBu4NPF6 | 80 | 12 | 0 |
| 8 | None | None | 80 | 12 | 0 |
We next turned our attention to the choice of solvent for the reaction (Table 2). As noted, one of the main drawbacks in using calcium as a reagent is its inherent insolubility in many organic solvents. This is partly circumvented through the use of the NTf2/nBu4NPF6 system, but it was noted early in this study that the reactions performed in 1,2-dichloroethane (DCE), upon prolonged reactions times, became heterogeneous. To combat this, several solvents were tested including dichloromethane (DCM), chlorobenzene, n-methyl-2-pyrrolidone (NMP) and dimethylformamide (DMF), however a reduction in isolated yield was observed in each case. We therefore focused our attention on using solvent mixtures employing DCE as the major constituent. After extensive experimentation, a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DCE:DME proved optimum and this was used throughout.
:1 mixture of DCE:DME proved optimum and this was used throughout.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Time (h) | Yield (%) |
| 1 | 1,2-DCE | 80 | 2 | 93 |
| 2 | DCM | 40 | 12 | 31 |
| 3 | DMF | 80 | 12 | 0 |
| 4 | NMP | 80 | 6 | 81 |
| 5 | DME | 80 | 6 | 71 |
| 6 | Et2O | 40 | 6 | 12 |
| 7 | THF | 80 | 6 | 39 |
| 8 | DCE:DME (1:1) |
80 | 2 | 86 |
| 9 | DCE:DME (4:1) |
80 | 2 | 92 |
| 10 |
DCE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :DME (10:1) :DME (10:1)
|
80 | 2 | 81 |
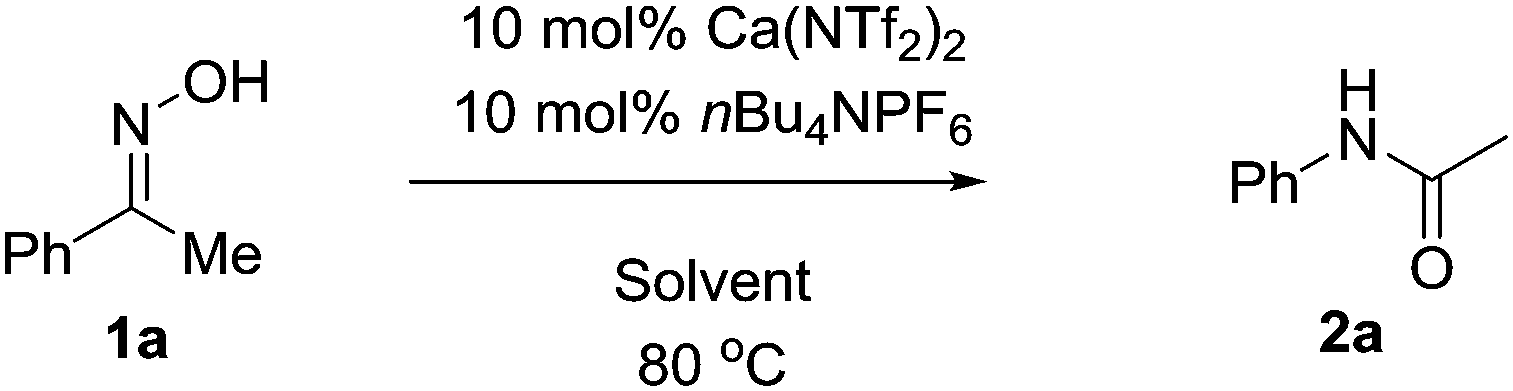
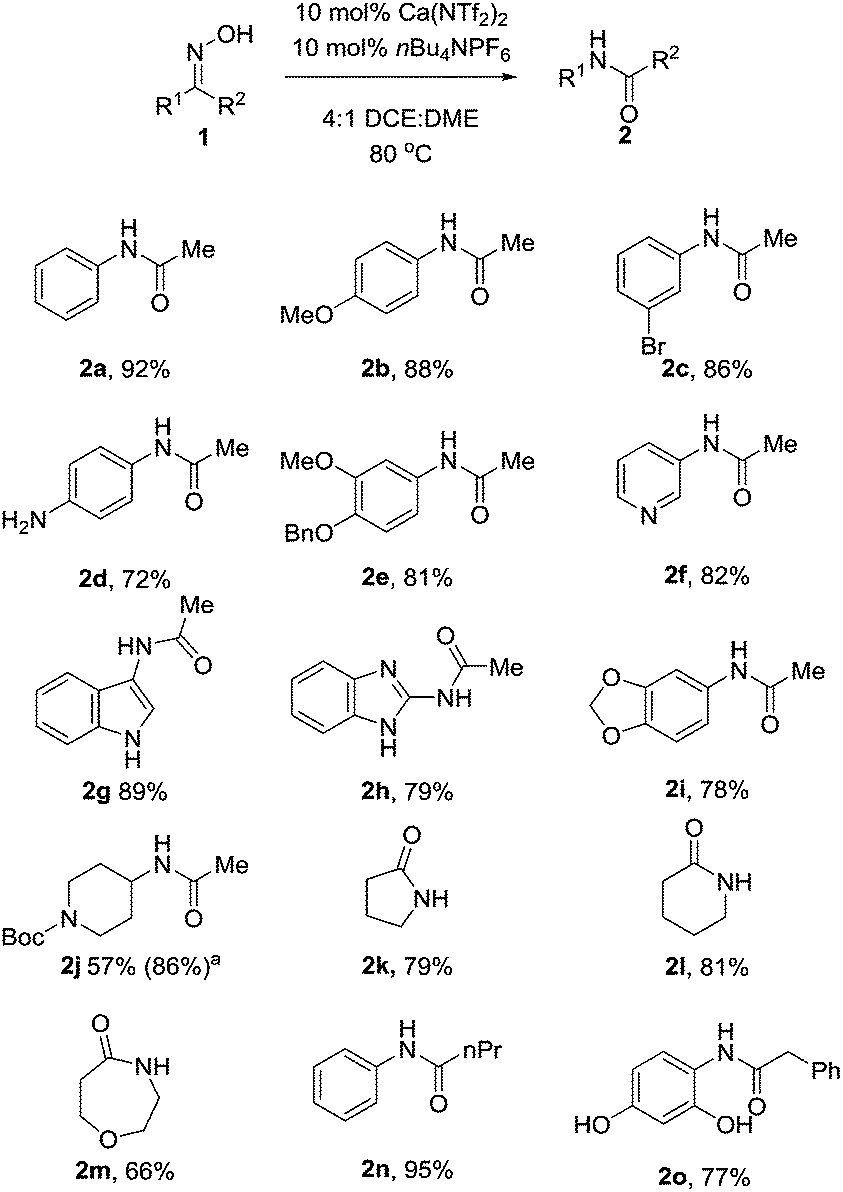
With these optimised conditions in hand, we probed the substrate scope of the reaction (Table 3). Substituted aryl groups underwent the rearrangement to afford the requisite amides in good yield with both electron donating (2b, 2e) and electron withdrawing groups (2c) being well tolerated. Similar to other reports on the Beckmann rearrangement, electron withdrawing groups resulted in a much slower reaction rate. Free amines (2d) also underwent the rearrangement in good yields. Heteroaryls containing basic nitrogen atoms such as pyridine (2f), indole (2g) and benzimidazole (2h) also underwent clean conversion to the product in excellent yields. Additionally, aryl acetal (2i) was also tolerated well, as was Boc-protected piperidine (2j) which underwent the reaction cleanly to afford the desired amide in good yields. This demonstrates the utility of our catalyst system toward acid labile functional groups. Our attention then turned to the cyclic, symmetrical oximes as these can provide ready access to a wide range of medicinally relevant scaffolds. Simple cycloalkyl compounds underwent the rearrangement (2k, 2l), cleanly affording the requisite lactam in high yields. Tetrahydropyranyl oxime also performed well, affording the oxazepanone (2m) in excellent yield. Finally we investigated changing the group at R2, with n-Pr (2n) and benzyl (2o) also cleanly rearranging to the requisite amides in good yield.
| a Reaction performed with 30 mol% catalyst Ca(NTf2)2/nBu4NPF6. All reactions were performed with anhydrous solvents however no precautions were taken to exclude air. |
|---|
|
We next wanted to demonstrate the synthetic utility of calcium catalyst in a complex molecular setting, and chose to perform the rearrangement on the oxime of Prasterone (3) and pregnenolone (5) acetate. Under the optimised conditions, the rearrangement proceeded smoothly furnishing the products in high yield (Scheme 2).
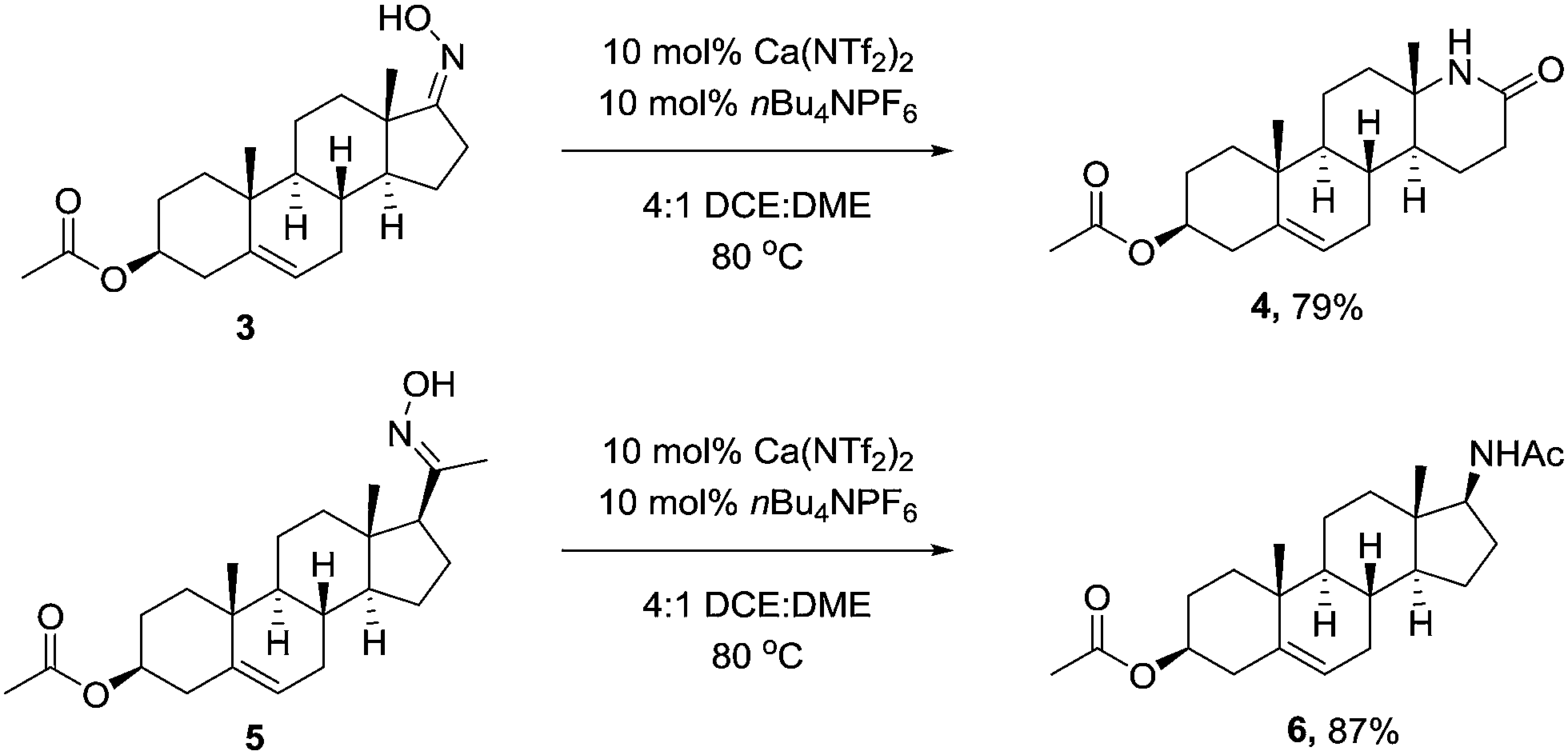 | ||
| Scheme 2 Beckmann rearrangement of prasterone and pregnenolone acetate. | ||
To ensure that our methodology could be applicable in the arena of total synthesis, we chose the well established synthesis of the macrolide antibiotic azithromycin from erythromycin, employing our catalyst system to mediate the key “trapped” Beckmann rearrangement.17 We were intially concerned that the number of hydroxyl groups on the oxime would interfere with the Beckmann, however we reasoned as none of them were benzylic or vinylic, the formation of the carbocation generated would be less favourable than the competing rearrangement. Subjecting erythromycin oxime (7) to the established conditions afforded the desired trapped intermediate (Scheme 3). Azithromycin (9) was then produced via the established reduction (8) and Eschweiler–Clarke methylation protocol.18
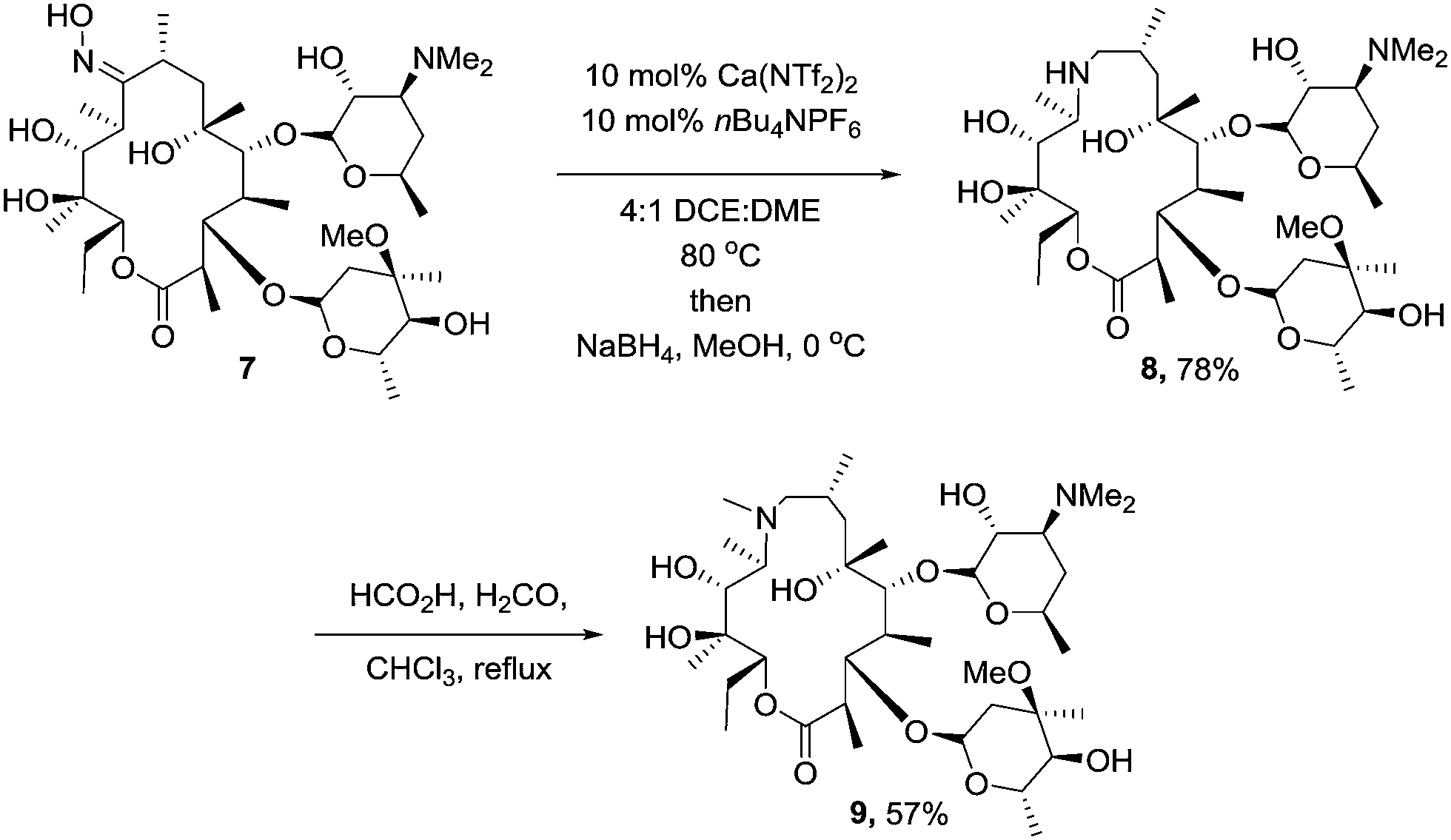 | ||
| Scheme 3 Synthesis of azithromycin via trapped Beckmann. | ||
Finally we turned our attention onto whether the reaction could be performed as a one pot procedure. After screening several inorganic bases (NaOAc, Na2CO3, K2CO3) with little success, we speculated that due to the high tolerance of the calcium catalyst to nitrogen heterocycles, pyridine may be the optimum choice. This proved to be the case, and the reaction proceeded smoothly to afford the amide in excellent yield. As shown, further examples also proved successful (Table 4).
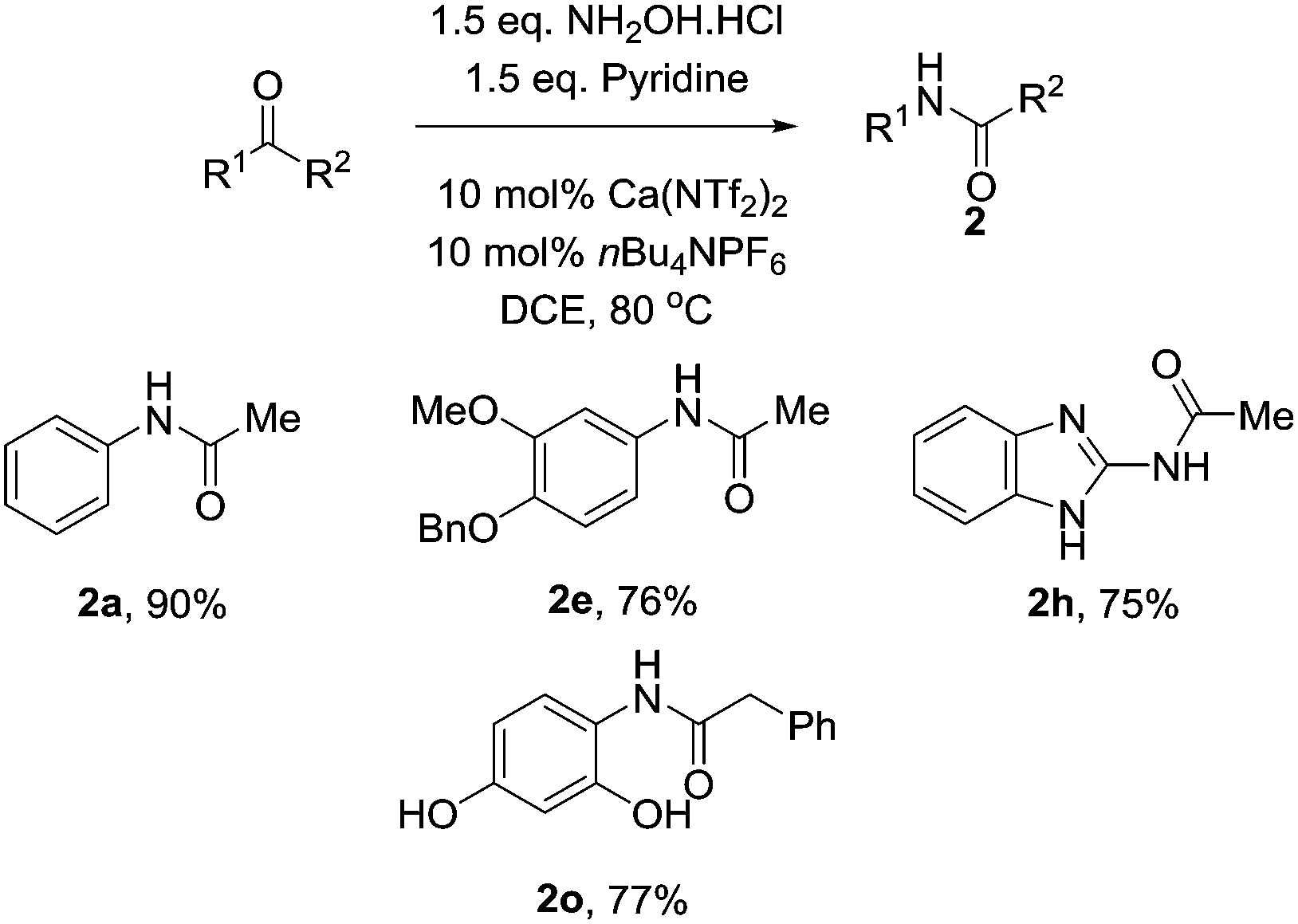
|
The initial mechanistic steps for the Beckmann rearrangements have been argued over and subjected to prolonged investigation over the past 50 years.19 It is generally accepted that after activation and dehydration of the oxime, a reactive nitrillium ion is formed. Consequently, the preformed water attacks this reactive intermediate, which following tautomerization, affords the requisite amide. We wanted to investigate the nature of the incoming nucleophile, and whether it was free water or a calcium-alkoxide type species. We therefore attempted the reaction with 1b, employing 4 Å molecular sieves to remove molecular water from the reaction, and observed full conversion to the amide in 2 hours. Based on this preliminary result, a plausible reaction mechanism is described below (Fig. 1). The calcium catalyst activates the hydroxyl moiety of the oxime which produces transient [HO− Ca2+ PF6−]. This potentially attacks the nitrillium to form intermediate 3a, and following loss of the amide, the active calcium species is regenerated through recomplexation with PF6−.
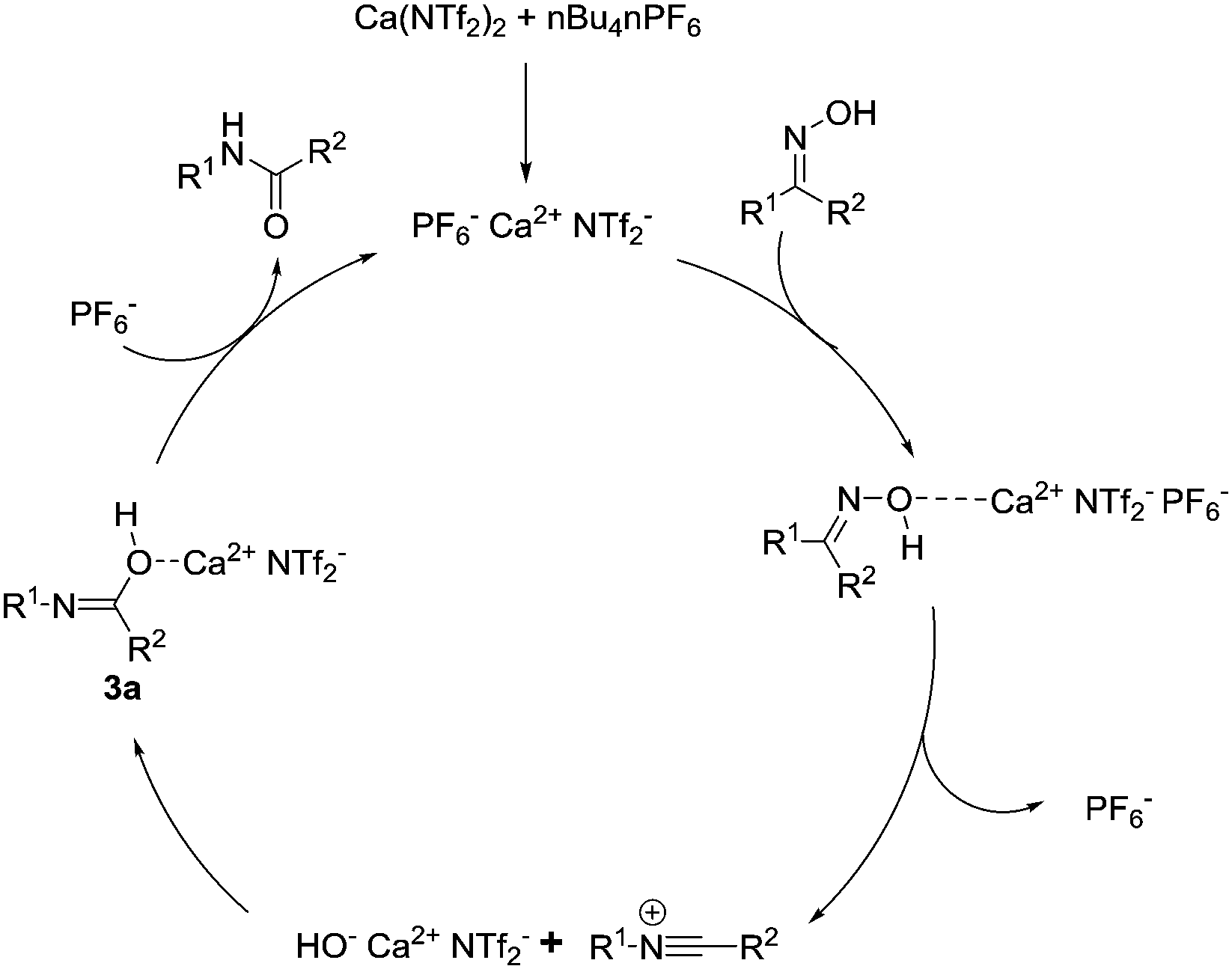 | ||
| Fig. 1 Plausible catalytic pathway. | ||
In conclusion we have developed a mild, catalytic Beckmann rearrangement employing a calcium(II) catalyst. Our system is tolerant of a range of functionalities pertinent to natural product synthesis and medicinal chemistry, as well as allowing the use of acid labile moieties. The synthetic utility of the reaction has been demonstrated, and we show that the reaction is ammenable to one pot. We propose a plausible mechanism based on preliminary investigations, with further detailed studies underway.
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z]. MGM wishes to thank MMU for startup funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. E. Gawley, in Organic Reactions, John Wiley & Sons, Inc., 1988, vol. 35, p. 1 Search PubMed; (b) J. Y. Shin, D. J. Jung and S.-G. Lee, ACS Catal., 2013, 3, 525–528 CrossRef CAS.
- L. De Luca, G. Giacomelli and A. Porcheddu, J. Org. Chem., 2002, 67, 6272–6274 CrossRef CAS PubMed.
- Y. Furuya, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 11240–11241 CrossRef CAS PubMed.
- M. Hashimoto, Y. Obora, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2008, 73, 2894–2897 CrossRef CAS PubMed.
- R. S. Ramón, J. Bosson, S. Díez-González, N. Marion and S. P. Nolan, J. Org. Chem., 2010, 75, 1197–1202 CrossRef PubMed.
- S. Chandrasekhar and K. Gopalaiah, Tetrahedron Lett., 2003, 44, 755–756 CrossRef CAS.
- C. Ramalingan and Y.-T. Park, J. Org. Chem., 2007, 72, 4536–4538 CrossRef CAS PubMed.
- N. C. Ganguly and P. Mondal, Synthesis, 2010, 3705–3709 CrossRef CAS.
- P. S. Mahajan, V. T. Humne, S. D. Tanpure and S. B. Mhaske, Org. Lett., 2016, 18, 3450–3453 CrossRef CAS PubMed.
- V. P. Srivastava, A. K. Yadav and L. D. S. Yadav, Synlett, 2014, 665–670 CAS.
- Y. Yan, Z. Zhang, Y. Wan, G. Zhang, N. Ma and Q. Liu, J. Org. Chem., 2017, 82, 7957–7963 CrossRef CAS PubMed.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- (a) J.-M. Begouin, F. Capitta, X. Wu and M. Niggemann, Org. Lett., 2013, 15, 1370–1373 CrossRef CAS PubMed; (b) S. Gao, T. Stopka and M. Niggemann, Org. Lett., 2015, 17, 5080–5083 CrossRef CAS PubMed; (c) V. J. Meyer, C. Ascheberg and M. Niggemann, Chem. – Eur. J., 2015, 21, 6371–6374 CrossRef CAS PubMed; (d) V. J. Meyer and M. Niggemann, Eur. J. Org. Chem., 2011, 3671–3674 CrossRef CAS; (e) M. Niggemann and M. J. Meel, Angew. Chem., Int. Ed., 2010, 49, 3684–3687 CrossRef CAS PubMed; (f) T. Stopka and M. Niggemann, Org. Lett., 2015, 17, 1437–1440 CrossRef CAS PubMed.
- (a) S. Kobayashi and Y. Yamashita, Acc. Chem. Res., 2011, 44, 58–71 CrossRef CAS PubMed; (b) S. Kobayashi, T. Tsubogo, S. Saito and Y. Yamashita, Org. Lett., 2008, 10, 807–809 CrossRef CAS PubMed; (c) S. Shimizu, T. Tsubogo, P. Xu and S. Kobayashi, Org. Lett., 2015, 17, 2006–2009 CrossRef CAS PubMed; (d) T. Tsubogo, Y. Kano, K. Ikemoto, Y. Yamashita and S. Kobayashi, Tetrahedron: Asymmetry, 2010, 21, 1221–1225 CrossRef CAS; (e) T. Tsubogo, S. Saito, K. Seki, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 13321–13332 CrossRef CAS PubMed; (f) T. Tsubogo, Y. Yamashita and S. Kobayashi, Angew. Chem., Int. Ed., 2009, 48, 9117–9120 CrossRef CAS PubMed; (g) T. Tsubogo, Y. Yamashita and S. Kobayashi, Top. Catal., 2014, 57, 935–939 CrossRef CAS.
- (a) M. C. Martin, M. J. Sandridge, C. W. Williams, Z. A. Francis and S. France, Tetrahedron, 2017, 73, 4093 CrossRef CAS; (b) M. J. Sandridge and S. France, Org. Lett., 2016, 18, 4218–4221 CrossRef CAS PubMed.
- S. P. Morcillo, M. Presset, S. Floquet, V. Coeffard, C. Greck, C. Bour and V. Gandon, Eur. J. Org. Chem., 2016, 2688–2694 CrossRef CAS.
- B. V. Yang, M. Goldsmith and J. P. Rizzi, Tetrahedron Lett., 1994, 35, 3025–3028 CrossRef CAS.
- R. R. Wilkening, R. W. Ratcliffe, G. A. Doss, R. T. Mosley and R. G. Ball, Tetrahedron, 1997, 53, 16923–16944 CrossRef CAS.
- (a) B.-X. Tian, N. An, W.-P. Deng and L. A. Eriksson, J. Org. Chem., 2013, 78, 6782–6785 CrossRef CAS PubMed; (b) R. Akimoto, T. Tokugawa, Y. Yamamoto and H. Yamataka, J. Org. Chem., 2012, 77, 4073–4078 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c7cc09491d |
| This journal is © The Royal Society of Chemistry 2018 |