
A stable free tetragermacyclobutadiene incorporating fused-ring bulky EMind groups†

Abstract
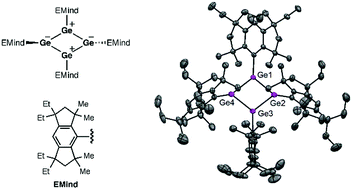
The first free cyclobutadiene (CBD) germanium analogue was obtained as room-temperature stable dark red crystals via the reaction of the bulky EMind-substituted 1,2-dichlorodigermene with lithium naphthalenide. The cyclic 4π-electron antiaromaticity is essentially stabilized by the polar Jahn–Teller distortion in the germanium CBD producing a planar rhombic-shaped charge-separated structure.
 Please wait while we load your content...
Please wait while we load your content...

